

Abstract
There is an increasing need to control the type, quantity, and timing of growth factors released during tissue healing. Sophisticated delivery systems offering the ability to deliver multiple growth factors with independently tunable kinetics are highly desirable. Here, a multilayered, mineral coated micro-particle (MCMs) platform that can serve as an adaptable dual growth factor delivery system is developed. Bone morphogenetic protein-2 (BMP-2) and vascular endothelial growth factor (VEGF) are bound to the mineral coatings with high binding efficiencies of up to 80%. BMP-2 is firstly bound onto a 1st mineral coating layer; then VEGF is bound onto a 2nd mineral coating layer. The release of BMP-2 is sustained over a period of 50 days while the release of VEGF is a typical two-phase release with rapid release in the first 14 days and more sustained release for the following 36 days. Notably, the release behaviors of both growth factors can be independently tailored by changing the intrinsic properties of the mineral coatings. Furthermore, the release of BMP-2 can be tuned by changing the thickness of the 2nd layer. This injectable microparticle based delivery platform with tunable growth factor release has immense potential for applications in tissue engineering and regenerative medicine.
1. Introduction
Large bone defect healing remains a significant clinical challenge worldwide due to the bone loss caused by traumatic injury, tumor resection, osteonecrosis, and infection.[1] Due to the limitations associated with conventional approaches such as autografts and allografts, tissue engineering strategies inspired by endogenous bone healing mechanisms have recently attracted more attention.[2-5] Osteogenesis and angiogenesis are considered to be equally important to a tissue engineering bone regeneration strategy since bone is a highly vascularized and mineralized tissue.[6,7] Insufficient angiogenesis during bone regeneration results in poor and unsustainable bone formation. Thus, both osteogenic and angiogenic growth factors have been used to promote these processes in order to enhance vascularized bone tissue formation. Bone morphogenetic protein-2 (BMP-2) has been identified as a potent osteogenic growth factor to induce bone formation. For example, BMP-2 has been shown to directly induce bone formation in both ectopic and orthotopic sites.[8,9] On the other hand, vascular endothelial growth factor (VEGF) is extensively used as a pro-angiogenic growth factor to enhance vascularization with bone defects.[10,11] Importantly, recent studies have demonstrated that sequential delivery of BMP-2 and VEGF could maximize their efficacy by inducing cooperative, synergistic effects that better mimic natural bone healing.[12,13] However, there remains a need for sophisticated delivery systems capable of precisely tuning the individual release kinetics of these growth factors.
The prevalence of growth factor signaling during wound healing has led to novel strategies to deliver growth factors to various tissues.[14,15] Bolus delivery of a single growth factor via direct injection is still a common approach used to regenerate a variety of tissues, but the success of this approach is limited to early animal studies and no significant efficacy in terms of new bone tissue formation has been achieved.[16,17] Various sustained growth factor delivery strategies have been developed to bypass the limitation of bolus delivery and provide localization of growth factors at the desired defect sites for extended timeframes.[18] Polymer sponges (e.g., collagen sponges), hydrogels, micro/nano particles, and thin films have been employed to successfully deliver growth factors in a sustained manner.[19-23] Controlled release of growth factors from these formulations has demonstrated promising outcomes in various aspects of tissue regeneration, such as preserving some protein activity, regulating stem cell behavior and promoting new tissue growth.[24,25] For instance, sustained release of BMP-2 has greatly enhanced its efficacy and improved its in vivo performance by localizing the morphogenetic stimulus.[26-28]
Although sustained release approaches have had an impact on the application of growth factors in biomedical applications, they often fail to mimic key characteristics of the natural bone healing process. For example, most current delivery systems have been limited to the controlled release of a single growth factor, even though spatiotemporal presentation of multiple growth factors is a hallmark of endogenous bone healing.[29] Recent studies have used advanced, multi-component materials processing strategies to demonstrate that delivery of multiple growth factors simultaneously can have synergistic effects, and enhance bone tissue regeneration.[30,31] Unfortunately, little work has been done to develop delivery systems that are capable of controlling the individual timing of different growth factors. Therefore, there is an increasing need to control the identity, quantity and timing of growth factors released during bone healing. Meanwhile, the development of these delivery systems also has to be simple and amenable to clinical translation in order to benefit patients in the short term.
We have developed a facile approach to bind growth factors onto injectable mineral coated microparticles for sustained release (Figure 1). More recently, we explored a series of approaches to control release kinetics of multiple proteins. Mechanisms for dual protein release have included controlling protein-mineral affinity, and creating multilayered mineral coatings on biomaterials.[32] Here we focus on multilayered, mineral coated microparticles (MCMs) for dual growth factor delivery. In order to mimic the vascularized bone regeneration process, we chose to release VEGF in a faster manner to promote angiogenesis at an early stage while we managed to release BMP-2 in a more sustained manner to drive osteogenesis during the bone healing. We hypothesized that BMP-2 and VEGF, bound on different mineral coating layers, would have distinct release kinetics from mineral coatings. Moreover, we reasoned that the release behaviors of both growth factors could be further tailored by changing the intrinsic properties (e.g., dissolution kinetics, thickness) of the mineral coatings.
Figure 1.
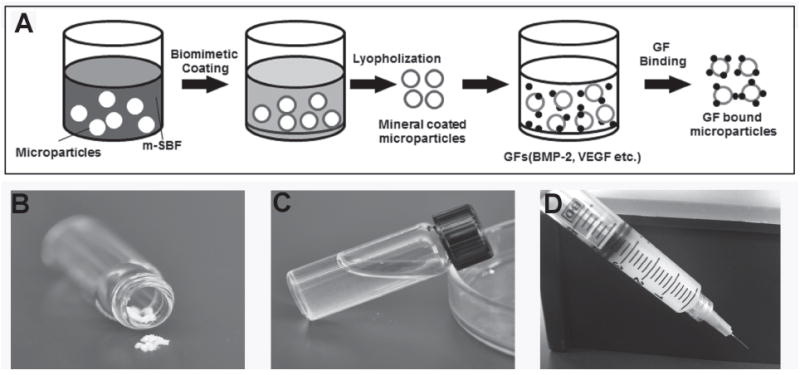
Growth factor binding on MCMs: A) Schematic of strategy for growth factor on MCMs. B) Lyophilized MCMs. C) MCMs resuspended in PBS. D) Injection of MCMs through a standard syringe.
2. Results
2.1. Control over BMP-2 Release by Accelerating Mineral Coating Dissolution
MCMs were uniformly covered by a layer of mineral coating with typical plate-like structure after incubating with mSBF for 7 days (Figure 2). The morphology of the mineral coatings was clearly influenced by the concentration of HCO3− in mSBF. As we increased the carbonate concentration in the mSBF from 4.2 mm (Low HCO3−), 25 mm (Mid HCO3−), to 100 mm (High HCO3−), the size of the nano-scale plates decreased from ≈1 μm to ≈100 nm (Figure 2B,D,F). The composition of the mineral coating was confirmed to be carbonate-substituted hydroxyapatite by XRD and FTIR (Figure S1, Supporting Information). Importantly, the dissolution of the mineral coatings was closely associated with the carbonate concentration in mSBF. The dissolution rate of the mineral coatings in TBS, which was characterized by the release of calcium ion, was proportional to the initial carbonate concentration in the mSBF during coating formation (Figure 2G).
Figure 2.
