

Abstract
Personalized medicine is the cornerstone of medical practice. It tailors treatments for specific conditions of an affected individual. The borders of personalized medicine are defined by limitations in technology and our understanding of biology, physiology and pathology of various conditions. Current advances in technology have provided physicians with the tools to investigate the molecular makeup of the disease. Translating these molecular make-ups to actionable targets has led to the development of small molecular inhibitors. Also, detailed understanding of genetic makeup has allowed us to develop prognostic markers, better known as companion diagnostics. Current attempts in the development of drug delivery systems offer the opportunity of delivering specific inhibitors to affected cells in an attempt to reduce the unwanted side effects of drugs.
Keywords: Personalized medicine, Targeted therapies, Hematology, Drug Delivery
1. Introduction
Personalized medicine attempts to identify individual tailored treatments based on the susceptibility profile of each individual. Precision medicine utilizes both conventional medicine and cutting edge technology to concur the disease proven to be resistant to conventional medical techniques. The borders of personalized medicine are defined by limitations in technology and our understanding of biology, and pathology of various conditions. Current advances in technology have enabled us to uncover the molecular makeup of diseases and translating these findings to actionable targets has led to the development of small molecular inhibitors. Monitoring disease outcome utilizing companion diagnostics has also assisted physicians in routine patient care. To date serious efforts are directed in increasing the efficacy of drug delivery to reduce the undesired side effects of medications (Fig. 1). Despite the current advances there are fundamental limitations on implementing personalized medicine into daily practice. Here we lay out the steps from bench to bedside for personalized therapeutics in hematology and explore the complex problems at each steps. We will first discuss the discovery platforms, and compare the existing technologies. Major discoveries utilizing these platforms will be discussed followed by summarizing the targeted inhibitors developed which are currently in clinical practice. Next we will briefly discuss the advantages of small molecular inhibitors over existing chemotherapeutic regimens and explore conditions that affect the drug response to these inhibitors (pharmacogenomics and pharmacovigilance). Finally we will discuss the drug delivery systems that could potentially enhance the outcome and limit the undesired effects of these medications.
Fig. 1.
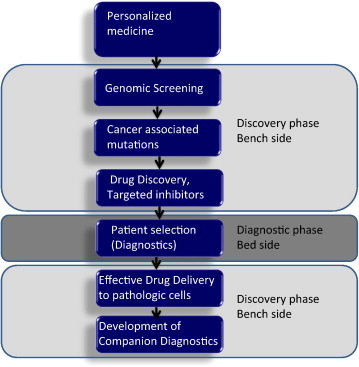
Implementation of personalized medicine requires combining discovery platforms and clinical practice. Early stage of discovery requires interrogation of large numbers of samples to uncover the somatic genomic alterations of tumor cells. Further studies on genomic mutations are conducted to demonstrate that the aberrations are driver mutations and therefore actionable. Small molecular inhibitors are developed to target proteins intercepting these alterations. Patients are screened in clinics to ensure that they carry the desired mutation targeted by small molecular inhibitors. Intermediate end-point biomarkers are identified and studied in the audit trail as early predictors of anti-tumor activity.
Advances in human genetics have clearly demonstrated the contribution of specific genes to certain malignancies [1–3]. Such genomic alterations are functionally manifested as dysfunctional proteins leading to aberrant signal transduction [4–7]. Consequently, discovering genomic alterations underlying various conditions is a fundamental step in implementing precision medicine. Selecting an appropriate screening technology is crucial for both discovery and diagnostics. In the era of genomic innovation, several platforms are available [8–11]. Mass spectrometric genotyping, allele-specific PCR-based technologies, hybrid-capture massively parallel sequencing technologies, and whole-genome sequencing are among the available platforms [12,13]. For discovery purposes, sequencing of the entire genome would be the preferred option, but, for diagnostic purposes, one must presently focus on cost-effective platforms that cover actionable cancer-associated mutations (MassArrays) [14] (Fig. 2).
Fig. 2.
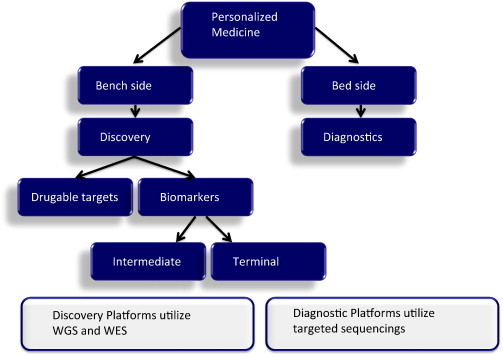
Steps from bench to bedside for personalized medicine: Discovery of drugable targets lay out the path for development of targeted inhibitors. Usually more comprehensive platforms, such as whole genome sequencing (WGS) and whole exome sequencing (WES) are used at this step. It is well established today that the sole presence of a target in tumor cells does not guarantee the drug response. To determine the best group of patients who would benefit from targeted inhibitors, both intermediate and terminal genomic biomarkers are in need. Again, comprehensive platforms (WGS and WES) are used for discovery purpose. Patient selection for targeted inhibitors (diagnostics) could be run using less expensive techniques, i.e. targeted sequencing.
Though personalized medicine appears to bring us ever closer to a step away from the cure for cancer, the reality is far more complicated. Despite current advances in genomics, there is still a long path to decoding all cancer-associated mutations, let alone the signaling pathway of new and novel mutations which would be an additional area to explore [15]. In both hematologic and solid tumors, a large fraction of affected proteins is represented by kinases, which are essential for physiological functions of cells, such as cellular growth and development [16–18]. Blocking these molecules usually drives the cells into developing compensatory mechanisms, and cancer cells eventually escape the inhibition, developing tumor resistance [19,20]. Another obvious challenge is excessive toxicity by nonselective inhibition of both mutant and wild-type proteins by some inhibitors [21–24]. Additional factors can affect efficacy of treatment. In particular, some genetic variations can alter the drug response of individuals, and this should be taken into consideration with drug dosing [25,26]. Therefore it is crucial to develop companion diagnostics by combining genomic information with proteomics as well as personal medical history and family history data to tailor the desired agent for targeting the neoplastic cells [26–29]. Consequently, it is essential to develop prognostic biomarkers to both screen the outcome of the treatment and screen for residual disease [30–33].
Another limited challenge in personalizing cancer therapy is the limited technologies in drug delivery [34]. It is essential to deliver the appropriate inhibitors to the affected cells; however, advancement in the development of nanoparticles that can achieve selective cellular targeting remains limited [35]. The use of nanoparticles has thus been restricted primarily to a reduction of drug toxicity, as evidenced by the success of Doxil (liposomal doxorubicin), which decreases cardiotoxicity [36–38]. Fortunately, our understanding of the interactions between nanoparticles and living cells continues to improve. A conceptual understanding of biological responses to nanoparticles is essential for developing safer targeted drug delivery in the future.
