

Abstract
Arsenic-containing lipids (arsenolipids) are novel natural products recently shown to be widespread in marine animals and algae. Research interest in these arsenic compounds lies in their possible role in the membrane chemistry of organisms and, because they occur in many popular seafoods, their human metabolism and toxicology. Progress has been restricted, however, by the lack of standard arsenolipids and of a quantitative method for their analysis. We report that the certified reference material CRM 7405-a (Hijiki) is a rich source of arsenolipids, and we describe a method based on HPLC-ICPMS/ESMS to quantitatively measure seven of the major arsenolipids present. Sample preparation involved extraction with DCM/methanol, a cleanup step with silica, and conversion of the (oxo)arsenolipids originally present to thio analogues by brief treatment with H2S. Compared to their oxo analogues, the thioarsenolipids showed much sharper peaks on reversed-phase HPLC, which facilitated their resolution and quantification. The compounds were determined by HPLC-ICPMS and HPLC-ESMS, which provided both arsenic-selective detection and high resolution molecular mass detection of the arsenolipids. In this way, the concentrations of two arsenic-containing hydrocarbons and five arsenosugar phospholipids are reported in the CRM Hijiki. This material may serve as a convenient source of characterized arsenolipids to delineate the presence of these compounds in seafoods and to facilitate research in a new era of arsenic biochemistry.
The presence of lipid-soluble arsenic (arsenolipids) in marine organisms has been known since the 1920s when Sadolin first reported arsenic concentrations of 3.0–4.5 μg g–1 in cod liver oil.1 Further work indicated the presence of at least two types of unidentified arsenolipids in the oil from herring and cod liver,2 and lipid-soluble arsenicals were also reported from marine and freshwater algae3 and in marine invertebrates.4 The first structure for an arsenolipid was provided in 1988 by the classic study of Morita and Shibata who identified, mainly by 1H NMR, an arsenosugar phospholipid in a marine alga (Wakame).5
Recent work has greatly extended the range of arsenolipids found in marine samples: arsenic-containing fatty acids (AsFA), first reported in cod liver oil,6 have now been found in a wide range of fish species7−10 in addition to algae;11 arsenic-containing hydrocarbons (AsHC), following the first report of their presence in capelin,12 have also been found in various fish13,14 and in two species of algae;15 and arsenosugar phospholipids (AsPL) have been found in algae.11,15 In total, about 55 arsenolipids have been identified so far, with over 40 of them being reported in the last two years. Most of these arsenolipids have been identified by analytical methods based on separations by HPLC and the complementary use of inductively coupled plasma mass spectrometry (ICPMS) and electrospray mass spectrometry (ESMS) for the detection of arsenic and molecular species, respectively. Figure 1 shows the structures of seven arsenolipids referred to in the work to be reported here.
Figure 1.

Seven arsenolipids found in algae and referred to in the current study.
Arsenolipids initially attracted research interest because of their novel structures, their possible involvement in membrane biochemistry and, because they are present in common seafoods, issues of human health and arsenic toxicity.16 Possible human health issues have recently been highlighted by the discovery that arsenic-containing hydrocarbons show toxicity comparable to that of the highly toxic arsenite in cytotoxicity tests with cultured human bladder and liver cells.17
Further progress in the biological chemistry and toxicology of arsenolipids has been hindered by the lack of standards and quantitative analytical methods. Although some of the AsHCs and AsFAs have recently been synthesized,18 there are no standards or reference compounds available for AsPLs. With the view of assisting research in the area of arsenolipids in biological chemistry, we report the characterization and quantification of seven of the major arsenolipids in the certified reference material NMIJ 7405-a (Hijiki). This CRM has already been certified for total arsenic and inorganic AsV.19
Experimental Section
Chemicals and Standards
Water was obtained from a Milli-Q system (18.2 MΩ cm, Millipore GmbH, Vienna, Austria). Methanol (≥99.9%, MeOH), dichloromethane (≥99.9%, DCM), chloroform (≥99.9%), methyl-tertiary-butyl ether (≥99.5%, MTBE), acetonitrile (≥99.9%, AcN), toluene (≥99.8%), formic acid (≥98%), and ammonia (25%) were obtained from Carl Roth GmbH (Karlsruhe, Germany); hexane (≥95%), ethyl acetate (≥99.5%, EtOAc), diethyl ether (≥99.5%, Et2O), and acetone (≥99.5%) were purchased from Sigma-Aldrich (Vienna, Austria); and ethanol (≥99.9%, EtOH) and silica gel 60 were obtained from Merck (Buchs, Switzerland). Arsenic(V) standard solution (998 ± 5 mg As L–1) was also obtained from Merck. The certified reference material (CRM) was NMIJ CRM 7405-a (Trace Elements and Arsenic Compounds in Seaweed—Hijiki) from the Natural Metrology Institute of Japan (Tsukuba, Ibaraki, Japan). Saturated hydrogen sulfide (H2S) solution was prepared by bubbling H2S gas (produced by a Kipp’s apparatus) through EtOH for 10 min. Standard compounds of AsHC332, AsHC360 and AsHC388 were synthesized in-house according to Taleshi et al.18 and prepared by dissolving 7.5 ± 0.2 μg (as As) in methanol (1 mL).