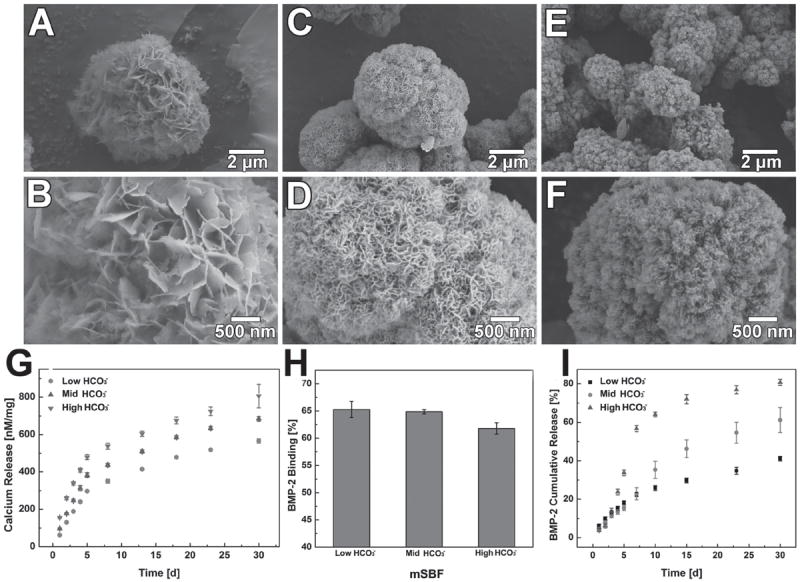
Morphology of MCMs with different carbonate concentrations: Low magnification: A) 4.2 mm (Low HCO3−), C) 25 mm (Mid HCO3−), E) 100 mm (High HCO3−). High magnification: B) 4.2 mm (Low HCO3−), D) 25 mm (Mid HCO3−), F) 100 mm (High HCO3−). G) Release of calcium ion from mineral coating with different carbonate concentrations. H) Binding efficiency of BMP-2 on MCMs with different carbonate concentrations I) BMP-2 release profiles from MCMs with varying carbonate concentrations.
BMP-2 was efficiently bound to MCMs via incubation in BMP-2-containing PBS. The amount of bound BMP-2 on the MCMs increased linearly as the concentration of BMP-2 in PBS increased from 100 to 1000 ng mL-1 (Figure S2, Supporting Information). The binding efficiencies of BMP-2 to mineral coated MCMs were 65% (±1%), 65% (±1%), and 62% (±2%) for mineral coatings with low, medium and high carbonate substitution (Figure 2H). Sustained release of BMP-2 was observed from the MCMs for over 30 days. Noticeably, the release kinetics of BMP-2 were strongly dependent on the carbonate content in the mineral coating, and were therefore readily controllable. 56% (±2%) of the BMP-2 was released in a relatively fast rate within the first seven days for the High HCO3− group while this amount decreased to 23% (±3%) and 22% (±2%) in Low and Mid HCO3− groups. By day 30, the percentage of released BMP-2 varied from 41% (±1%) in the Low HCO3− condition to 80% (±2%) in the High HCO3− condition (Figure 2I). Thus, the release of BMP-2 from the mineral coated microparticles could be modulated by destabilizing the mineral coating via carbonate substitution.
2.2. Control over BMP-2 Release by Decelerating Mineral Coating Dissolution
To decelerate the mineral coating dissolution on MCMs, fluoride was chosen to be incorporated into the mineral since fluoridated apatite has previously shown lower solubility compared to hydroxyapatite in various solution environments.[33] 1.0 mm sodium fluoride was added into three different mSBF systems: Low HCO3− (4.2 mm), High HCO3− (100 mm), and No magnesium (No Mg2+) (Detailed mSBFs formulation shown in Table S-1, Supporting Information). Both the morphology and stability of the mineral coatings was substantially affected by the incorporation of fluoride (Figure 3). Instead of forming the typical plate-like nanostructure, the fluoride-doped coatings formed in each of the three groups showed a needle-like structure (Figure 3A–F), which is a typical fluoridated apatite morphology.[34] The width of the needle-like structure was about 100 nm. The addition of fluoride significantly decelerated the dissolution of the mineral coating. In fluoride-containing Low HCO3− and No Mg2+ groups, the release of calcium from the coating in TBS decreased by 52% and 67%, respectively, when compared to fluoride-free conditions (Figure 3G,I). In the case of the fluoride-containing High HCO3− group, this impact was not as prominent (Figure 3H), likely due to the extremely high carbonate concentration in the system and multiple types of carbonate substitution (both type A and B) during mineral coating formation.[35]
Figure 3.
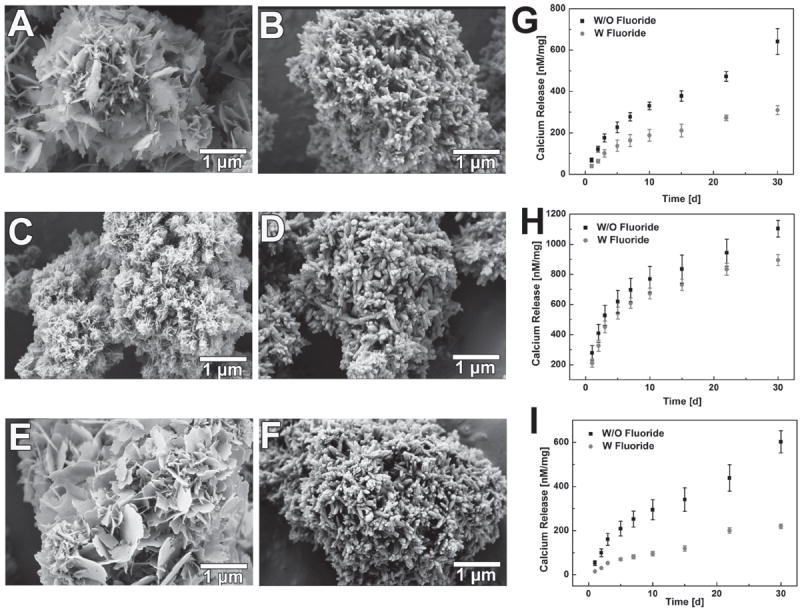
Impact of fluoride incorporation on mineral coating morphology and dissolution kinetics: SEM images of mineral coating without fluoride: A) 4. 2 mm (Low HCO3−), C) 100 mm (High HCO3−), E) No Mg2+; SEM images of mineral coating with fluoride: B) 4. 2 mm (Low HCO3−), D) 100 mm (High HCO3−), F) No Mg2+; Dissolution kinetics of mineral coating in term of calcium ion release: G) 4. 2 mm (Low HCO3−), H) 100 mm (High HCO3−), I) No Mg2+.
Incorporation of fluoride into the mineral coating substantially affected the binding efficiency of BMP-2. Compared to the mineral coatings without fluoride incorporation (Figure 4A,C,E), the binding efficiency of BMP-2 decreased in each type of fluoride-doped mineral coating. In the fluoride-containing Low HCO3− group, BMP-2 binding efficiency dropped from 66% (±3%) to 54% (±0.6%) when compared to the fluoride-free condition (Figure 4A). The fluoride-containing High HCO3− and no Mg2+ groups also showed decreases in the BMP-2 binding efficiency when compared to the fluoride-free groups (Figure 4C,E). The release profiles of BMP-2 from the mineral coatings were also impacted by the incorporation of fluoride into the coatings (Figure 4B,D,F). The release percentage of BMP-2 after 30 days from the fluoride-free Low HCO3− coating was 41% (±0.5%), while the release percentage from fluoride-doped Low HCO3− coatings was only 28% (±1%) (Figure 4B). Similar results were also obtained for the No Mg2+ group (Figure 4F). However, the incorporation of fluoride in the High HCO3− group did not show a significant impact on BMP-2 release (Figure 4D). This result also matches with the aforementioned coating stability test, in which fluoride did not influence the stability of the High HCO3− mineral coatings. In summary, binding and release of BMP-2 could be regulated by decelerating the coating dissolution by the addition of fluoride into the mineral coatings.
Figure 4.