2. Discovery platforms
Almost a decade after the completion of the first genome sequencing, genome research composes the main core of discovery in various cancers [39–41]. The classical discovery platform used for sequencing the human genome was a capillary based electrophoresis (CE) system [42–44]. Although this system was developed by Fredrick Sanger in the late 70s, it was the most widely used technique for over two decades. The high cost of sample processing along with the restriction on clinical scalability led to the emergence of new technologies based on hybrid capture and massively parallel sequencing (MPS) [45–47], better known as next generation sequencings (NGS) [48–50]. These profiling platforms enable the investigators to detect point mutations, copy number alterations, and chromosomal aberrations using a single run and a small amount of DNA input [51,53,54]. These platforms are highly sensitive (i.e. they detect genetic alterations in small allele fractions) and fairly scalable (i.e. they can be tuned for resolution and coverage) [52]. Last but not least, these platforms are rather affordable and they have brought down the cost of the sequencing of the entire genome to 5000 USD per samples. It is suggested that these new sequencing instruments could sequence several samples in less than a day [52]. Table 1 summarizes the advantages and disadvantages of Sanger and NGS.
Table 1.
Comparison of Sanger sequencing with next generation sequencing.
| Advantages | Disadvantages | |
|---|---|---|
| Sanger |
|
|
| NGS |
|
|
Several discoveries in field of hematology were made utilizing the discovery platforms. For instance ALK, PDGFR and FGFR are all discovered using sequencing platforms (Sanger/NGS). BRAF-V600E discovery in LCH and ECD was based on targeted sequencing. The cutting edge medications developed targeting genomic alterations are discussed in the next section.
3. Diagnostic platforms: PCR based technologies and massively parallel sequencing
It is well known that most hematologic malignancies are caused by genomic alteration (point mutation, chromosomal aberrations, copy number variations), and therefore complete understanding of these diseases can only be achieved by comprehensive screening of a large number of clinical samples. Despite the fact that the cost of sequencing of the whole genome has dropped significantly in the past decade (from 3 billion USD to roughly 5000 USD), screening a large number of clinical samples could still impose economic challenges. Also, most of the information provided by whole genome sequencing (WGS) cannot be fully interpreted [55,56]. Despite the fact that there are over 800 new small molecules on developmental pipeline [57–59], the number of drugable targets currently available in clinics is less than 30 [59,60]. The economic impact of a large scale application of WGS along with limited clinical applicability of information obtained from whole genome is suggestive for the utilization of alternative tools for diagnostic purposes.
One of the first platforms developed for high throughput screening of clinical samples in oncology was a mass spectrometric base genotyping platform developed by Garraway and colleagues for the detection of cancer-associated mutations [61]. They relied on the fact that a large subset of cancer-associated deriver mutations affects hotspot amino acids. This led to the development of multiplex allele-specific PCR platforms [61–63]. This platform enabled us to detect a BRAF-V600E mutation in Langerhans cell histiocytosis (LCH), a disease which, until this observation, was known as a reactive-inflammatory one [64–66]. Despite the high sensitivity of this platform, it had a very limited coverage and was biased to a subset of genes. It was also unable to detect chromosomal aberrations and large indels [67]. To overcome these shortcomings, NGS platforms were adapted for enrichment of subsets of the human genome. By scaling these platforms and enriching a subset of genome, e.g. on exons or targeted genomic sequences for drugable targets and predictive biomarkers for drug response, several questions could be addressed for a fraction of the cost of WGS (Table 2) [68–71,132].
Table 2.
Comparison of PCR based technologies with massively parallel sequencing technologies.
| Advantages | Disadvantages | |
|---|---|---|
| Genotyping platforms (PCR based technologies) |
|
|
| Massively parallel sequencing (hybrid capture techniques) |
|
|
Whole genome sequencing (WGS) enables identification of coding mutations and copy number alterations (amplifications and deletions), but its ability to detect chromosomal translocation in commercially available probes is rendered due to the lack of intronic sequences in capturing probes [72]. Stromal contaminations and genomic heterogeneity could also complicate the interpretation of data. On the other hand, targeted sequencing can achieve a larger depth of coverage and consequently higher sensitivity at a comparable cost [52]. This approach could be very useful for clinical samples with low preservation (samples derived from formalin fixed tissue) and high stromal contamination, or in the case of hematologic tumors, contamination by bystander cells [73–75].
On the other hand, to fully understand the genetic background of a disease we must know the extent of gene expression as well. Chromatin immunoprecipitations (Chip-Seq) could unravel the mutation and methylation statuses of gene regulatory sites and determine the activation status of genes, and consequently gene expression [76,78]. Transcriptome sequencing (RNA-seq) [76] captures the expressed genome of cancer cells enabling robust detection of deregulated genes [77], and gene fusions [78–80]. Combining expression data with genomic findings could shed light on the pathomechanisms of the disease and facilitate the design of targeted inhibitors (Table 3). Clinical applicability of these techniques is quite important and FDA has recently approved several NGS instruments for clinical applications.
Table 3.
Cancer genome profiling techniques.
| WGS | Target capture | RNA-Seq | |
|---|---|---|---|
| Scale | Whole genome | Targeted areas of genome (whole exome, actionable genome) | Transcribed region of genome |
| Substrate | DNA | DNA or cDNA | cDNA |
| Application | Research | Diagnostic | Diagnostic, research |
| Limitations | Cost, ability to interpret the data | Unable to detect chromosomal aberrations Biased to gene within the targeted region |
Sensitivity limited to transcribed genome |
| Advantages | A single platform give information on point mutations, chromosomal aberrations and CNV | High sequencing debt on a given cost, adaptable to individual needs | Detects novel transcripts with low level of expression Detects non-mutational events |
CNV: copy number variations, cDNA: complementary DNA.
4. Targeted therapies and current drugs
Targeted therapies or small molecular inhibitors block the proliferation of cancer cells by intercepting their specific target [81–84]. Since their range of action is smaller than general chemotherapy agents, the adverse effect caused by these inhibitors is smaller as well and they are better tolerated by patients [85–88]. This seems to be a success story, but the final picture is complicated. These inhibitors are not curative, and disease relapse remains a fairly common complication in these malignancies. Several reasons could be attributed to disease relapse, among which is the escape of tumors cells which obtain new surviving mutations and the evolution of new neoplastic populations due to weakened immune response. On the other hand, there are obvious caveats in targeting cancer cells with very specific molecular inhibitors. First of all, most of the small molecular inhibitors currently available in clinics are targeting protein kinases [89–91]. These are an essential cellular component and blocking these molecules could result in cellular compensation. This could manifest either as overproduction of the inhibited protein (upregulation on the gene level), or escalating alternative compensatory pathways for survival. Second, the delivery of these molecules to the affected cells is limited by tissue vascularization and cellular uptake. This forces physicians to escalate the dose of inhibitor in compensation, leading to undesirable side effects. Currently there are several small molecular inhibitors in clinical practice. Table 4 summarizes the current small molecular inhibitors in clinical practice [92–96,130,131].
Table 4.