Instrumentation
Solvents were evaporated on a centrifugal lyophilizer (Maxi Dry Plus, Heto Holten, Allerød, Denmark). Acid digestion of samples was performed with an Ultraclave microwave system (MLS GmbH, Leutkirch, Germany). HPLC separations were performed on an Agilent 1100 series system prior to online ICPMS measurements with an Agilent 7500ce series instrument (Agilent Technologies, Waldbronn, Germany). The ICPMS was equipped with an Ari Mist HP nebulizer (Burgerner, Mississauga, Canada) and an ESI PC3 Peltier cooled cyclonic spray chamber (Elemental Scientific, Omaha, USA). High Resolution-ESMS measurements were carried out on a Q-Exactive Hybrid Quadrupole-Orbitrap MS after HPLC performed on a Dionex Ultimate 3000 series instrument (Thermo Fischer Scientific, Erlangen, Germany). The ESMS was equipped with an atmospheric pressure ionization source employing electrospray nebulization with nitrogen as nebulizer gas. Measurements were performed in positive mode, with a drying gas temperature of 350 °C, a spray voltage of 3.2 kV and a resolution of 70 000. The mass range was set to m/z 300–1100 without additional fragmentation.
Total Arsenic Determination—Acid Digestion and ICPMS
The CRM 7405-a (15 mg), a portion of all sample extracts (100 μL), and the AsHC standards (50 μL of the prepared 7.5 ± 0.2 mg As L–1 standards) were analyzed for total arsenic content in triplicate. The samples were weighed into quartz tubes (12 mL) and solvents, if present, were evaporated (10 mbar, room temperature; Maxi Dry Plus). Water (2 mL) and HNO3 (2 mL) were added to the samples before the tubes were covered with Teflon caps, transferred to a Teflon rack in the microwave system which then was closed. Argon (40 bar) was applied and the following temperature program was started: 0–10 min, 80 °C; 10–30 min, 150 °C; 30–45 min, 250 °C; 45–65 min, 250 °C. The digest solutions were then allowed to cool to room temperature, transferred to polypropylene tubes (15 mL) and diluted with water to 9.9 mL. Finally, 100 μL of a solution containing 1 mg L–1 Ge, In, and Te was added to all digested samples as internal standards, giving a final concentration of 10 μg L–1 Ge, In, and Te.
Determination of arsenic in the digested samples was carried out by ICPMS in collision cell mode (He, 5 mL min–1) to avoid polyatomic interferences from argon chloride (40Ar35Cl) on arsenic (75As). Standards for calibration were prepared in 20% HNO3 for matrix matching and contained 10 μg L–1 Ge, In and Te as internal standards. The total arsenic determination was validated against the certified reference material CRM 7405-a (Hijiki) with a certified value for arsenic of 35.8 ± 0.9 μg As g–1; we obtained 35.6 ± 0.9 μg As g–1 (n = 3).
Optimization of the Extraction Procedure
The following parameters were varied to optimize the extraction process for arsenolipids from the CRM 7405-a; all experiments were performed in triplicate using 100 mg of CRM Hijiki. (i) Extraction solutions: The extractants tested were hexane (5 mL) or a solvent/MeOH mixture (always 2 + 1 v/v, 5 mL) where the solvents were DCM, chloroform, toluene, acetone, Et2O, MTBE, EtOAc, or AcN. (ii) For DCM/MeOH (2 + 1) volume of extraction solution tested was 3, 5, and 7 mL. (iii) Sequential extraction was tested for the DCM/MeOH mixture (4 × 5 mL). (iv) Recoveries were tested by spiking CRM at levels comparable to the natural level of two arsenolipids with standards of AsHC332 (750 ng As g–1) and AsHC360 (60 ng As g–1) prior to extraction. (v) The efficiency of the cleanup step (silica column) was tested by comparing presilica (crude) and postsilica fractions; for the presilica fraction, the crude extract (1 mL) was taken directly after the centrifugation step, evaporated to dryness and redissolved in EtOH (500 μL).