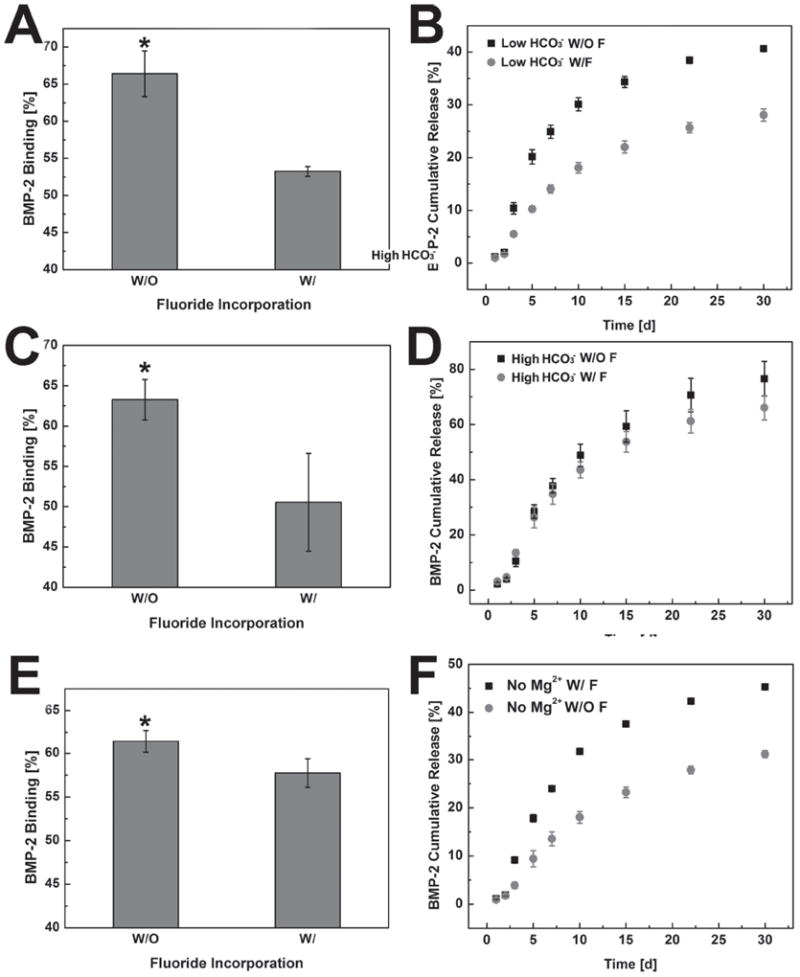
Impact of fluoride incorporation on BMP-2 binding and release from MCMs: Binding efficiency of BMP-2 on MCMs with/without fluoride: A) 4. 2 mm (Low HCO3−), C) 100 mm (High HCO3−), E) No Mg2+; BMP-2 release profiles from MCMs with/without fluoride: B) 4. 2 mm (Low HCO3−), D) 100 mm (High HCO3−), F) No Mg2+. * represents significant difference compared to fluoride-containing conditions.
2.3. Binding and Release of Two Distinct Growth Factors from Multilayered MCMs
The distinct protein release kinetics from mineral coatings with different degradation kinetics allowed for development of a controllable dual growth factor delivery system. Figure 5 shows a schematic illustration of a VEGF/BMP-2 dual growth factor delivery system using multilayered MCMs, which allows for relatively fast release of VEGF and more sustained release of BMP-2. By changing the formulation of the mSBF used to form the multilayered mineral coatings, we obtained mineral coatings with controllable morphology on layer 1 (“L1”) and layer 2 (“L2”). A plate-like nanostructure was observed (Figure 6A) when high carbonate mSBF was used, and typical needle-shaped morphology was obtained when fluoride was incorporated (Figure 6B). These morphologies were consistent with the morphologies observed for the corresponding single layer coatings described above (Figures 2,3). This kind of coating morphology was also switchable between L1 and L2 depending on the intended growth factor release kinetics. As shown in Figure 6A, when we switch coating formation condition for L1 and L2, coatings with needle-like structure were obtained for L1 (Figure 6C) while coatings with plate-like structure were obtained for L2 (Figure 6D).
Figure 5.
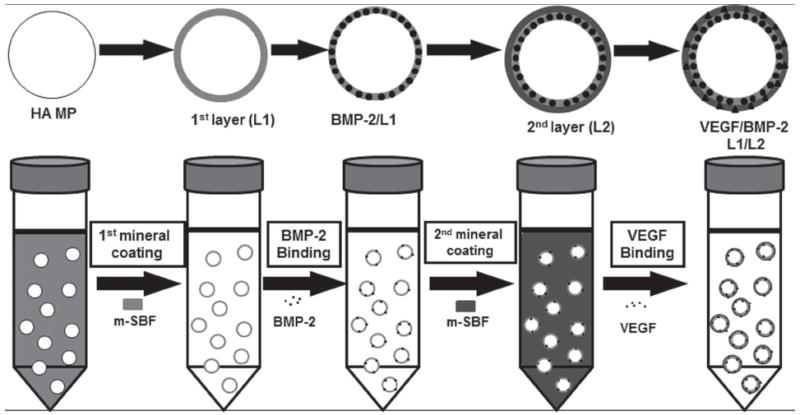
Schematic of dual growth factor delivery system using multilayered MCMs. HA MPs were coated with a layer of mineral, then BMP-2 was bound on the 1st layer mineral; the 2nd layer of mineral coating was formed on top of the 1st layer by incubating the HA MPs in mSBF for 7 days; then VEGF was bound on the 2nd layer of mineral coating.
Figure 6.
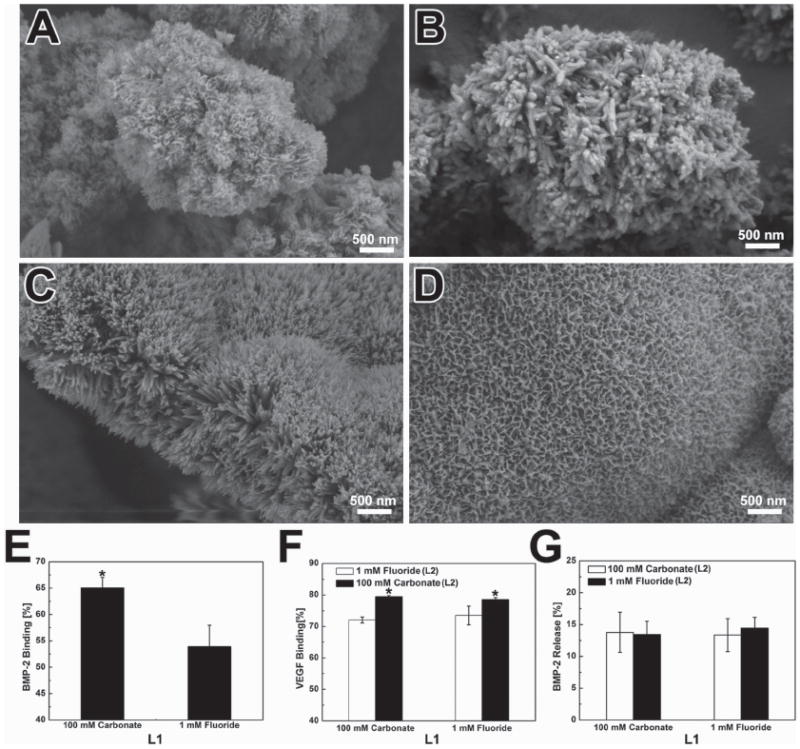
Growth factor binding on multilayered MCMs: SEM image of individual mineral coating layer: A) 100 mm carbonate (L1), B) 1.0 mm fluoride (L1), C) 1.0 mm fluoride (L2), D) 100 mm carbonate (L2); E) BMP-2 binding on L1 of MCMs. F) VEGF binding on L2 of MCMs. G) Loss of BMP-2 during the formation of L2.