Targeted inhibitors used in treatment of hematologic malignancies.
| Gene | Genetic alterations | Tumor type | Targeted agent |
|---|---|---|---|
| Receptor tyrosine kinase | |||
| ALK | Mutation, CNV | Anaplastic large cell lymphoma | Crizotinib |
| FGFR1 | Translocation | CML, myelodysplastic disorders | Imatinib methylase |
| FGFR3 | Translocation, mutation | Multiple myeloma [113] | PKC412, BIBF-1120 |
| FLT3 | CNV | AML | Lestaurtinib, XL999 |
| PDGFRB | Translocation, mutation | CML | Sunitinib, sorafenib, imatinib, nilotinib |
| Non-receptor tyrosine kinase | |||
| ABL | Translocation (BCR-ABL) | CML. AML | Dasatinib, nilotinib, bosutinib |
| JAK2 ERK1/2 |
Mutation (V617F) translocation Mutation |
CML, myeloproliferative disorders Mantle cell lymphoma, CLL |
Lestaurtinib, INCB018424 Ibrutinib |
| Serine–threonin kinase | |||
| Aurora A and B kinase | CNV | Leukemia | MK5108 |
| BRAFV600E | Mutation | LCH, ECD [110], hairy cell leukemia [112] | Vemurafenib (PLX4032) |
| Polo like kinase | Mutation | Lymphoma | B12536 |
| Non-kinase targets | |||
| PARP | Mutation, CNV | Advanced hematologic malignancies, CLL, mantle cell lymphoma | BMN 673 |
| Antibodies | |||
| CD20 | Hodgkin lymphoma | Rituximab | |
| CD52 | B-cell chronic lymphocytic leukemia | Alemtuzumab | |
| CD20 | Non-Hodgkin lymphoma | Ibritumomab tiuxetan | |
| Apoptotic agents | |||
| Proton pump inhibitors | Multiple myeloma, mantle cell lymphoma, peripheral T-cell lymphoma | Bortezomib, pralatrexate | |
CNV: copy number variations, AML: acute myeloid leukemia, CML: chronic myeloid leukemia, LCH: Langerhans cell histiocytosis, ECD: Erdheim Chester disease.
Data adapted from NCI: http://www.cancer.gov/cancertopics/factsheet/Therapy/targeted.
“In the future we should drive our focus on enhancing the patient's response based on their unique genetic makeup using appropriate companion diagnostics (pharmacogenomics) along with targeting the driver event”. Also, to maximize the benefits of small molecular inhibitors, we must deliver the targeted agents to a susceptible population based on individuals' susceptibility profiles determined by companion diagnostics. Eventually, a better outcome will be achieved by matching the right therapy to the right parties, taking us a step closer to a potential cure for these malignancies. Last, but not least combining well designed collaborations between private sectors and academics will expedite the drug discovery process [130,131].
5. Pharmacogenomics and drug response
Pharmacogenomics is the science that studies the role of inherited and acquired genetic variations to drug response. It correlates gene expression and single nucleotide polymorphism (SNP) with both drug toxicity and efficacy to optimize therapeutic regimens for each individual [97,98]. One of the classical examples of pharmacogenomics is the effect of CytP450 variants on the dosing of warfarin [99–101,111].
There are several fundamental differences between cytotoxic chemotherapies and small molecular inhibitors. Dose-related toxicities have traditionally been considered key end points of Phase I trials and the maximum tolerated dose (MTD) was regarded as the optimal dose providing the best efficacy with manageable toxicity. Recently, development of targeted inhibitors has challenged the paradigms used in cytotoxic chemotherapy trial design. In precision medicine pharmacokinetic (PK) and pharmacodynamic (PD) end points tend to take a backseat to toxicity. Molecularly targeted agents do not always maintain the same dose–toxicity relationship as cytotoxic agents and tend to produce minimal organ toxicity. Furthermore, molecular therapeutic agents usually result in prolonged disease stabilization and provide clinical benefit without tumor shrinkage, a characteristic seen with cytotoxic agents, therefore necessitating alternative measures of anti-tumor efficacy. These end points include biologically relevant drug exposures, PD biomarker measures of target inhibition, and intermediate end-point biomarkers, such as molecular biomarkers (Fig. 3).
Fig. 3.
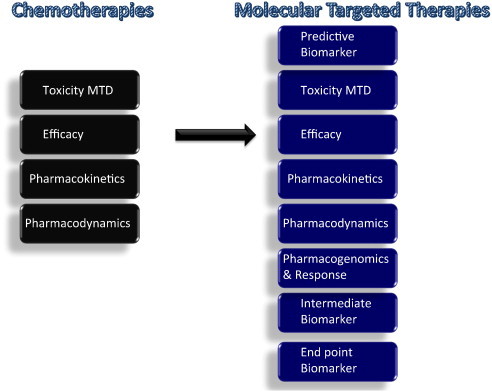
Comparison of standard chemotherapy with novel molecular targeted therapies: Dose-related toxicities have traditionally been considered key end points of Phase I trials and the maximum tolerated dose (MTD) is regarded as the optimal dose that provides the best efficacy with manageable toxicity. Pharmacokinetic (PK) and pharmacodynamic (PD) end points tend to take a backseat to toxicity. Recently, development of targeted inhibitors has challenged the paradigms used in cytotoxic chemotherapy trial design. Molecularly targeted agents do not always maintain the same dose–toxicity relationship as cytotoxic agents and tend to produce minimal organ toxicity. Furthermore, molecular therapeutic agents usually result in prolonged disease stabilization and provide clinical benefit without tumor shrinkage, a characteristic seen with cytotoxic agents, therefore necessitating alternative measures of anti-tumor efficacy. These end points include biologically relevant drug exposures, PD biomarker measures of target inhibition, intermediate end-point biomarkers, such as circulating tumor cells and other molecular biomarkers, including functional imaging.
In the field of cancer, pharmacogenomics is complicated by the fact that two genomes are involved: the germline genome of the patient and the somatic genome of tumor, the latter of which is of primary interest [102]. This genome predicts whether specific targeting agents will have a desired effect in the individual. On the other hand, germline pharmacogenetics can identify patients likely to demonstrate severe toxicities when given cytotoxic treatments. For example, germline SNPs in the gene encoding the enzyme thiopurine S-methyltransferase (TPMT) can result in increased sensitivity to mercaptopurine as a result of decreased drug metabolism, whereas the number of TA repeats in the promoter region of UGT1A1 can increase the toxic effects of irinotecan again as a result of decreased drug metabolism. Therefore, understanding the variable response to drugs is quite pressing in oncology where cytotoxic agents have narrow therapeutic indices and severe side effects [103,108,109]. Table 5 summarizes the companion diagnostics developed by the FDA for the treatment of hematologic malignancies.
Table 5.
Companion diagnostics and anticancer treatments in hematology.
| Anticancer treatments approved by FDA carrying companion diagnostics | |
|---|---|
| Biomarker with pharmacokinetic effect | TPMT (mercaptopurine, thioguanine) |
| UGR1A1 (irinotecan, nilotinib) | |
| Biomarkers with pharmacodynamic effect | EGFR (cetuximab, erlotinib, gefitinib, panitumumab, afatinib) |
| KRAS (cetuximab, panitumumab) | |
| ABL (imatinib, dasatinib, nilotinib) | |
| BCR-ABL (bosutinib, busulfan) | |
| ALK (crizotinib) | |
| C-kit (imatinib) | |
| HER2/neu (lapatinib, trastuzumab) | |
| ER (tamoxifen, anastrozole) | |
Genes in bold are used for companion diagnostics of the drugs mentioned in the brackets.