Extraction Procedure for Arsenic Speciation Analysis
Triplicates of CRM 7405-a (∼100 mg, weighed to a precision of 0.1 mg) were extracted with 5 mL DCM/MeOH (2 + 1, v/v) on a rotatory cross for 1 h at room temperature. The mixture was centrifuged (10 min, 2100 G) to separate the pellet from the liquid phase. A portion of the supernatant (4 mL) was transferred to a silica column (glass Pasteur pipet, 150 × 5 mm, filled to a height of 4 cm with silica gel 60), conditioned with MeOH/acetone (1 + 1, v/v) containing 1% formic acid (5 mL); the column was washed with the “conditioning” mixture (4 mL), then MeOH (2 mL), and finally MeOH containing 1% NH3 (8 mL). H2S (in EtOH, 100 μL) was added to the alkaline fraction and mixed prior to complete evaporation of the solvent (10 mbar, room temperature). The obtained pellet was redissolved in EtOH (500 μL) with ultrasonication and vortexing at room temperature. All steps in the procedure were performed with glassware.
Chemical Conversion Procedure—Formation and Stability of Thio Arsenolipids
A solution containing standards of AsHC332, AsHC360, and AsHC388 (300 μg As L–1 each) was prepared in EtOH to a final volume of 1 mL and measured with RP-HPLC-HR-ESMS (t = 0). Then, a saturated H2S solution (100 μL) was added, the mixture was shaken and immediately measured again (t = ca. 0.5 min); repeated measurements (RP-HPLC-HR-ESMS) were made at the following times: t = 11, 22, 32, and 122 min and 1 and 7 days.
Identification and Quantification of Arsenic Species (RP-HPLC-ICPMS/HR-ESMS)
Separation was carried out by reversed-phase HPLC using a ZORBAX Eclipse XDB-C8 column (4.6 × 150 mm, 5 μm particle size). Elution was performed with an aqueous solution containing 0.1% formic acid and EtOH containing 0.1% formic acid with the following gradient: 0–3 min, 70% EtOH; 3–10 min, 90% EtOH; and 10–20 min, 90% EtOH. The flow rate was 1.0 mL min–1 and the injection volume was 20 μL for ICPMS and 5 μL for HR-ESMS measurements. The HPLC effluent was split, whereby 20% was transferred to the detection unit and 80% to waste (to avoid destabilization of the plasma caused by overloading with organic solvent) using a passive splitter (Analytical Scientific Instruments, Richmond, USA). For ICPMS measurements, a support flow of water containing 1% formic acid and 10 μg L–1 Ge and Te (1.0 mL min–1) was introduced through a T-piece after the splitter. Carbon compensation20 was performed by continuous addition of water/EtOH (4 + 1, v/v) delivered with an ISIS pump (0.02 rpm) to ensure a constant carbon content reaching the plasma. ESMS data were obtained in positive scan mode at m/z 300–1100. Data evaluation was done with chromatographic software Xcalibur Version 3.0.63 (Thermo Scientific, San Jose, USA). ICPMS signals were recorded at m/z 75 (75As and 40Ar35Cl) and m/z 77 (40Ar37Cl, for possible chloride interferences) at dwell times of 300 ms, and for internal standards at m/z 74 (74Ge) and m/z 125 (125Te) at dwell times of 100 ms. A summary of the used techniques and parameters is given in Table 1. Data evaluation was carried out with chromatographic software MassHunter Version B.01.01 (Agilent Technologies, Waldbronn, Germany). Quantification was based on peak areas against external calibration with standard AsHC332, AsHC360 and AsHC388.
Table 1. Parameters for the Quantification and Speciation of Arsenolipids with RP-HPLC-ICPMS/ESMS.
| HPLC | Agilent 1100 |
|---|---|
| column | ZORBAX Eclipse XDB-C8 (4.6 × 150 mm, 5 μm) |
| column temperature | 30 °C |
| injection volume | 20 μL |
| flow rate | 1 mL min–1 |
| mobile phase | A: 0.1% formic acid in water |
| B: 0.1% formic acid in ethanol | |
| gradient | 0–3 min, 70% B; 3–10 min, 90% B; 10–20 min, 90% B |
| splitter | 20% delivered to ICPMS or ESMS |
| support flow | 1% formic acid in water (containing internal standard: Te and Ge; 1 mL min–1) (ICPMS only) |
| gradient compensation | 20% EtOH in water (delivered with ISIS; 0.02 rpm) (ICPMS only) |
| ICPMS (total) | Agilent 7500ce |
|---|---|
| mode | full quant |
| RF power | 1550 W |
| carrier gas | 0.79 mL min–1 |
| reaction mode | 5 mL min–1 He |
| masses recorded | m/z 72 (Ge), m/z 75 (As), m/z 77 (Se), m/z 125 (Te) |
| ICPMS (species) | Agilent 7500ce |
|---|---|
| mode | time resolved analysis |
| RF power | 1550 W |
| carrier gas | 0.71 mL min–1 |
| optional gas | 5% O2 |
| masses recorded | m/z 72 (Ge), m/z 75 (As), m/z 77 (Se), m/z 125 (Te) |
| ESMS | Thermo Fisher—Q Exactive Hybrid Quadrupole-Orbitrap |
|---|---|
| mode | positive |
| spray voltage | 3.2 kV |
| capillary temperature | 350 °C |
| resolution (FWHM) | 70 000 |
| scan range | m/z 300–1100 |
Results and Discussion
We aimed to develop a method based on HPLC-ICPMS/ESMS for the quantitative determination of arsenolipids applicable to the certified reference material CRM 7405-a (Hijiki), which could then be used as a characterized source of arsenolipids. The method development involved the following steps: (i) choice of solvent and extraction conditions, (ii) cleanup step using silica, (iii) conversion of the (oxo) arsenolipids to their thio derivatives, and (iv) identification and quantification of the major arsenolipids in CRM Hijiki.