The binding of growth factors in the dual delivery system was very similar to the protein binding in the single delivery system. BMP-2 binding on fluoride-incorporated mineral coatings was lower than high carbonate mineral coatings (53 ± 2%) versus (65 ± 4%) (Figure 6E). A similar trend was also observed when VEGF was bound on L2. Specifically, the binding efficiency of VEGF on fluoride-containing coatings was 72% (±1%) while it rose to 79% (±1%) on high carbonate coatings. In addition, the binding efficiency of VEGF on L2 (Figure 6F) was higher than on the single layer mineral coatings (Figure S4, Supporting Information), which is perhaps due to the increase of surface area after the second layer of mineral coating. Only 12% (±3%) of the bound BMP-2 was lost during the formation of second mineral layer, and this minimal loss is attributable to the initial release of this protein during the second mSBF incubation (Figure 6G).
Two distinct release profiles of the incorporated growth factors were observed. VEGF bound on the second layer showed a typical two-phase release profile with fast release over the first two weeks and more sustained release over the next five weeks. The release of BMP-2 bound on the first layer exhibited near zero-order release kinetics with no “burst” release (Figure 7). Furthermore, the release of BMP-2 could be tuned by changing the dissolution kinetics of both layers of the coating. When we switched L1 from high carbonate (high dissolution rate) to fluoride-doped (low dissolution rate), the release of BMP-2 was decreased by about 5% (±2) regardless of the composition of L2. The composition of L2 layer had a more profound impact on BMP-2 release. When we switched L2 from high carbonate to fluoride-doped, the release of BMP-2 dropped by almost 2-fold (from 32 ± 2% to 13% ± 2%, respectively). Similar effects of the second layer on BMP-2 release results were observed whether the first layer was high carbonate or fluoride-doped (Figure 7A,B).
Figure 7.
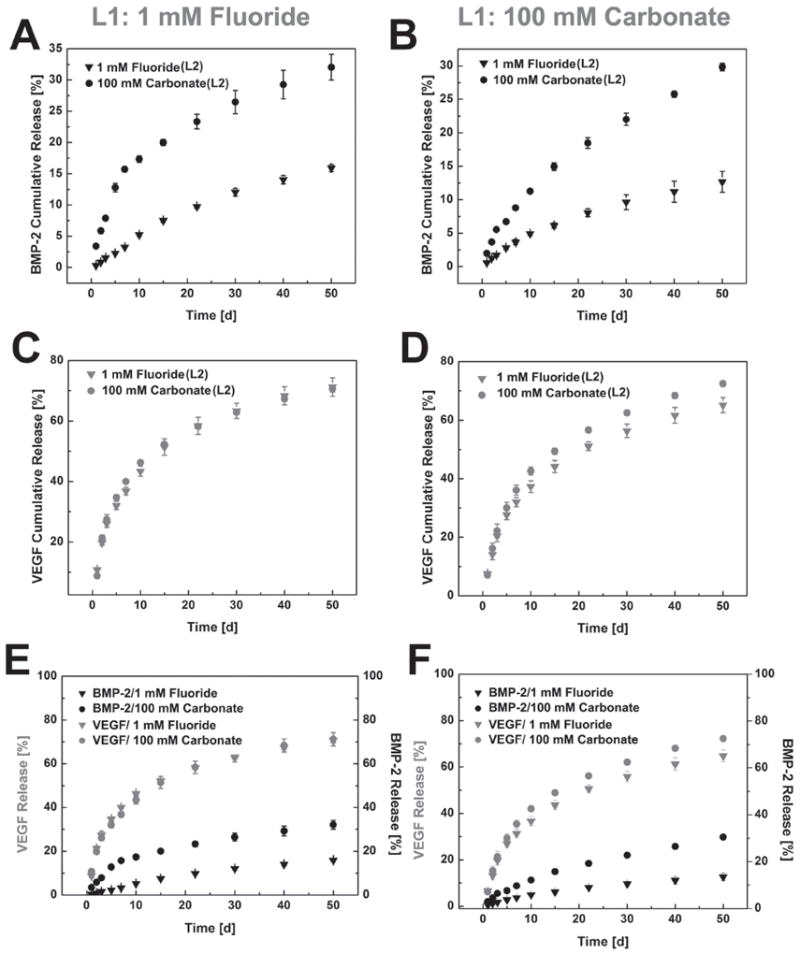
Dual growth factor release from multilayered MCMs: 1 mm fluoride mineral coating as L1: A) BMP-2 release from L1, C) VEGF release from L2, E) BMP-2/VEGF dual release; 100 mm carbonate mineral coating as L1: B) BMP-2 release from L1, D) VEGF release from L2, F) BMP-2/VEGF dual release; For each condition, both 1 mm fluoride and 100 mm carbonate condition were tested as the 2nd layer mineral coating.
In contrast, the release of VEGF from L2 was not significantly affected by the change in composition of the coating. Neither the compositional change of L1 nor L2 had a significant impact on VEGF release (Figure 7C&D). Notably, at the end of the 50 day release period, 72% (±2) of the initially bound VEGF was released, while only 12% (±2) of the BMP-2 was released (Figure 7E,F). Growth of the 2nd layer further eliminated the small burst release of BMP-2, and allowed for relatively fast release of VEGF during the initial stages of delivery. These dual release results showed precise yet controllable delivery of multiple growth factors using multilayered, mineral-coated microparticles.
2.4. Dual Growth Factor Release from Coatings of Varying Thickness
We next investigated the influence of the thickness of L2 mineral layer on growth factor release, as the creation of a 2nd layer had an obvious impact on growth factor release. We hypothesized that the nanostructure created during the formation of L2 could serve as a barrier during the release of protein from L1, thus a thicker 2nd layer would result in slower release of growth factor (Figure 8A). To address this hypothesis, the thickness of L2 was controlled by changing the coating time in mSBF. After the initial binding of BMP-2 on L1, the BMP-2-loaded MCMs were incubated in low carbonate mSBF for 0, 0.5, and 5 days respectively to obtain L2s with different thicknesses. SEM observation showed an increase of thickness of L2 as the incubation time was prolonged (Figure 8B). The binding efficiency of BMP-2 on L1 did not change, as expected since the binding occurred before the formation of L2. However, the binding efficiency of VEGF on the 2nd layer increased with increased coating time, which is likely due to the increased surface area after the application of the 2nd layer (Figure 8C,E).
Figure 8.
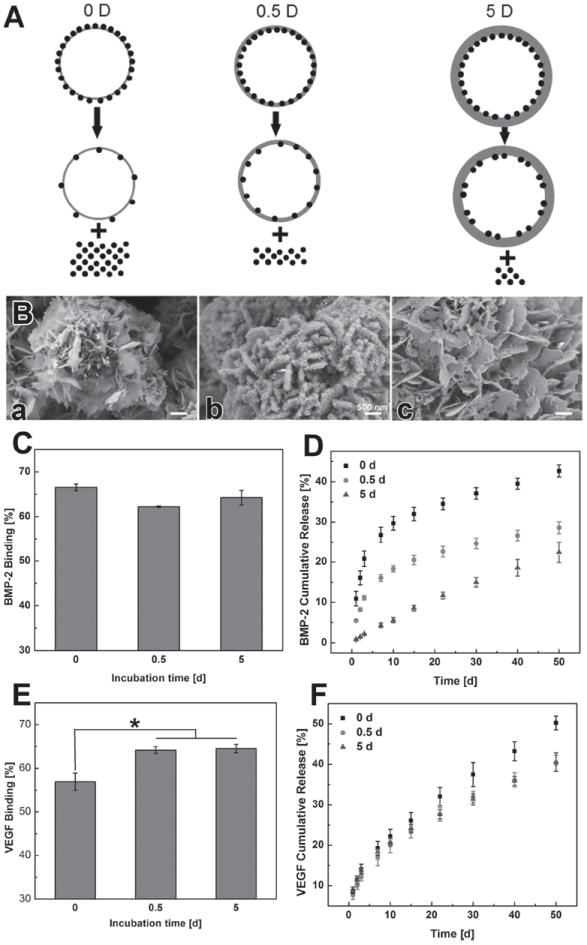
Impact of mineral coating thickness on dual growth factor release from multilayered MCMs: A) schematic of growth factor release from mineral coating with different thickness. B) SEM image of L2 with different thicknesses (different incubation time). C) BMP-2 binding on L1. D) BMP-2 release from multilayered MCMs with different thickness L2. E) VEGF 2 binding on L2 with different thicknesses. F) VEGF release from L2 with different thicknesses.