Data are obtained from FDA pharmacogenomics website (http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm).
Generalization and clinical application of pharmacogenetics are rather challenging in precision medicine. Most of the affected individuals have unique profiles in their tumors in addition to the fact that every individual has a unique SNP profile at a germline level. If a certain type of cancer carries several driver mutations then the choice of targeted therapy becomes complicated. In disseminated tumors, the picture would be further complicated by inter-tumor and intra-tumor heterogeneity of cancer [104–107]. Therefore, a greater understanding of the complexities of multiple gene modifiers of outcome, and the statistical challenge of understanding such data, will be needed before individualized therapy can be applied on a routine basis.
Consequently, tumor heterogeneity makes the use of combination therapies attractive. If an individual carries several driver mutations which inhibitors should be prescribed? What would be the appropriate dosing of each? How will drug interactions affect the picture? How can we increase the therapeutic index? Addressing these questions seems particularly pressing in the era of abundance of targeting inhibitors and the enormous economic pressures on healthcare providers.
6. Drug delivery
Effective drug delivery could substantially increase the efficacy of small molecule inhibitors in cancer. Currently, several nanoparticulate platforms are under investigation [114]. A desirable carrier would be able to incorporate and release drugs with defined kinetics, should have stable formulation for extended shelf life, should be highly specific for its target, and should be bioinert [115]. Biological materials such as albumin, phospholipids, synthetic polymers, and even solid components can be used as substrates for nanoparticles [116,117] (Table 6).
Table 6.
Structure and applications of nanoparticles.
| Particle class | Material | Application |
|---|---|---|
| Natural material | Chitosan Dextran Gelatin Liposome Starch |
Gene delivery Small molecule delivery |
| Silica variants | Silica nanoparticles | Gene delivery |
| Dendrimers | Branched polymers | Drug delivery, gene delivery |
| Polymer carriers | Polylactic acid Poly(cyano)acrylates Polyethyleneimine Block copolymers Polycaprolactone |
Drug delivery, gene delivery Small molecule delivery |
Ideally, the particles could be readily conjugated with a targeting ligand to facilitate specific uptake by target cells [118]. This would result in increased efficacy by increasing drug concentration in the intended target cells as well as in decreased systemic toxicity by reducing non-specific uptake [119]. Unfortunately several drug delivery matrix (nanoparticles) used by the pharmaceutical industry imposed risk to the patients [120,121]. These toxicities varied depending on the surface properties of nanoparticles [122,123], chemical composition [119,124], their half life [125] and distribution [126]. Among the in vivo side effects of nanoparticles, pulmonary inflammation (PSP), pulmonary neoplasia (PSP), immune response (polystyrene, CB, DEP), and platelet aggregations (PM, latex-aggregate surface) are well established [127,128].
In order to achieve enhanced delivery, reduced toxicity, and eventually enhanced therapeutic index, development of long-circulating and target-specific nanoparticles is needed. A conceptual understanding of biological responses to nanomaterials is necessary for development and safe application of nanomaterials in drug delivery in the future. Furthermore, a close collaboration between those working in drug delivery and particle toxicology is necessary for the exchange of concepts, methods, and know-how to move this issue ahead.
To date the most common vehicle used for targeted drug delivery is the liposomes. These molecules are non-toxic, non-hemolytic, and non-immunogenic even upon repeated injections. Liposomes are biodegradable and can be designed with various half-lives. Liposomes are currently used in cancer therapies (metastatic breast cancer, advanced melanoma, colorectal cancers) but their high cost creates severe limitations [129].
7. Future directions
Currently, there are huge amounts of screening data available at the genomic level. One of the shortcomings is our limited understanding of the functional importance of these findings. It is curtailed to distinguish driver genomic events from passenger ones. Today, there are over 800 new drugs (targeted inhibitors) in the development pipeline. At this point, our shortcoming is not the availability of targeted inhibitors but rather our limitations on the delivery of these molecules to the affected cells with a high degree of specificity. Next, we must improve our ability to get the targeted inhibitors designed for malfunctioning cellular components into the affected cells. By increasing the efficacy of targeted drug delivery, we will both reduce the unwanted side effects of antineoplastic agents on healthy cells and increase their cytotoxicity on affected cells. Lastly, we should be aware of the economic effects of precision medicine. An outstanding high cost will not be sustainable in the long term, so development of technologies for cost reduction should not be ignored.
Acknowledgment
I would like to express my gratitude to Professor Barrett Rollins for his mentorship, and Dr. Paul VanHummelen and Dr. Michael Goldberg for the scientific discussion.
References
- 1.Lawrence M.S., Stojanov P. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. Jan 5 2014 doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexandrov L.B., Nik-Zainal S. Signatures of mutational processes in human cancer. Nature. Aug 22 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Badalian-Very G., Vergilio J.A. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. Sep 16 2010;116(11):1919–1923. doi: 10.1182/blood-2010-04-279083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ylipää A., Yli-Harja O., Zhang W., Nykter M. Characterization of aberrant pathways across human cancers. BMC Syst Biol. 2013;7(Suppl. 1):S1. doi: 10.1186/1752-0509-7-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paul I., Bhattacharya S. Current understanding on EGFR and Wnt/β-catenin signaling in glioma and their possible crosstalk. Genes Cancer. Nov 2013;4(11–12):427–446. doi: 10.1177/1947601913503341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koul H.K., Pal M., Koul S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer. Sep 2013;4(9–10):342–359. doi: 10.1177/1947601913507951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gäbler K., Behrmann I. JAK2 mutants (e.g., JAK2V617F) and their importance as drug targets in myeloproliferative neoplasms. JAKSTAT. Jul 1 2013;2(3):e25025. doi: 10.4161/jkst.25025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chmielecki J., Meyerson M. DNA sequencing of cancer: what have we learned? Annu Rev Med. Nov 20 2013;65:63–79. doi: 10.1146/annurev-med-060712-200152. [DOI] [PubMed] [Google Scholar]