Extraction of Arsenolipids
Methods for extracting lipids from biological samples have traditionally used methanol/chloroform mixtures,21 whereas more recent work has demonstrated the practical advantages of using methanol/MTBE.22 We tested the extraction efficiency of hexane (polarity index =0) and eight solvents as mixtures with methanol (2 + 1, v/v) by measuring the total arsenic extracted (Table 2) and, individually, the seven main arsenolipids in the extracts (Figure 2 and Supporting Information, Table S-1). Hexane extracted less than 3% (0.97 μg g–1) of the total arsenic in the original CRM (35.6 μg g–1). Much higher extraction efficiencies (∼18%) were obtained with mixtures of methanol with toluene (6.58 μg g–1), DCM (6.24 μg g–1), or chloroform (6.21 μg g–1); the other five solvent mixtures returned lower extraction efficiencies of ∼10–13%.
Table 2. Extraction of Arsenic from Hijiki CRM 7405-a (Initial Arsenic Content = 35.6 ± 0.9 μg As g–1) by Various Extraction Solutionsa.
| solvent | total As [μg As g–1] | % of total As | relative polarity of pure solvents23 |
|---|---|---|---|
| hexane (pure solvent) | 0.97 ± 0.21 | 2.7 ± 0.6 | 0.009 |
| toluene | 6.58 ± 0.19 | 18.5 ± 0.5 | 0.099 |
| ethyl ether | 4.22 ± 0.05 | 11.9 ± 0.2 | 0.117 |
| MTBE | 4.50 ± 0.12 | 12.6 ± 0.4 | 0.124 |
| ethyl acetate | 4.72 ± 0.18 | 13.3 ± 0.5 | 0.228 |
| chloroform | 6.21 ± 0.31 | 17.4 ± 0.9 | 0.259 |
| DCM | 6.24 ± 0.05 | 17.5 ± 0.2 | 0.309 |
| acetone | 4.42 ± 0.14 | 12.4 ± 0.4 | 0.355 |
| acetonitrile | 3.74 ± 0.11 | 10.5 ± 0.3 | 0.460 |
Except for hexane, which was used alone, extraction solution was a mixture of solvent/MeOH (2 + 1, v/v). In all cases, 5 mL of solvent was used with 100 mg of CRM Hijiki (n = 3).
Figure 2.
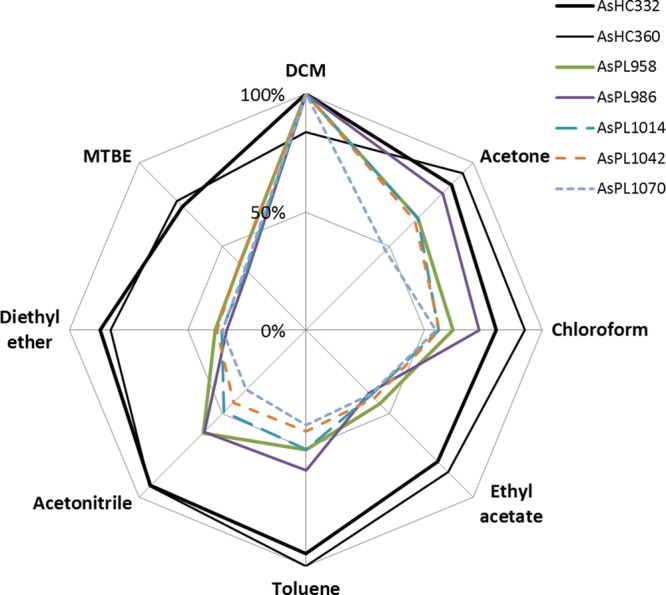
Mean relative extraction efficiencies (n = 3) for seven arsenolipids depending on solvent mixture (100 mg CRM extracted with 5 mL solvent/MeOH (2 + 1, v/v). For each arsenolipid, values are recorded as the amount of arsenolipid extracted by a particular solvent relative to the amount extracted by the most efficient solvent (expressed as %); detailed data are presented in Supporting Information, Table S-1).