Release of BMP-2 from L1 heavily relied on the thickness of L2. When there was no 2nd layer on MCMs (“0 d” condition), the release profile of BMP-2 was a typical two-phase release profile (Figure 8D), which is consistent with the results observed in the single growth factor release study (Figure 2J). However, the release of BMP-2 was slowed after applying the 2nd coating layer even for the shortest timeframe studied (0.5 d). Specifically, the overall release of BMP-2 dropped from 42% (±2%) to 29% (±2%) after the formation of the 0.5 d 2nd coating layer. Importantly, when L2 was formed on top of L1 for a longer timeframe (5 d), the release of BMP-2 showed near zero-order release kinetics without showing any initial burst (Figure 8D). The total BMP-2 release decreased to 22% (±3%) for the thickest (5 d) L2 condition when compared with 0 d and 0.5 d L2 groups. In contrast, no significant change of VEGF release from L2 was observed when we tuned the coating time for L2 formation (Figure 8F).
3. Discussion
In this study, we developed multilayered, mineral coated microparticles that can serve as an adaptable dual growth factor delivery system. Two distinct growth factors could be released with controllable kinetics taking advantage of the intrinsic properties of the mineral coating layers. In particular, the release of the growth factors was controlled by binding on different mineral coating layers with tunable dissolution kinetics. Distinct release profiles of two different growth factors (BMP-2/VEGF) could be obtained when these growth factors were bound on different layers of mineral coating (Figures 7,8D,F). Moreover, the release rate of these growth factors could be further tailored by changing the intrinsic properties of the coating on each layer. There is an increasing need to develop sophisticated systems for multiple growth factor release to orchestrate the complicated cellular activities during tissue regeneration. Recent studies have addressed this need by encapsulating multiple proteins into hierarchically organized polymer carriers (scaffolds, microspheres, etc.), which often include rapidly dissolving and slowly dissolving materials in distinct locations within the carrier.[15,36-39] The binding of multiple proteins to mineral coatings, described here, provides a relatively simple and efficient way to deliver growth factors with temporal control over release kinetics. The adaptability of mineral coating synthesis allows us to define relationships between the structure of the mineral layer and its function in controllable growth factor release. This feature results in high levels of control over growth factor release kinetics. The injectable formulation of these mineral-coated microparticles may also allow for minimally invasive administration in future therapeutic applications.
Protein binding on mineral coatings proceeds through electrostatic interactions between calcium phosphate mineral surfaces within the nano-structured coating and the side chains on proteins.[40-43] The highly nano-porous structure of the mineral coatings allowed for efficient protein binding due to the large surface area provided by the plate-like or needle-like nano-structure.[44] In particular, the binding efficiencies of BMP-2 and VEGF obtained by this approach were 65% (±1%) and 79% (±1%) respectively (Figures 4,6). The encapsulation efficiencies of most particulate drug delivery systems are below 70%, so the binding efficiencies here are comparable to optimal drug delivery systems formed using other techniques.[45-47] High binding efficiency is critical for growth factor delivery, as in vivo applications typically require supraphysiological dosages to achieve efficacy while the cost of growth factors is extremely high (usually over $5000 mg−1).[48] For example, clinical approaches that use collagen scaffolds to deliver BMP-2 require milligram quantities of BMP-2, which is six orders of magnitude higher than the amount of BMP-2 present in a typical healing bone tissue.[49] This example and others indicate that the efficiency of protein incorporation is a critical parameter in growth factor delivery strategies. Additionally, many other approaches expose growth factors to several processing and storage-related stresses, which can result in significant loss of protein activity.[50] The mineral binding approach only involves simple buffer incubation, and we have shown previously that the bioactivity of biological molecules (e.g., growth factors) is not significantly compromised during binding and release.[50-53]
It is noteworthy that the technique described here can be easily adapted to other microparticle platforms, such as polyester microspheres, alginate beads, and chitosan microspheres.[54,55] For some of these substrates (e.g., alginate,[56] mineral coatings similar to those described here can be formed in an identical manner to our current study. In other cases (e.g., polyester microspheres,[21,57] a one-step surface modification such as alkaline treatment allows for surface functionalization, which in turn enables mineral coating nucleation and growth. For example, Jongpaiboonkit et al. formed mineral coatings on poly(lactide-co-glycolide) (PLG) microspheres and achieved efficient protein binding and sustained release.[21,57] Our data also indicate that the size of microparticle did not significantly change after the formation of the 1st layer mineral coating (Figure S3, Supporting Information), and the mineral-coated microparticles could still be uniformly re-suspended in aqueous solution after growth factor incorporation. This characteristic may allow growth factor-loaded microparticles to be directly injected into defect sites to provide localized growth factor delivery.[58] Therefore, our dual growth factor deliver strategy offers simplicity, adaptability and injectability.
Growth factor release from the MCMs was directly regulated by the mineral dissolution rate, and this could be an important feature to tune the release kinetics of various growth factors. Two approaches were explored here to customize mineral dissolution: i) substitution of carbonate in the mineral coating to accelerate coating dissolution; and ii) addition of fluoride in the mineral coating to decelerate coating dissolution. Carbonate and fluoride incorporation impacted the coating morphology observed by SEM as well as the degradation rate of the coatings (Figure 2, 3). These observations are consistent with previous reports that substitution of carbonate and fluoride in hydroxyapatite can influence crystal lattice structure and effect the dissolution rate.[59-61] Earlier studies from our group have also shown that the release of protein from calcium phosphate based mineral coatings is dependent on mineral dissolution.[19,62] Here, our data show that the release of BMP-2 closely correlated with the mineral coating dissolution rate. More specifically, the release of BMP-2 was proportional to the carbonate content used in mSBF, as 80% (±2) of bound BMP-2 was released from coatings formed in high carbonate mSBF after 30 days of release (Figure 2J). In contrast, the total release of BMP-2 from fluoride-doped mineral coatings was as low as 28% (±1%) after 30 days of release (Figure 4B). The ability to tailor BMP-2 release dosage and kinetics over a broad range has potential in bone tissue regeneration, since it allows the release system to meet desired orthobiologic needs for clinical applications.[25]
The multilayered mineral coating strategy allowed the binding of different growth factors on different coating layers, followed by release in an independent yet controllable manner. The realization of BMP-2/VEGF dual growth factor release likely relied on two key variables. One variable is the binding location of different growth factors, which determines the release pathway of the growth factors as they leave the mineral coating. Our data showed that release of BMP-2 from 1st (inner) layer was much slower than release from the 2nd (outer) layer. While a minor “burst release” of BMP-2 could still be observed as it was released directly from the 1st layer, it was completely eliminated after the formation of the 2nd (outer) layer (Figure 8D). The more sustained release of BMP-2 could be attributed to the longer transport pathway, as BMP-2 molecules would either need to traverse multiple layers of mineral nano-structure during transport out of the microparticles, or be liberated till the 2nd layer is dissolved. This mechanism is further supported by release studies in which the thickness of the 2nd mineral layer was varied, and a thicker 2nd layer resulted in much slower BMP-2 release (Figure 8D). A second key variable influencing growth factor release was the dissolution rate of the mineral coating, which was strictly controlled by the substitution of carbonate or fluoride during the formation of the coatings. The total BMP-2 amount released from fluoride-doped coatings was less than half of the amount released from the highly carbonate-substituted coatings after 50 days (Figure 4). The switch from carbonate-substituted to fluoride-doped coatings also helped to reduce the initial burst release. The combination of these two variables allows precise control over the release of multiple growth factors, and this capability could be useful in applications where precise presentation of multiple proteins is required at distinct stages of tissue healing.[48,63]
The release profiles of dual growth factors demonstrated in our study could be valuable in the context of vascularized bone regeneration. The appropriate development of blood vessels in bone defects is an essential components of bone healing.[6] The vasculature transports nutrients, oxygen, and soluble factors which make it possible for osteoprogenitor and other related cell types to survive, proliferate, and differentiate.[64] Pro-angiogenic growth factors such as VEGF peak earlier than osteogenic growth factors like BMP-2 during normal bone healing.[65-67] Thus, a temporal release pattern of growth factors mimicing their natural expression might be favorable for the regeneration of bone tissue. In our study, we released 72% (±2) of the VEGF while only 12% (±2) of the BMP-2 was liberated during the same time period (Figure 6). This release result mimics the expression pattern of these two growth factors during natural bone healing.[68] We also showed our capability to further tailor the release kinetics of dual growth factors by manipulating the intrinsic properties of the multilayered coating system. These findings might lead to new delivery systems fitting into different needs for bone tissue engineering such as intramembranous ossification and endochondral ossification.[69]
4. Conclusions
Sophisticated protein delivery systems offering the ability to deliver multiple growth factors with distinct release kinetics in a tunable manner are desirable in the field of tissue engineering and regenerative medicine. A multilayered, mineral coated microparticle platform was developed that can serve as an adaptable growth factor delivery system. Growth factors could be released with controllable kinetics based on the intrinsic properties of the mineral coating layers. Controlled release features that were achieved by varying coating properties included: i) elimination of burst release, ii) control over the release kinetics of single growth factors, and iii) independently tunable control over release of two growth factors from the same delivery system. This injectable microparticle-based delivery platform has potential for use in numerous applications in the field of tissue engineering and regenerative medicine.