- 9.Leggett R.M., Ramirez-Gonzalez R.H., Ramirez-Gonzalez R.H., Clavijo B.J., Waite D., Davey R.P. Sequencing quality assessment tools to enable data-driven informatics for high throughput genomics. Front Genet. Dec 17 2013;4:288. doi: 10.3389/fgene.2013.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hou L., Zhao H. A review of post-GWAS prioritization approaches. Front Genet. Dec 9 2013;4:280. doi: 10.3389/fgene.2013.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borha A. Emerging paradigms in genomics-based crop improvements. Sci World J. Nov 17 2013;2013:585467. doi: 10.1155/2013/585467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carter S.L., Cibulskis K. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. May 2012;30(5):413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacConaill L.E., Van Hummelen P., Meyerson M., Hahn W.C. Clinical implementation of comprehensive strategies to characterize cancer genome: opportunities and challenges. Cancer Discov. Sep 2011;1(4):297–311. doi: 10.1158/2159-8290.CD-11-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagle N., Berger M.F., Davis M.J. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. Jan 2012;2(1):82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding L., Getz G. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. Oct 23 2008;455(7216):1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tournier C. The 2 faces of JNK signaling in cancer. Genes Cancer. Sep 2013;4(9–10):397–400. doi: 10.1177/1947601913486349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebelt N.D., Cantrell M.A. c-Jun N-terminal kinases mediate a wide range of targets in the metastatic cascade. Genes Cancer. Sep 2013;4(9–10):378–387. doi: 10.1177/1947601913485413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jang Y.N., Baik E.J. JAK–STAT pathway and myogenic differentiation. JAKSTAT. Apr 1 2013;2(2):e23282. doi: 10.4161/jkst.23282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Francipane M.G., Lagasse E. mTOR pathway in colorectal cancer: an update. Oncotarget. Jan 15 2014;5(1):49–66. doi: 10.18632/oncotarget.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haroche J., Cohen-Aubart F., Van Den Berg C.L. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim–Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. Feb 28 2013;121(9):1495–1500. doi: 10.1182/blood-2012-07-446286. [Epub 2012 Dec 20] [DOI] [PubMed] [Google Scholar]
- 21.Arora A., Sholar E.M. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. Dec 2005;315(3):971–979. doi: 10.1124/jpet.105.084145. (Epub 2005 Jul 7) [DOI] [PubMed] [Google Scholar]
- 22.Hartmann J.T., Haap M. Tyrosine kinase inhibitors — a review on pharmacology, metabolism and side effects. Curr Drug Metab. Jun 2009;10(5):470–481. doi: 10.2174/138920009788897975. [DOI] [PubMed] [Google Scholar]
- 23.Druker B.J., Sawyers C.L. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. Apr 5 2001;344(14):1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 24.Force T., Krause D.S. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. May 2007;7(5):332–344. doi: 10.1038/nrc2106. [DOI] [PubMed] [Google Scholar]
- 25.Wang L., McLeod H.L., Weinshilboum R.M. Genomics and drug response. N Engl J Med. Mar 24 2011;364(12):1144–1153. doi: 10.1056/NEJMra1010600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hudson K.L. Genomics, health care, and society. N Engl J Med. Sep 15 2011;365(11):1033–1041. doi: 10.1056/NEJMra1010517. [DOI] [PubMed] [Google Scholar]
- 27.Simon R. Drug-diagnostics co-development in oncology. Front Oncol. Dec 23 2013;3:315. doi: 10.3389/fonc.2013.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Utsugi T. New challenges and inspired answers for anticancer drug discovery and development. Jpn J Clin Oncol. Oct 2013;43(10):945–953. doi: 10.1093/jjco/hyt131. [Epub 2013 Sep 5] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montagna R.A. Meeting the technical challenges of personalized medicine and companion diagnostics. MLO Med Lab Obs. Jan 2012;44(1):16. [18, 20 passim] [PubMed] [Google Scholar]
- 30.Sweet K., Zhang L., Pinilla-Ibarz J. Biomarkers for determining the prognosis in chronic myelogenous leukemia. J Hematol Oncol. Jul 19 2013;6(1):54. doi: 10.1186/1756-8722-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ashariati A., Ugroseno S. Profile of BCR-ABL transcript levels based on Sokal prognostic score in chronic myeloid leukemia patients treated with imatinib. Acta Med Indones. Apr 2013;45(2):107–113. [PubMed] [Google Scholar]
- 32.Martin H., Mali R.S. Pak and Rac GTPases promote oncogenic KIT-induced neoplasms. J Clin Invest. Oct 1 2013;123(10):4449–4463. doi: 10.1172/JCI67509. [Epub 2013 Sep 16] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berger F., Reiser M.F. Micro-RNAs as potential new molecular biomarkers in oncology: have they reached relevance for the clinical imaging sciences? Theranostics. Nov 30 2013;3(12):943–952. doi: 10.7150/thno.7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stenvang J., Kümler I. Biomarker-guided repurposing of chemotherapeutic drugs for cancer therapy: a novel strategy in drug development. Front Oncol. Dec 25 2013;3:313. doi: 10.3389/fonc.2013.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldberg M.S., Hook S.S. Biotargeted nanomedicines for cancer: six tenets before you begin. Nanomedicine (Lond) Feb 2013;8(2):299–308. doi: 10.2217/nnm.13.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kraft J.C., Freeling J.P., Wang Z., Ho RJ. Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J Pharm Sci. Jan 2014;103(1):29–52. doi: 10.1002/jps.23773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldberg M.S. siRNA delivery for the treatment of ovarian cancer. Methods. Sep 15 2013;63(2):95–100. doi: 10.1016/j.ymeth.2013.01.007. [Epub 2013 Feb 10] [DOI] [PubMed] [Google Scholar]
- 38.Lee S.G., Jeong J.H. Nanostructured lipid carrier-loaded hyaluronic acid microneedles for controlled dermal delivery of a lipophilic molecule. Int J Nanomedicine. 2014;9:289–299. doi: 10.2147/IJN.S54529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacConaill L.E., Campbell C.D. Profiling critical cancer gene mutations in clinical tumor samples. PLoS One. Nov 18 2009;4(11):e7887. doi: 10.1371/journal.pone.0007887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matulonis U.A., Hirsch M. High throughput interrogation of somatic mutations in high grade serous cancer of the ovary. PLoS One. 2011;6(9):e24433. doi: 10.1371/journal.pone.0024433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ojesina A.I., Lichtenstein L. Landscape of genomic alterations in cervical carcinomas. Nature. Dec 25 2013 doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanger F. The early days of DNA sequences. Nat Med. Mar 2001;7(3):267–268. doi: 10.1038/85389. [DOI] [PubMed] [Google Scholar]