There were also differences, depending on solvent mixture, in the extraction efficiencies for the two major groups of arsenolipids in CRM Hijiki, namely arsenosugar phospholipids (AsPLs) and arsenic-containing hydrocarbons (AsHCs). All eight solvent mixtures showed comparable ability to extract the AsHCs giving values of 80–95% of that obtained for the most efficient solvent mixture (DCM/methanol for AsHC332 and toluene/methanol for AsHC360). The AsPLs, however, showed a clear preference for the DCM/methanol mixture which was the most efficient solvent in all cases: the other seven solvent mixtures showed efficiencies of 35–80% relative to DCM/methanol (Figure 2). Thus, further method development was performed with the DCM/methanol mixture.
Up to this point, extractions had been performed with 100 mg of (dry) CRM Hijiki and a single extraction with 5 mL of solvent mixture. The extraction efficiency was tested for the DCM/methanol mixture also for volumes of 3 and 7 mL (100 mg CRM Hijiki) and found to have no significant effect on extraction efficiency; the 5 mL volume was the most convenient for handling. Sequential extraction, whereby 100 mg CRM Hijiki was treated with 4 × 5 mL of the DCM/methanol mixture, showed that ca. 90% was removed in the first extraction (Supporting Information, Tables S-2 and S-3). On the basis of these results, a single extraction with 5 mL of the DCM/methanol mixture was used in the subsequent steps of method development.
Cleanup Step using Silica
Preliminary attempts to analyze the crude extract from CRM Hijiki showed that although HPLC-ICPMS data were reasonable, the data from HPLC-ESMS were strongly matrix-affected. Thus, a previously described cleanup step15 was incorporated whereby a portion (4 mL) of the extract was applied to a small silica column and the column was washed with MeOH/acetone (1 + 1, v/v, containing 1% formic acid) and then MeOH to remove matrix components. By virtue of the interaction between the basic O=AsR3 group of the arsenolipids and silica, the arsenolipids are retained on the small column whereas normal lipids are not. To remove the arsenolipids, the column was then washed with MeOH containing 1% NH3. In this way, the arsenolipids were recovered quantitatively, while ∼98% (by mass) of the original matrix was removed.
Conversion of the (Oxo) Arsenolipids to their Thio Derivatives
Similar to many naturally occurring water-soluble arsenicals, arsenolipids contain arsenic in the oxo form, that is, O=AsR3. Our early attempts to effect separation of the arsenolipids in CRM Hijiki were hampered by broad poorly resolved peaks, which we presumed was due to the polar oxo arsenic group interacting with the silica backbone of the HPLC-column. However, the thio analogues of naturally occurring oxo arsenicals (i.e., containing S=AsR3), which are readily formed from the oxo compounds, are much less polar. Our previous experience24,25 with the separation of water-soluble arsenic compounds demonstrated that the thio analogues are better resolved than the oxo compounds. We found similar effects for arsenolipids–for the three standard compounds tested, the thio analogues were better resolved and gave sharper peaks compared with the oxo analogues (Figure 3). Resolution was further improved by running the HPLC gradients with mixtures containing ethanol rather than methanol. The higher elution power of ethanol offered a wider scope of gradient conditions compared to methanol which required 100% solvent for long periods to elute all compounds.
Figure 3.
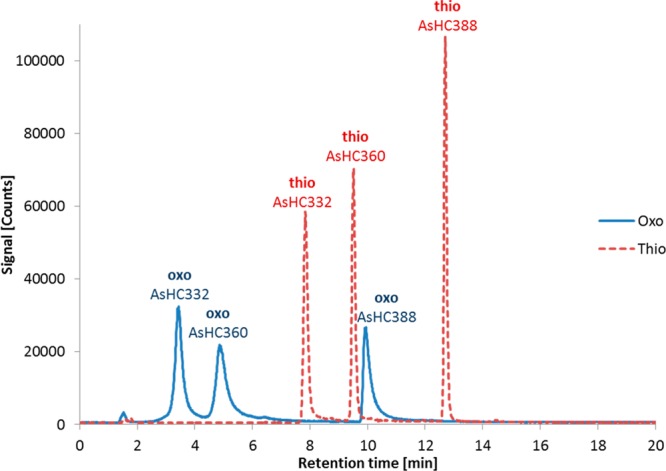
RP-HPLC-ICPMS chromatograms of the three arsenolipid standard compounds AsHC332, AsHC360, and AsHC388 as oxo and thio analogues (300 μg As L–1 of each compound in EtOH). ZORBAX Eclipse XDB-C8 (4.6 × 150 mm, 5 μm); mobile phase, water/ethanol gradient (70–90% EtOH, incl. 0.1% formic acid); flow rate, 1 mL min–1; column temperature, 30 °C; injection volume, 20 μL.