5. Experimental Section
Mineral Coated Microparticles (MCMs) Preparation in mSBF
Hydroxyapatite microparticles (HA MPs) were obtained from Plasma Biotal LTD (Derbyshire, UK). The particle size of the HA MPs was between 3–5 μm in diameter. To form the mineral coating on HA MPs, the particles were incubated in modified simulated body fluid (mSBF) at 37 °C with 4.2, 25 or 100 mm HCO3− for 7 days with continuous rotation. The mSBFs were prepared by adding the following reagents into deionized water in the order shown: 141 mm NaCl, 4.0 mm KCl, 0.5 mm MgSO4, 1.0 mm MgCl2, 4.2, 25, or 100 mm NaHCO3, 20.0 mm HEPES, 5.0 mm CaCl2, and 2.0 mm KH2PO4 (all detailed mSBF formulations are shown in Table S-1, Supporting Information). The pH of mSBFs was then adjusted to 6.80. In particular, each 100 mg HA MP particles was incubated in 50 mL mSBF in a conical tube to form the mineral coatings. The mSBFs were refreshed daily throughout the entire coating process in order to maintain consistent ion concentrations for mineral coating growth. After 7 days of incubation, the MCMs were rinsed with deionized water and lyophilized (Figure 1).
Fluoride-doped MCMs were prepared by modifying the mSBF with sodium fluoride. Three different mSBF formulations were used to tailor the intrinsic properties of the coatings: 4.2 mm NaHCO3 mSBF, 100 mm NaHCO3 m-SBF, and 4.2 mm NaHCO3 mSBF without magnesium. For each formulation, 500 μL NaF (100 mm) were added into 50 mL mSBF to obtain 1 mm NaF in the final solution. The fluoride-containing mSBFs were then used to form the fluoride-doped MCMs by incubating the particles in various mSBFs for 7 days with daily mSBF refresh. The fluoride-free MCMs were collected and lyophilized after washing with deionized water.
MCMs Characterization
The morphology and composition of the MCMs were examined by scanning electron microscopy (SEM), Fourier transform infrared (FTIR) spectroscopy, and X-ray diffraction (XRD). The surface morphology of the MCMs was observed by LEO 1530 field emission scanning microscopy (FESEM, Zeiss, Germany) after sputter-coating with gold. FTIR was employed to analyze the composition of the mineral coating on MCMs using an Equinox 55 spectrometer (Bruker AXS, Germany). The FTIR spectra of the samples were recorded from 400 to 2000 cm−1 in transmission mode using potassium bromide pellets. The phase composition of the mineral coating was identified by XRD with a Hi-Star X-ray diffractometer with CuKα radiation.
The stability of the MCMs was evaluated by monitoring the amount of calcium released into Tris buffered saline (TBS, pH 7.40). Briefly, 5.0 mg of the MCMs was incubated in 1.0 mL TBS in a 1.5 mL Eppendorf tube at 37 °C. The releasing buffer was collected from the tube at various predetermined time points and 1.0 mL fresh TBS was refilled into the tube. The calcium amount released from the particles was quantified by an Arsenazo III based assay. Briefly, 5 μL of sample was mixed with 195 μL of 0.4 mm Arsenazo III in 20 mm Tris buffer (pH 7.40). The amount of calcium was then detected by measuring absorbance at 615 nm using a Molecular Device microplate reader, and the concentration was calculated by a set of standards with predetermined calcium concentrations.
Binding and Release of BMP-2 from MCMs
Radiolabeled BMP-2 (125I labeled BMP-2, Perkin Elmer, Waltham, MA) was used in this study to determine the binding and subsequent release of BMP-2 from the MCMs. Briefly, 5.0 mg of each type of MCMs was incubated in 1.0 mL solutions with 1000 ng mL-1 BMP-2 (0.5% 125I BMP-2) in PBS at 37 °C for 4.0 h with rotation. The MCMs were then centrifuged at 12000 RPM for 2 min and washed with 1.0 mL PBS once. The radioactivity in the supernatant and washing PBS was measured by a Packard Cobra II Gamma Counter (Perkin Elmer, Waltham, MA) to determine the BMP-2 amount in these solutions. The binding efficiency on each type of microparticles was calculated from the protein concentration change before and after binding. For BMP-2 release, the BMP-2 bound MCMs (5.0 mg) from binding assays were incubated in 1.0 mL Tris buffered saline (TBS, pH 7.40) at 37 °C and the released protein amount was determined by measuring the radioactivity of the release medium using the Gamma Counter at predetermined time points.
Dual Growth Factor Incorporation into MCMs
To incorporate dual growth factor into MCMs, BMP-2 is bound to the 1st mineral coating layer (L1) on the microparticles; then a 2nd layer of mineral coating (L2) is formed on top of L1 after binding of BMP-2; VEGF is then bound on the surface of L2. Briefly, HA MPs were incubated in designated mSBF to create the 1st layer of mineral coating by incubating 10 mg of HA MPs in 50 mL mSBF with varying ionic compositions at pH 6.80 and 37 °C for 7 days. The MCMs were then washed and lyophilized for growth factor binding. To bind BMP-2 on the 1st layer coating, 5.0 mg of the MCMs were incubated in 1.0 mL 1000 ng mL-1 BMP-2 in PBS at 37 °C for 4 h to allow binding of BMP-2 on the 1st mineral layer. The BMP-2 bound MCMs were then incubated in 50 mL designed mSBF for 5 days with daily mSBF refresh to generate the 2nd layer of mineral coating. The double layered MCMs were then washed with an excess volume of PBS and collected by centrifugation. The second growth factor VEGF was then bound to the double layered MCMs by incubating with 1.0 mL PBS containing 100 ng VEGF for 4 h at 37 °C. After incubation period, the MCMs were collected after washing with PBS.