- 43.Sanger F., Nicklen S. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. Dec 1977;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanger F., Coulson A.R. Cloning in single-stranded bacteriophage as an aid to rapid DNA sequencing. J Mol Biol. Oct 25 1980;143(2):161–178. doi: 10.1016/0022-2836(80)90196-5. [DOI] [PubMed] [Google Scholar]
- 45.MacConaill L.E. Existing and emerging technologies for tumor genomic profiling. J Clin Oncol. May 20 2013;31(15):1815–1824. doi: 10.1200/JCO.2012.46.5948. [Epub 2013 Apr 15] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verma M., Khoury M.J., Ioannidis J.P. Opportunities and challenges for selected emerging technologies in cancer epidemiology: mitochondrial, epigenomic, metabolomic, and telomerase profiling. Cancer Epidemiol Biomarkers Prev. Feb 2013;22(2):189–200. doi: 10.1158/1055-9965.EPI-12-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Enkemann S.A. Standards affecting the consistency of gene expression arrays in clinical applications. Cancer Epidemiol Biomarkers Prev. Apr 2010;19(4):1000–1003. doi: 10.1158/1055-9965.EPI-10-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pickrell W.O., Rees M.I., Chung S.K. Next generation sequencing methodologies—an overview. Adv Protein Chem Struct Biol. 2012;89:1–26. doi: 10.1016/B978-0-12-394287-6.00001-X. [DOI] [PubMed] [Google Scholar]
- 49.McArt D.G., Dunne P.D. Connectivity mapping for candidate therapeutics identification using next generation sequencing RNA-Seq data. PLoS One. Jun 26 2013;8(6):e66902. doi: 10.1371/journal.pone.0066902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marquardt J.U., Andersen J.B. Next-generation sequencing: application in liver cancer—past, present and future? Biology (Basel) Aug 31 2012;1(2):383–394. doi: 10.3390/biology1020383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu B., Morrison C.D. Computational methods for detecting copy number variations in cancer genome using next generation sequencing: principles and challenges. Oncotarget. Nov 2013;4(11):1868–1881. doi: 10.18632/oncotarget.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Illimina. An introduction to next generation sequencing. http://res.illumina.com/documents/products/illumina_sequencing_introduction.pdf
- 53.Qian X., Ba Y., Zhuang Q., Zhong G. RNA-Seq technology and its application in fish transcriptomics. OMICS. Dec 31 2013;18(2):98–110. doi: 10.1089/omi.2013.0110. [Epub 2013 Dec 31] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El-Metwally S., Hamza T. Next-generation sequence assembly: four stages of data processing and computational challenges. PLoS Comput Biol. Dec 2013;9(12):e1003345. doi: 10.1371/journal.pcbi.1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sims D., Ilott N.E. CGAT: computational genomics analysis toolkit. Bioinformatics. Jan 5 2014;30(9):1290–1291. doi: 10.1093/bioinformatics/btt756. [Epub 2014 Jan 5] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ayers K.L., Cordell H.J. Identification of grouped rare and common variants via penalized logistic regression. Genet Epidemiol. Sep 2013;37(6):592–602. doi: 10.1002/gepi.21746. [Epub 2013 Jul 8] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phimister E.G., Feero W.G., Guttmacher A.E. Realizing genomic medicine. N Engl J Med. Feb 23 2012;366(8):757–759. doi: 10.1056/NEJMe1200749. [DOI] [PubMed] [Google Scholar]
- 58.Feero W.G., Guttmacher A.E., Collins F.S. Genomic medicine—an updated primer. N Engl J Med. May 27 2010;362(21):2001–2011. doi: 10.1056/NEJMra0907175. [DOI] [PubMed] [Google Scholar]
- 59.Chang F., Li M.M. Clinical application of amplicon-based next-generation sequencing in cancer. Cancer Genet. Oct 11 2013 doi: 10.1016/j.cancergen.2013.10.003. [pii: S2210-7762(13)00142-7] [DOI] [PubMed] [Google Scholar]
- 60.Labidi-Galy SI, Clauss A. Elafin drives poor outcome in high-grade serous ovarian cancers and basal-like breast tumors. Oncogene. 2014 Jan 27 doi: 10.1038/onc.2013.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Macconaill L.E., Garraway L.A. Clinical implications of the cancer genome. J Clin Oncol. Dec 10 2010;28(35):5219–5228. doi: 10.1200/JCO.2009.27.4944. [Epub 2010 Oct 25] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shin S.C., Kim G., Yang H.B., Park K.W., Kang B.C., Park H.G. Application of the ASLP technology to a novel platform for rapid and noise-free multiplexed SNP genotyping. Biosens Bioelectron. Nov 20 2013;54C:687–694. doi: 10.1016/j.bios.2013.10.071. [DOI] [PubMed] [Google Scholar]
- 63.Rowe L.R., Bentz B.G., Bentz J.S. Detection of BRAF V600E activating mutation in papillary thyroid carcinoma using PCR with allele-specific fluorescent probe melting curve analysis. J Clin Pathol. Nov 2007;60(11):1211–1215. doi: 10.1136/jcp.2006.040105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Badalian-Very G., Vergilio J.A., Fleming M., Rollins B.J. Pathogenesis of Langerhans cell histiocytosis. Annu Rev Pathol. Jan 24 2013;8:1–20. doi: 10.1146/annurev-pathol-020712-163959. [Epub 2012 Aug 6] [DOI] [PubMed] [Google Scholar]
- 65.Badalian-Very G., Vergilio J.A., Degar B.A., Rodriguez-Galindo C., Rollins B.J. Recent advances in the understanding of Langerhans cell histiocytosis. Br J Haematol. Jan 2012;156(2):163–172. doi: 10.1111/j.1365-2141.2011.08915.x. [Epub 2011 Oct 24] [DOI] [PubMed] [Google Scholar]
- 66.Satoh T., Smith A. RAF mutant alleles associated with Langerhans cell histiocytosis, a granulomatous pediatric disease. PLoS One. 2012;7(4):e33891. doi: 10.1371/journal.pone.0033891. [Epub 2012 Apr 10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thomas R.K., Baker A.C. High-throughput oncogene mutation profiling in human cancer. Nat Genet. Mar 2007;39(3):347–351. doi: 10.1038/ng1975. [Epub 2007 Feb 11] [DOI] [PubMed] [Google Scholar]
- 68.Badalian-Very G. A common progenitor cell in LCH and ECD. Blood. 2014 Aug 14;124(7):991–992. doi: 10.1182/blood-2014-06-581736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cancer genome atlas research network, Weinstein J.N. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. Oct 2013;45(10):1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Turner E.H., Lee C., Ng S.B., Nickerson D.A., Shendure J. Massively parallel exon capture and library-free resequencing across 16 genomes. Nat Methods. May 2009;6(5):315–316. doi: 10.1038/nmeth.f.248. [Epub 2009 Apr 6] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tewhey R., Warner J.B. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat Biotechnol. Nov 2009;27(11):1025–1031. doi: 10.1038/nbt.1583. [Epub 2009 Nov 1] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lonigro R.J., Grasso C.S. Detection of somatic copy number alterations in cancer using targeted exome capture sequencing. Neoplasia. Nov 2011;13(11):1019–1025. doi: 10.1593/neo.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Benson A.B., III Data acquisition, tumor heterogeneity and precision medicine: future challenges for oncologic comparative effectiveness research. J Comp Eff Res. Jan 2013;2(1):17–21. doi: 10.2217/cer.12.76. [DOI] [PubMed] [Google Scholar]