We then investigated the rate of the oxo/thio conversion, and the stability of the thio products, by measuring with HPLC-ESMS both the appearance of the thio analogue and the disappearance of the oxo analogue following the addition of H2S. At the first time measurement point (t = 0.5 min) the reaction was already ∼60% complete, and at t = 11 min, it was >99% complete (Supporting Information, Table S-4). Even after 24 h, the signal for the thio compound remained undiminished. Thus, the reaction was quantitative and fast (<11 min), and the product was stabile in solution for at least 1 day. In view of the ease of the derivatization and the ensuing chromatographic advantages, further steps in the method development were performed on the thio derivatives of the arsenolipids.
Identification and Quantification of Seven Major Arsenolipids in CRM Hijiki
The optimized method incorporating DCM/MeOH extraction, silica cleanup and thio derivatization was then applied to the CRM Hijiki. We checked the recovery of the whole procedure by spiking the CRM Hijiki with two standard compounds AsHC332 and AsHC360, and found recoveries of 101 ± 6% and 85 ± 13% (n = 3), respectively (Figure 4). The lower precision obtained for AsHC360 reflected its much lower concentration in Hijiki compared with AsHC332 (90 ± 5 ng As g–1 versus 1073 ± 44 ng As g–1, each n = 3). In addition to the two arsenic hydrocarbons, five arsenosugar phospholipids were also identified by HPLC-high resolution MS (Figure 5). The advantage of the silica column cleanup step is demonstrated by the improvement in the chromatogram (Figure 5a and 5b). The compounds were concurrently quantified by ICPMS detection and in the case of the two AsHCs, for which standards were available, also by ESMS detection (Table 3). The lack of standards for AsPLs precluded our obtaining quantitative data by using HPLC-ESMS, which could have been used to support the ICPMS data. Although quantification by ICPMS can be performed without the specific arsenolipid standard (since signal response depends primarily on the arsenic content, independent of species), the values for the AsPLs might be compromised by possible coelution with other arsenic-containing species.
Figure 4.
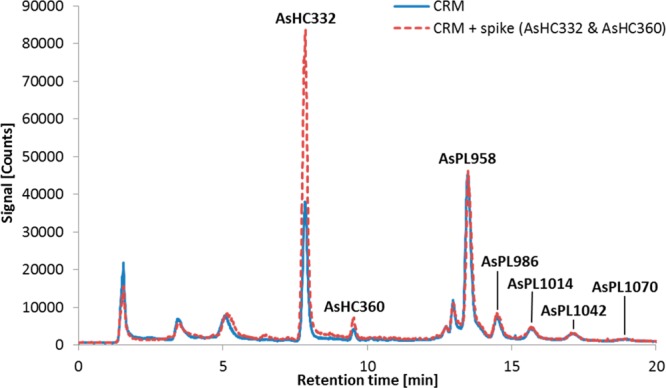
RP-HPLC-ICPMS chromatograms of the unspiked and spiked CRM 7405-a (Hijiki) after extraction with DCM/MeOH (2 + 1; 5 mL), silica cleanup, and thio conversion. ZORBAX Eclipse XDB-C8 (4.6 × 150 mm, 5 μm); mobile phase, water/ethanol gradient (70–90% EtOH, incl. 0.1% formic acid); flow rate, 1 mL min–1; column temperature, 30 °C; injection volume, 20 μL.
Figure 5.