Dual Growth Factor Release from MCMs
Two separate experiments were performed to study the binding and dual release of BMP-2 and VEGF, respectively. To monitor BMP-2 binding and release, 0.5% 125I BMP-2 of the total BMP-2 was used as a tracer when regular VEGF without radiolabel tracer was bound on the 2nd layer mineral. In another set of experiments, 1.0% 125I VEGF of the total VEGF was used as a tracer to study VEGF binding and release on the 2nd layer while un-labeled BMP-2 was incorporated on the 1st layer mineral. Specifically, 5.0 mg mineral coated HA MPs bound with two growth factors (1.0% 125I BMP-2/VEGF or BMP-2/0.5% 125I VEGF) were incubated in 1.0 mL TBS at 37 °C and in an incubator. At each predetermined time point, the amount of growth factor released was measured by counting the radioactivity in the release medium using a Gamma Counter as described previously. In addition, the loss of BMP-2 during the formation of the 2nd layer of coating was tracked by measuring the radioactivity of mSBF collected during coating formation.
Modulating BMP-2/VEGF Release from Multilayered Mineral Coating by Varying the Coating Thickness
The thickness of the 2nd layer mineral coating was varied by changing the incubation time in mSBF during the creation of this layer. After the binding of BMP-2 on the 1st mineral layer, the BMP-2-bound MCMs were incubated in 4.2 mm mSBF for additional 0 (no second layer), 0.5, and 5 days to form the 2nd layers of mineral coating with different thicknesses. The morphology of coating with different thicknesses were observed using LEO 1530 field emission scanning microscopy (FESEM, Zeiss, Germany) after being sputter-coated with gold. Then, a series of binding and release studies were performed as described previously.
Statistical Analysis
All quantitative results are expressed as means ± standard deviations. Statistical differences were analyzed using analysis of variance (ANOVA), and p < 0.05 was considered significant.
Supplementary Material
Acknowledgments
The authors would like to thank financial support from the AO Foundation, the National Institutes of Health (R01AR059916), and the National Science Foundation (Collaborative Research Proposal DMR 1106165/1105591). A.K. gratefully acknowledges financial support from NIGMS (5T32GM08349).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Xiaohua Yu, Department of Biomedical Engineering, University of Wisconsin, 1111 Highland Ave, Madison, WI, 53705, USA
Mr. Andrew Khalil, Department of Biomedical Engineering, University of Wisconsin, 1111 Highland Ave, Madison, WI, 53705, USA
Phuong Ngoc Dang, Department of Biomedical Engineering and Orthopaedic Surgery, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH, 44106, USA.
Prof. Eben Alsberg, Department of Biomedical Engineering and Orthopaedic Surgery, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH, 44106, USA AO Foundation Collaborative Research Center, Clavadelerstrasse 8, Davos, 7270, Switzerland.
Prof. William L. Murphy, Email: wlmurphy@wisc.edu, Department of Biomedical Engineering, University of Wisconsin, 1111 Highland Ave, Madison, WI, 53705, USA; AO Foundation Collaborative Research Center, Clavadelerstrasse 8, Davos, 7270, Switzerland; Department of Orthopedics and Rehabilitation, 1300 University Ave, University of Wisconsin, Madison, WI, 53705, USA.
References
- 1.Rezwan K, Chen QZ, Blaker JJ, Boccaccini AR. Biomaterials. 2006;27:3413. doi: 10.1016/j.biomaterials.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 2.Salgado AJ, Coutinho OP, Reis RL. Macromol Biosci. 2004;4:743. doi: 10.1002/mabi.200400026. [DOI] [PubMed] [Google Scholar]
- 3.Chen F-M, Zhang J, Zhang M, An Y, Chen F, Wu Z-F. Biomaterials. 2010;31:7892. doi: 10.1016/j.biomaterials.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 4.Chen F-M, Wu L-A, Zhang M, Zhang R, Sun H-H. Biomaterials. 2011;32:3189. doi: 10.1016/j.biomaterials.2010.12.032. [DOI] [PubMed] [Google Scholar]
- 5.Adam C. Cell Tissue Res. 2012;347:489. doi: 10.1007/s00441-011-1234-2. [DOI] [PubMed] [Google Scholar]
- 6.Giannoudis PV, Jones E, Einhorn TA, Hankenson KD, Dishowitz M, Gray C, Schenker M. Injury. 2011;42:556. doi: 10.1016/j.injury.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kleinheinz J, Stratmann U, Joos U, Wiesmann H-P. J Oral Maxillofac Surg. 2005;63:1310. doi: 10.1016/j.joms.2005.05.303. [DOI] [PubMed] [Google Scholar]
- 8.Patel ZS, Young S, Tabata Y, Jansen JA, Wong MEK, Mikos AG. Bone. 2008;43:931. doi: 10.1016/j.bone.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeon O, Song SJ, Kang S-W, Putnam AJ, Kim B-S. Biomaterials. 2007;28:2763. doi: 10.1016/j.biomaterials.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 10.Carmeliet P, Jain RK. Nature. 2000;407:249. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 11.Ferrara N, Gerber H-P, LeCouter J. Nat Med. 2003;9:669. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 12.Patel ZS, Young S, Tabata Y, Jansen JA, Wong MEK, Mikos AG. Bone. 2008;43:931. doi: 10.1016/j.bone.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Young S, Patel ZS, Kretlow JD, Murphy MB, Mountziaris PM, Baggett LS, Ueda H, Tabata Y, Jansen JA, Wong M, Mikos AG. Tissue Eng Part A. 2009;15:2347. doi: 10.1089/ten.tea.2008.0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tayalia P, Mooney DJ. Adv Mater. 2009;21:3269. doi: 10.1002/adma.200900241. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Jia X, Han F, Zhao J, Zhao Y, Fan Y, Yuan X. Biomaterials. 2013;34:2202. doi: 10.1016/j.biomaterials.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 16.King WJ, Krebsbach PH. Adv Drug Delivery Rev. 2012;64:1239. doi: 10.1016/j.addr.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Einhorn TA. J Bone Jt Surg. 2003;85-A:82. doi: 10.2106/00004623-200300003-00014. [DOI] [PubMed] [Google Scholar]
- 18.Liechty WB, Kryscio DR, Slaughter BV, Peppas NA. Ann Rev Chem Biomol Eng. 2010;1:149. doi: 10.1146/annurev-chembioeng-073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suárez-González D, Barnhart K, Migneco F, Flanagan C, Hollister SJ, Murphy WL. Biomaterials. 2012;33:713. doi: 10.1016/j.biomaterials.2011.09.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burdick JA, Mason MN, Hinman AD, Thorne K, Anseth KS. J Control Release. 2002;83:53. doi: 10.1016/s0168-3659(02)00181-5. [DOI] [PubMed] [Google Scholar]
- 21.Jongpaiboonkit L, Franklin-Ford T, Murphy WL. Adv Mater. 2009;21:1960. doi: 10.1021/am9001716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De M, Ghosh PS, Rotello VM. Adv Mater. 2008;20:4225. [Google Scholar]
- 23.Fan D, De Rosa E, Murphy MB, Peng Y, Smid CA, Chiappini C, Liu X, Simmons P, Weiner BK, Ferrari M, Tasciotti E. Adv Funct Mater. 2012;22:282. [Google Scholar]