- 74.Badalian G., Barbai T., Derecskei K., Szendrôi M., Tímár J. Phenotype of bone metastases of non-small cell lung cancer: epidermal growth factor receptor expression and K-RAS mutational status. Pathol Oncol Res. 2007;13(2):99–104. doi: 10.1007/BF02893484. [Epub 2007 Jul 3] [DOI] [PubMed] [Google Scholar]
- 75.Michor F., Polyak K. The origins and implications of intratumor heterogeneity. Cancer Prev Res (Phila) Nov 2010;3(11):1361–1364. doi: 10.1158/1940-6207.CAPR-10-0234. [Epub 2010 Oct 19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. Jan 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hammerman P.S., Lawrence M.S. Comprehensive genomic characterization of squamous cell lung cancers. Cancer genome atlas research network. Nature. Sep 27 2012;489(7417):519–525. doi: 10.1038/nature11404. [Epub 2012 Sep 9] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maher C.A., Palanisamy N. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc Natl Acad Sci U S A. Jul 28 2009;106(30):12353–12358. doi: 10.1073/pnas.0904720106. [Epub 2009 Jul 10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Padilla J., Jenkins N.T. Transcriptome-wide RNA sequencing analysis of rat skeletal muscle feed arteries. Part II: impact of exercise training in obesity. J Appl Physiol (1985) Jan 9 2014;7(2):e00225. doi: 10.1152/japplphysiol.01234.2013. eCollection 2014 Feb 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.FANTOM Consortium, Suzuki H. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat Genet. May 2009;41(5):553–562. doi: 10.1038/ng.375. [Epub 2009 Apr 19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fujisawa S., Nakamae H. Efficacy and safety of dasatinib versus imatinib in Japanese patients with newly diagnosed chronic-phase chronic myeloid leukemia (CML-CP): subset analysis of the DASISION trial with 2-year follow-up. Int J Hematol. Dec 20 2013;99(2):141–153. doi: 10.1007/s12185-013-1470-1. [Epub 2013 Dec 20] [DOI] [PubMed] [Google Scholar]
- 82.Schiffer C.A. Which TKI should be recommended as initial treatment for CML in chronic phase? Oncology (Williston Park) Oct 2012;26(10):912. [914] [PubMed] [Google Scholar]
- 83.Zhao R., Follows G.A. Inhibition of the Bcl-xL deamidation pathway in myeloproliferative disorders. N Engl J Med. Dec 25 2008;359(26):2778–2789. doi: 10.1056/NEJMoa0804953. [DOI] [PubMed] [Google Scholar]
- 84.Jabbour E., Cortes J., Ravandi F., O'Brien S., Kantarjian H. Targeted therapies in hematology and their impact on patient care: chronic and acute myeloid leukemia. Semin Hematol. Oct 2013;50(4):271–283. doi: 10.1053/j.seminhematol.2013.09.006. (Epub 2013 Oct 3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Verheul H.M., Pinedo H.M. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat Rev Cancer. Jun 2007;7(6):475–485. doi: 10.1038/nrc2152. [DOI] [PubMed] [Google Scholar]
- 86.Belum V.R., Fontanilla Patel H. Skin toxicity of targeted cancer agents: mechanisms and intervention. Future Oncol. Aug 2013;9(8):1161–1170. doi: 10.2217/fon.13.62. [DOI] [PubMed] [Google Scholar]
- 87.Balagula Y., Rosen S.T., Lacouture ME. The emergence of supportive oncodermatology: the study of dermatologic adverse events to cancer therapies. J Am Acad Dermatol. Sep 2011;65(3):624–635. doi: 10.1016/j.jaad.2010.06.051. [Epub 2011 Jul 20] [DOI] [PubMed] [Google Scholar]
- 88.Boone S.L., Rademaker A., Liu D., Pfeiffer C., Mauro D.J., Lacouture M.E. Impact and management of skin toxicity associated with anti-epidermal growth factor receptor therapy: survey results. Oncology. 2007;72(3-4):152–159. doi: 10.1159/000112795. [Epub 2007 Dec 21] [DOI] [PubMed] [Google Scholar]
- 89.Shaw A.T., Hsu P.P., Awad M.M., Engelman J.A. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer. Nov 2013;13(11):772–787. doi: 10.1038/nrc3612. [Epub 2013 Oct 17] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Soverini S., Branford S. Implications of BCR-ABL1 kinase domain-mediated resistance in chronic myeloid leukemia. Leuk Res. Jan 2014;38(1):10–20. doi: 10.1016/j.leukres.2013.09.011. [Epub 2013 Sep 23] [DOI] [PubMed] [Google Scholar]
- 91.Zhang J., Yang P.L. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. Jan 2009;9(1):28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 92.Yang S.H. Molecular basis of drug resistance: epidermal growth factor receptor tyrosine kinase inhibitors and anaplastic lymphoma kinase inhibitors. Tuberc Respir Dis (Seoul) Nov 2013;75(5):188–198. doi: 10.4046/trd.2013.75.5.188. [Epub 2013 Nov 29] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gomez G.G., Wykosky J., Zanca C., Furnari F.B., Cavenee W.K. Therapeutic resistance in cancer: microRNA regulation of EGFR signaling networks. Cancer Biol Med. Dec 2013;10(4):192–205. doi: 10.7497/j.issn.2095-3941.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dibb N.J., Dilworth S.M. Switching on kinases: oncogenic activation of BRAF and the PDGFR family. Nat Rev Cancer. Sep 2004;4(9):718–727. doi: 10.1038/nrc1434. [DOI] [PubMed] [Google Scholar]
- 95.Greuber E.K., Smith-Pearson P., Wang J., Pendergast A.M. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer. Aug 2013;13(8):559–571. doi: 10.1038/nrc3563. [Epub 2013 Jul 11] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Seton-Rogers S. Oncogenes: direct hit on mutant RAS. Nat Rev Cancer. Jan 2014;14(1):8–9. doi: 10.1038/nrc3650. [Epub 2013 Dec 5] [DOI] [PubMed] [Google Scholar]
- 97.Ross J.S., Symmans W.F., Pusztai L., Hortobagyi G.N. Pharmacogenomics and clinical biomarkers in drug discovery and development. Am J Clin Pathol. Dec 2005;124:S29–S41. doi: 10.1309/XYQAFANAPYNC6X59. [Suppl.] [DOI] [PubMed] [Google Scholar]
- 98.Marton M.J., Weiner R. Practical guidance for implementing predictive biomarkers into early phase clinical studies. Biomed Res Int. 2013;2013:891391. doi: 10.1155/2013/891391. [Epub 2013 Oct 22] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee M.Y., Borgiani P. High warfarin sensitivity in carriers of CYP2C9*35 is determined by the impaired interaction with P450 oxidoreductase. Pharmacogenomics J. Dec 10 2013 doi: 10.1038/tpj.2013.41. [DOI] [PubMed] [Google Scholar]
- 100.Ye C., Jin H., Zhang R. Variability of warfarin dose response associated with CYP2C9 and VKORC1 gene polymorphisms in Chinese patients. J Int Med Res. Feb 2013;42(1):67–76. doi: 10.1177/0300060513499094. [Epub 2013 Nov 28] [DOI] [PubMed] [Google Scholar]