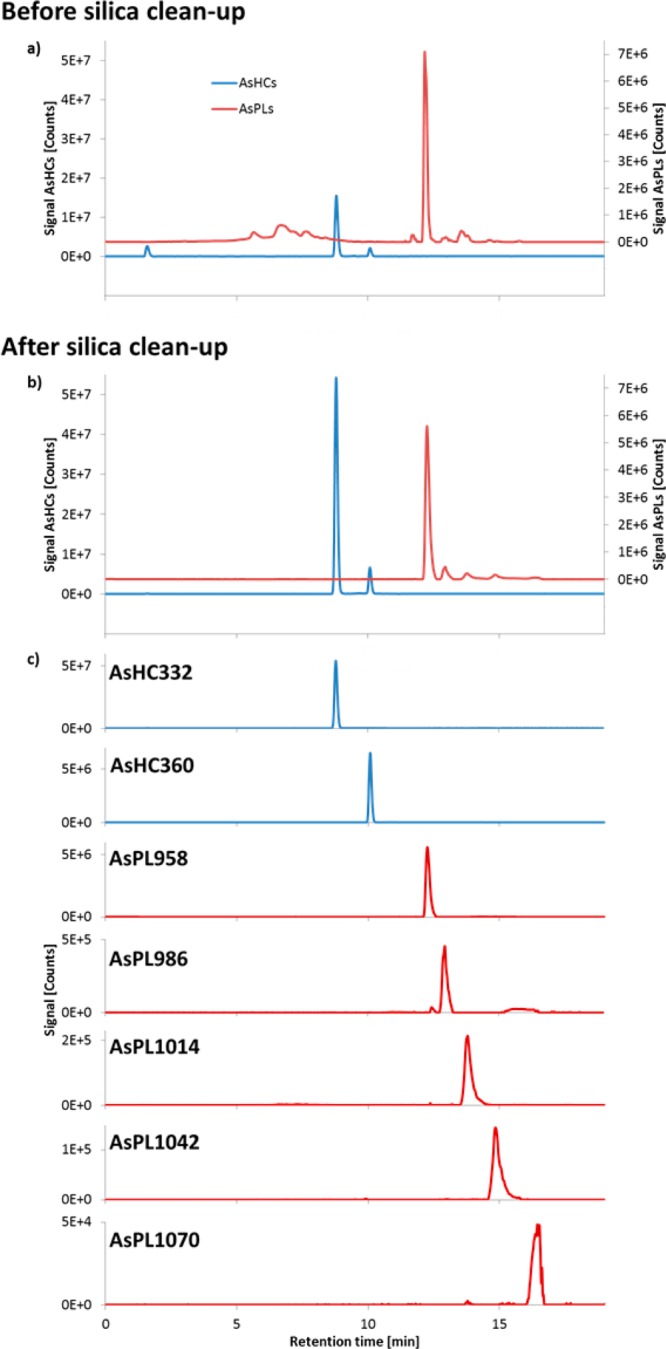
RP-HPLC-HR-ESMS chromatograms of CRM 7405-a (Hijiki) after extraction with DCM/MeOH (2 + 1): (a) the crude extract, (b) extract after silica cleanup, and (c) extracted m/z for each arsenolipid species after silica cleanup; all as thio analogues. ZORBAX Eclipse XDB-C8 (4.6 × 150 mm, 5 μm); mobile phase, water/ethanol gradient (70–90% EtOH, incl. 0.1% formic acid); flow rate, 1 mL min–1; column temperature, 30 °C; injection volume, 5 μL. The following ions (m/z), as their thio derivatives, were extracted: AsHC332–349.1914; AsHC360–377.2228; AsPL958–975.4993; AsPL986–1003.5327; AsPL1014–1031.5647; AsPL1042–1059.5949; AsPL1070–1087.6228.
Table 3. Concentrations of Seven Arsenolipid Species Present in CRM 7405-a (Hijiki)a.
| concentration
in CRM 7405-a (Hijiki) [ng As g–1], mean ± SD, n = 3 |
||
|---|---|---|
| arsenolipid | RP-HPLC-ICPMS | RP-HPLC-HR-ESMS |
| AsHC332 | 1073 ± 44 | 1194 ± 62 |
| AsHC360 | 90 ± 5 | 130 ± 8 |
| AsPL958 | 1587 ± 31 | n.q. |
| AsPL986 | 304 ± 6 | n.q. |
| AsPL1014 | 208 ± 9 | n.q. |
| AsPL1042 | 114 ± 3 | n.q. |
| AsPL1070 | 43 ± 2 | n.q. |
Quantities are based on triplicate analyses incorporating a single extraction with DCM/MeOH (2 + 1, v/v), followed by silica clean-up, formation of the thio analogues, and quantification by RP-HPLC-ICPMS/High-Resolution-ESMS (n.q. = not quantified because of lack of standards).
In summary, we report a method for the quantification of arsenolipids in the reference material CRM 7405-a (Hijiki), which has been previously certified for total arsenic and inorganic arsenic(V). The CRM is a readily available, characterized source of arsenolipids to assist research requiring the analysis of these new and fascinating marine natural products, which occur in many common types of seafood.
Acknowledgments
This research has been supported by the Austrian Science Fund (FWF) project number 23761-N17. We thank also NAWI Graz and the Styrian Government for supporting the Graz Central Lab—Metabolomics. Dr Tomohiro Narukawa kindly provided the CRM Hijiki.