- 24.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Proc Natl Acad Sci U S A. 2003;100:5413. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lienemann PS, Lutolf MP, Ehrbar M. Adv Drug Delivery Rev. 2012;64:1078. doi: 10.1016/j.addr.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 26.Li C, Vepari C, Jin HJ, Kim HJ, Kaplan DL. Biomaterials. 2006;27:3115. doi: 10.1016/j.biomaterials.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 27.Johnson MR, Lee H-J, Bellamkonda RV, Guldberg RE. Acta Biomater. 2009;5:23. doi: 10.1016/j.actbio.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lutolf MP, Weber FE, Schmoekel HG, Schense JC, Kohler T, Muller R, Hubbell JA. Nat Biotechnol. 2003;21:513. doi: 10.1038/nbt818. [DOI] [PubMed] [Google Scholar]
- 29.Kolambkar YM, Boerckel JD, Dupont KM, Bajin M, Huebsch N, Mooney DJ, Hutmacher DW, Guldberg RE. Bone. 2011;49:485. doi: 10.1016/j.bone.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah NJ, Macdonald ML, Beben YM, Padera RF, Samuel RE, Hammond PT. Biomaterials. 2011;32:6183. doi: 10.1016/j.biomaterials.2011.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen F-M, Zhang M, Wu Z-F. Biomaterials. 2010;31:6279. doi: 10.1016/j.biomaterials.2010.04.053. [DOI] [PubMed] [Google Scholar]
- 32.Lee JS, Suarez-Gonzalez D, Murphy WL. Adv Mater. 2011;23:4279. doi: 10.1002/adma.201100060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Budz JA, Nancollas GH. J Cryst Growth. 1988;91:490. [Google Scholar]
- 34.Iijima M, Moradian-Oldak J. Biomaterials. 2005;26:1595. doi: 10.1016/j.biomaterials.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 35.LeGeros RZ. Chem Rev. 2008;108:4742. doi: 10.1021/cr800427g. [DOI] [PubMed] [Google Scholar]
- 36.Martins A, Duarte ARC, Faria S, Marques AP, Reis RL, Neves NM. Biomaterials. 2010;31:5875. doi: 10.1016/j.biomaterials.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Jaklenec A, Wan E, Murray ME, Mathiowitz E. Biomaterials. 2008;29:185. doi: 10.1016/j.biomaterials.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 38.Kim S, Kang Y, Krueger CA, Sen M, Holcomb JB, Chen D, Wenke JC, Yang Y. Acta Biomater. 2012;8:1768. doi: 10.1016/j.actbio.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kempen DHR, Lu L, Heijink A, Hefferan TE, Creemers LB, Maran A, Yaszemski MJ, Dhert WJA. Biomaterials. 2009;30:2816. doi: 10.1016/j.biomaterials.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 40.Luong LN, Hong SI, Patel RJ, Outslay ME, Kohn DH. Biomaterials. 2006;27:1175. doi: 10.1016/j.biomaterials.2005.07.043. [DOI] [PubMed] [Google Scholar]
- 41.Kandori K, Miyagawa K, Ishikawa T. J Colloid Interface Sci. 2004;273:406. doi: 10.1016/j.jcis.2004.01.069. [DOI] [PubMed] [Google Scholar]
- 42.Yu X, Qu H, Knecht D, Wei M. J Mater Sci Mater Med. 2009;20:287. doi: 10.1007/s10856-008-3571-6. [DOI] [PubMed] [Google Scholar]
- 43.Yu X, Wei M. J Biomed Mater Res Part B Appl Biomater. 2011;97B:345. doi: 10.1002/jbm.b.31820. [DOI] [PubMed] [Google Scholar]
- 44.Ginebra M-P, Canal C, Espanol M, Pastorino D, Montufar EB. Adv Drug Deliv Rev. 2012;64:1090. doi: 10.1016/j.addr.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 45.Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. J Control Release. 2008;125:193. doi: 10.1016/j.jconrel.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 46.Carrasquillo KG, Stanley AM, Aponte-Carro JC, De Jésus P, Costantino HR, Bosques CJ, Griebenow K. J Controlled Release. 2001;76:199. doi: 10.1016/s0168-3659(01)00430-8. [DOI] [PubMed] [Google Scholar]
- 47.Kissel T, Li YX, Volland C, Görich S, Koneberg R. J Controlled Release. 1996;39:315. [Google Scholar]
- 48.Mehta M, Schmidt-Bleek K, Duda GN, Mooney DJ. Adv Drug Delivery Rev. 2012;64:1257. doi: 10.1016/j.addr.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang EA, Rosen V, D’Alessandro JS, Bauduy M, Cordes P, Harada T, Israel DI, Hewick RM, Kerns KM, LaPan P. Proc Natl Acad Sci U S A. 1990;87:2220. doi: 10.1073/pnas.87.6.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suárez-González D, Lee JS, Lan Levengood SK, Vanderby R, Murphy WL. Acta Biomater. 2012;8:1117. doi: 10.1016/j.actbio.2011.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu Y, Markel MD, Nemke B, Lee JS, Graf BK, Murphy WL. Arthroscopy. 2009;25:1427. doi: 10.1016/j.arthro.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi S, Yu X, Jongpaiboonkit L, Hollister SJ, Murphy WL. Sci Rep. 2013;3:1567. doi: 10.1038/srep01567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu Y, Lee JS, Nemke B, Baer G, Graf BK, Murphy WL, Markel MD. Curr Orthop Pract. 2011;22:425. [Google Scholar]
- 54.Wei G, Ma PX. Biomaterials. 2004;25:4749. doi: 10.1016/j.biomaterials.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 55.Ethirajan A, Musyanovych A, Chuvilin A, Landfester K. Macromol Chem Phys. 2011;212:915. [Google Scholar]
- 56.Suárez-González D, Barnhart K, Saito E, Vanderby R, Jr, Hollister SJ, Murphy WL. J Biomed Mater Res Part A. 2010;95:222. doi: 10.1002/jbm.a.32833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jongpaiboonkit L, Franklin-Ford T, Murphy WL. ACS Appl Mater Interfaces. 2009;1:1504. doi: 10.1021/am9001716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kretlow JD, Young S, Klouda L, Wong M, Mikos AG. Adv Mater. 2009;21:3368. doi: 10.1002/adma.200802009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okazaki M, Takahashi J, Kimura H, Aoba T. J Biomed Mater Res. 1982;16:851. doi: 10.1002/jbm.820160610. [DOI] [PubMed] [Google Scholar]
- 60.Driessens FC. Nature. 1973;243:420. doi: 10.1038/243420a0. [DOI] [PubMed] [Google Scholar]
- 61.Barralet J, Best S, Bonfield W. J Biomed Mater Res. 1998;41:79. doi: 10.1002/(sici)1097-4636(199807)41:1<79::aid-jbm10>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 62.Lee JS, Lu Y, Baer GS, Markel MD, Murphy WL. J Mater Chem. 2010;20:8894. [Google Scholar]
- 63.Putney S, Burke P. Nat Biotechnol. 1998;16:153. doi: 10.1038/nbt0298-153. [DOI] [PubMed] [Google Scholar]
- 64.Kanczler JM, Oreffo ROC. Eur Cell Mater. 2008;15:100. doi: 10.22203/ecm.v015a08. [DOI] [PubMed] [Google Scholar]
- 65.Uchida S, Sakai A, Kudo H, Otomo H, Watanuki M, Tanaka M, Nagashima M, Nakamura T. Bone. 2003;32:491. doi: 10.1016/s8756-3282(03)00053-x. [DOI] [PubMed] [Google Scholar]
- 66.Niikura T, Hak DJ, Reddi AH. J Orthop Res. 2006;24:1463. doi: 10.1002/jor.20182. [DOI] [PubMed] [Google Scholar]
- 67.Bacic M, Edwards NA, Marill MJ. Proc Natl Acad Sci U S A. 2002;99:9656. [Google Scholar]
- 68.Cho T-J, Gerstenfeld LC, Einhorn TA. J Bone Miner Res. 2002;17:513. doi: 10.1359/jbmr.2002.17.3.513. [DOI] [PubMed] [Google Scholar]
- 69.Olsen BR, Reginato AM, Wang W. Annu Rev Cell Dev Biol. 2000;16:191. doi: 10.1146/annurev.cellbio.16.1.191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.