- 101.Lee M.T., Klein T.E. Pharmacogenetics of warfarin: challenges and opportunities. J Hum Genet. Jun 2013;58(6):334–338. doi: 10.1038/jhg.2013.40. [Epub 2013 May 9] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dotson W.D., Douglas M.P. Prioritizing genomic applications for action by level of evidence: a horizon-scanning method. Clin Pharmacol Ther. Nov 14 2013 doi: 10.1038/clpt.2013.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Emadi A., Karp J.E. The clinically relevant pharmacogenomic changes in acute myelogenous leukemia. Pharmacogenomics. Aug 2012;13(11):1257–1269. doi: 10.2217/pgs.12.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lawrence M.S., Stojanov P. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. Jul 11 2013;499(7457):214–218. doi: 10.1038/nature12213. [Epub 2013 Jun 16] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lôrincz T., Tóth J., Badalian G., Tímár J., Szendrôi M. HER-2/neu genotype of breast cancer may change in bone metastasis. Pathol Oncol Res. 2006;12(3):149–152. doi: 10.1007/BF02893361. [Epub 2006 Sep 23] [DOI] [PubMed] [Google Scholar]
- 106.Badalian G., Derecskei K., Szendroi A., Szendroi M., Timar J. EGFR and VEGRFR protein expression in bone metastases of clear cell renal cancer. Anticancer Res. Mar-Apr 2007;27(2):889–894. [PubMed] [Google Scholar]
- 107.Marusyk A., Polyak K. Cancer. Cancer cell phenotypes, in fifty shades of grey. Science. Feb 1 2013;339(6119):528–529. doi: 10.1126/science.1234415. [DOI] [PubMed] [Google Scholar]
- 108.Lynch T.J., Jr., Kim E.S., Eaby B., Garey J., West D.P., Lacouture M.E. Epidermal growth factor receptor inhibitor-associated cutaneous toxicities: an evolving paradigm in clinical management. Oncologist. May 2007;12(5):610–621. doi: 10.1634/theoncologist.12-5-610. [DOI] [PubMed] [Google Scholar]
- 109.Scripture C.D., Figg W.D., Sparreboom A. Peripheral neuropathy induced by paclitaxel: recent insights and future perspectives. Curr Neuropharmacol. Apr 2006;4(2):165–172. doi: 10.2174/157015906776359568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Haroche J., Charlotte F. High prevalence of BRAF V600E mutations in Erdheim–Chester disease but not in other non-Langerhans cell histiocytoses. Blood. Sep 27 2012;120(13):2700–2703. doi: 10.1182/blood-2012-05-430140. [Epub 2012 Aug 9] [DOI] [PubMed] [Google Scholar]
- 111.Scripture C.D., Sparreboom A., Figg W.D. Modulation of cytochrome P450 activity: implications for cancer therapy. Lancet Oncol. Oct 2005;6(10):780–789. doi: 10.1016/S1470-2045(05)70388-0. [DOI] [PubMed] [Google Scholar]
- 112.Allen C.E., McClain K.L. Erdheim–Chester: beyond the lesion. Blood. Mar 10 2011;117(10):2745–2746. doi: 10.1182/blood-2011-01-330233. [DOI] [PubMed] [Google Scholar]
- 113.Seton-Rogers S. Multiple myeloma: destruction of Ikaros. Nat Rev Cancer. Dec 23 2013;14(1):9. [Google Scholar]
- 114.Valencia P.M., Farokhzad O.C., Rhee M., Langer R., Farokhzad O.C., Karnik R. Microfluidic technologies for accelerating the clinical translation of nanoparticles. Nat Nanotechnol. Oct 2012;7(10):623–629. doi: 10.1038/nnano.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Valencia P.M., Pridgen E.M. Microfluidic platform for combinatorial synthesis and optimization of targeted nanoparticles for cancer therapy. ACS Nano. Dec 23 2013;7(12):10671–10680. doi: 10.1021/nn403370e. [Epub 2013 Nov 11] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hydbring P., Badalian-Very G. Version 3. F1000Res; 2013 Jun 6. Clinical applications of microRNAs. [revised 2013 Oct 8];2:136. http://dx.doi.org/10.12688/f1000research.2-136.v3 eCollection 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Goldberg M., Langer R., Jia X. Nanostructured materials for applications in drug delivery and tissue engineering. J Biomater Sci Polym Ed. 2007;18(3):241–268. doi: 10.1163/156856207779996931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Salvador-Morales C., Valencia P.M., Gao W., Karnik R., Farokhzad O.C. Recent developments in multifunctional hybrid nanoparticles: opportunities and challenges in cancer therapy. Front Biosci (Elite Ed) Jan 1 2012;4:529–545. doi: 10.2741/e398. [DOI] [PubMed] [Google Scholar]
- 119.Ferrari R., Lupi M. Integrated multiplatform method for in vitro quantitative assessment of cellular uptake for fluorescent polymer nanoparticles. Nanotechnology. Jan 7 2014;25(4):045102. doi: 10.1088/0957-4484/25/4/045102. [DOI] [PubMed] [Google Scholar]
- 120.De Jong W.H., Borm P.J. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133–149. doi: 10.2147/ijn.s596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gerber A., Bundschuh M., Klingelhofer D., Groneberg D.A. Gold nanoparticles: recent aspects for human toxicology. J Occup Med Toxicol. Dec 11 2013;8(1):32. doi: 10.1186/1745-6673-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vandebriel R.J., De Jong W.H. A review of mammalian toxicity of ZnO nanoparticles. Nanotechnol Sci Appl. Aug 15 2012;5:61–71. doi: 10.2147/NSA.S23932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Elsabahy M., Zhang S. Surface charges and shell crosslinks each play significant roles in mediating degradation, biofouling, cytotoxicity and immunotoxicity for polyphosphoester-based nanoparticles. Sci Rep. Nov 22 2013;3:3313. doi: 10.1038/srep03313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Polat B.E., Blankschtein D., Langer R. Low-frequency sonophoresis: application to the transdermal delivery of macromolecules and hydrophilic drugs. Expert Opin Drug Deliv. Dec 2010;7(12):1415–1432. doi: 10.1517/17425247.2010.538679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ferrari R., Colombo C. Synthesis of surfactant free PCL–PEG brushed nanoparticles with tunable degradation kinetics. Int J Pharm. Sep 10 2013;453(2):551–559. doi: 10.1016/j.ijpharm.2013.06.020. [Epub 2013 Jun 21] [DOI] [PubMed] [Google Scholar]
- 126.Gao W., Hu C.M., Fang R.H., Zhang L. Liposome-like nanostructures for drug delivery. J Mater Chem B Mater Biol Med. Dec 2013;28:1(48). doi: 10.1039/C3TB21238F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Guadagnini R., Moreau K., Hussain S., Marano F., Boland S. Toxicity evaluation of engineered nanoparticles for medical applications using pulmonary epithelial cells. Nanotoxicology. Nov 29 2013 doi: 10.3109/17435390.2013.855830. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 128.Kwon S., Yang Y.S. Nasal and pulmonary toxicity of titanium dioxide nanoparticles in rats. Toxicol Res. Dec 2012;28(4):217–224. doi: 10.5487/TR.2012.28.4.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Scott R., Crabbe D., Krynska B., Ansari R., Kiani M.F. Aiming for the heart: targeted delivery of drugs to diseased cardiac tissue. Expert Opin Drug Deliv. Nov, 2013;5(4):459–470. doi: 10.1517/17425247.5.4.459. [DOI] [PubMed] [Google Scholar]
- 130.Vaudana Elisabetta. The innovative medicine initiative: a public private partnership model to foster drug discovery. Comput Struct Biotechnol J. 2013;6(7) doi: 10.5936/csbj.201303017. [e.201303017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lounnas Valere, Ritschel Tina, Kelder J., McGuire R., Bywater R.P., Foloppe N. Current progress in structure-based rational drug design marks a new mindset in drug discovery. Comput Struct Biotechnol J. 2013;5(6) doi: 10.5936/csbj.201302011. [e.201302011] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hopkins A.L., Groom C.R. The drugable genome. Nat Rev Discov. Sep 2002;1(9):727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]