Supporting Information Available
Extraction efficiency of seven arsenolipids depending on solvents, extraction efficiencies according to sequential extraction, amount of solvent, and silica cleaning, conversion of arsenolipids from the oxo to the thio analogue with time after adding H2S, and extracted amounts of seven arsenolipids present in CRM 7405-a depending on the amount of solvent. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Sadolin E. Biochem. Z. 1928, 201, 323–331. [Google Scholar]
- Lunde G. J. Am. Oil Chem. Soc. 1968, 45, 331–332. [DOI] [PubMed] [Google Scholar]
- Lunde G. Acta Chem. Scand. 1972, 26, 2642–2644. [Google Scholar]
- Vaskovsky V. E.; Korotchenko O. D.; Kosheleva L. P.; Levin V. S. Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol. 1972, 41, 777–784. [Google Scholar]
- Morita M.; Shibata Y. Chemosphere 1988, 17, 1147–1152. [Google Scholar]
- Rumpler A.; Edmonds J. S.; Katsu M.; Jensen K. B.; Goessler W.; Raber G.; Gunnlaugsdottir H.; Francesconi K. A. Angew. Chem., Int. Ed. 2008, 47, 2665–2667. [DOI] [PubMed] [Google Scholar]
- Amayo K. O.; Petursdottir A.; Newcombe C.; Gunnlaugsdottir H.; Raab A.; Krupp E. M.; Feldmann J. Anal. Chem. 2011, 83, 3589–3595. [DOI] [PubMed] [Google Scholar]
- Lischka S.; Arroyo-Abad U.; Mattusch J.; Kuehn A.; Piechotta C. Talanta 2013, 110, 144–152. [DOI] [PubMed] [Google Scholar]
- Sele V.; Sloth J. J.; Holmelid B.; Valdersnes S.; Skov K.; Amlund H. Talanta 2014, 121, 89–96. [DOI] [PubMed] [Google Scholar]
- Amayo K. O.; Raab A.; Krupp E. M.; Marschall T.; Horsfall M.; Feldmann J. J. Trace Elem. Med. Biol. 2014, 28, 131–137. [DOI] [PubMed] [Google Scholar]
- Raab A.; Newcombe C.; Pitton D.; Ebel R.; Feldmann J. Anal. Chem. 2013, 85, 2817–2824. [DOI] [PubMed] [Google Scholar]
- Taleshi M. S.; Jensen K. B.; Raber G.; Edmonds J. S.; Gunnlaugsdottir H.; Francesconi K. A. Chem. Commun. 2008, 39, 4706–4707. [DOI] [PubMed] [Google Scholar]
- Taleshi M. S.; Edmonds J. S.; Goessler W.; Ruiz-Chancho M. J.; Raber G.; Jensen K. B.; Francesconi K. A. Environ. Sci. Technol. 2010, 44, 1478–1483. [DOI] [PubMed] [Google Scholar]
- Arroyo-Abad U.; Mattusch J.; Mothes S.; Moeder M.; Wennrich R.; Elizalde-González M. P.; Matysik F.-M. Talanta 2010, 82, 38–43. [DOI] [PubMed] [Google Scholar]
- García-Salgado S.; Raber G.; Raml R.; Magnes C.; Francesconi K. A. Environ. Chem. 2012, 9, 63–66. [Google Scholar]
- Francesconi K. A. Pure Appl. Chem. 2010, 82, 373–381. [Google Scholar]
- Meyer S.; Matissek M.; Müller S. M.; Taleshi M. S.; Ebert F.; Francesconi K. A.; Schwerdtle T. Metallomics 2014, 6, 1023–1033. [DOI] [PubMed] [Google Scholar]
- Taleshi M. S.; Seidler-Egdal R. K.; Jensen K. B.; Schwerdtle T.; Francesconi K. A. Organometallics 2014, 33, 1397–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narukawa T.; Inagaki K.; Zhu Y.; Kuroiwa T.; Narushima I.; Chiba K.; Hioki A. Anal. Bioanal. Chem. 2012, 402, 1713–1722. [DOI] [PubMed] [Google Scholar]
- Raber G.; Raml R.; Goessler W.; Francesconi K. A. J. Anal. At. Spectrom. 2010, 25, 570–576. [Google Scholar]
- Blight E. G.; Dyer W. J. Can. J. Biochem. Physiol. 1959, 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Matyash V.; Liebisch G.; Kurzchalia T. V.; Shevchenko A.; Schwudke D. J. Lipid Res. 2008, 49, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt C.Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Raml R.; Goessler W.; Francesconi K. A. J. Chromatogr. A 2006, 1128, 164–170. [DOI] [PubMed] [Google Scholar]
- Raber G.; Raml R.; Goessler W.; Francesconi K. A. J. Anal. At. Spectrom. 2010, 25, 570–576. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.