

Summary
Apert syndrome (AS) is a rare genetic and congenital disease characterized by craniosynostosis and syndactly of hands and feet. AS patients generally require lifelong management, however there are still no effective treatment methods except surgery. In recent years, research has made great progress in the pathogenesis of AS. FGFR2 mediates extracellular signals into cells and the mutations in the FGFR2 gene cause AS occurrence. Activated FGFs/FGFR2 signaling disrupt the balance of cell proliferation, differentiation and apoptosis via its downstream signal pathways. However, how the pathways transform the balance is not well understood and contradictions have occurred in different studies. In this review, we'll focus on these problems to get a better understanding of AS pathogenesis.
Keywords: Apert syndrome, FGFR2 gene, pathogenesis, signal pathways
1. Introduction
Apert syndrome (AS) is one of the most severe craniosynostosis syndromes, accounting for about 4.5% of all craniosynostosis, with a prevalence of 1 in 65,000 individuals (1,2). AS was first described by Wheaton in 1894 and then reviewed extensively by the French physician Apert (3). AS has a dominant inheritance patern, but cases most are sporadic and exhibit a paternal effect. More than 98% of AS cases are caused by FGFR2 de novo mutations (S252W and P253R) (4,5).
Because there are no effective treatment alternatives, many AS patients must have surgery to correct both facial and hand/foot anomalies (6). Great progress have been made in understanding the molecular and cellular basis of AS. The disturbance of the FGFs/FGFR2 signal, which changes the balance of proliferation, differentiation and apoptosis of osteoblasts, is ascribed to the development of AS (7). Signal pathways which play a vital role in bone formation are also involved in this process (8). In this review, we summarize recent studies of AS to have a better and comprehensive understanding of AS pathogenesis.
2. AS clinical features
AS also known as acrocephalosyndactyly type 1, though its clinical features are distinctive, acrobrachycephaly is presented in almost all AS cases. In addition, AS affects many other organs and exhibits tissue clinical manifestations including craniofacial, oral, skeletal, cutaneous, respiratory and visceral features.
Based on a clinical study of 136 AS patients, Cohen et al. concluded the craniofacial features of AS, including hyperacrobrachycephaly, craniofacial asymmetry, steep wide forehead, flat occiput, downslanting palpebral fissures, divergent upgaze, eso-tropic downgaze, marked depression of the nasal bridge, ocular hepertelorism and proptosis, short and wide nose with a bulbous tip and reduced anterior facial height (9).
Oral anomalies are also common conditions for AS patients. Cleft palate or bifid uvula was present in approximately 75% of AS cases (10). The intraoral features included impacted teeth, delayed eruption, ectopic eruption, supernumerary teeth and thick gingiva (11). An anterior and posterior open bite and crossbite were also observed by occlusal examination (12).
AS patients also presented skeletal defects, of which severe syndactyly of hands and feet, which frequently affected the second, third and fourth fingers or toes, was the most constant. Other skeletal features included decreased glenohumeral mobility, complete glenohumeral ankylosis, short humerus, dramatically short humerus, limited elbow mobility, radiohumeral synostosis, pectus excavatum, flattening of the chest wall, asymmetric chest wall, spina bifida, hemivertebrae, spinal fusions, scoliosis, lordosis, wide interpubic distance, genua valga and osseous ankylosis at the knees (13).
Cutaneous manifestations of AS are hyperhidrosis, oily skin, resistant acne, interrupted eyebrows, excessive forehead wrinkling, lateral plantar hyperkeratosis, skin dimpling over joints and oculocutaneous hypopigmentation (14,15).
Respiratory complications are presented in about 33% of AS patients with anesthesia complications, which could be severe enough to cause cancellation of surgery (16). Based on 12 autopsies of AS patients, reported anomalies of the respiratory system occurred at a lower frequency (1.5%). Of AS visceral features, cardiovascular and genitourinary anomalies are the most common, occurring in 10% and 9.6%, respectively (17).
Mental retardation or central nervous system (CNS) anomalies occur in about 55.6% of AS patients and may be partly due to brain malformations or high intracranial pressure (18). It also includes megalencephaly, ventriculomegaly, corpus callosum anomalies, hippocampal hypoplasia or dysplasia, hypoplasia or dysplasia of the septum pellucidum, cerebral cortex dysplasia and anomalies of gyral patterning (19).
3. The gene mutation of As
FGFR2 is one of the transmembrane tyrosine kinase receptors FGFRs which are composed of three immunoglobulin-like (Ig) domains in the extracellular region, a transmembrane region and a cytoplasmic tyrosine kinase domain (20). It mediates signal transduction from the extracellular into the intracellular area and regulates cell activities through its downstream pathways. FGFR2 is expressed in a wide variety of tissues, for example it is expressed embryonically in early cartilage condensations, proliferating osteoprogenitors, limb mesenchyme, the lungs, brain and skin. Two alternative gene products have been characterized: the IIIb isoform expressed in epithelia and IIIc isoform expressed in mesenchyme and neural tissue. Both isoforms have specific FGF ligands, which control normal development of the organ (21).
The FGFR2 gene is located in 10q26, whose mutations cause AS occurrence. In 1995, it was found that two mutations in the gene: ser252trp of 755C>G and pro253arg of 758C>G in cDNA, which were located in the inner region of IgII and IgIII of FGFR2 (22). Patients with the FGFR2 S252W mutation accounted for about 2/3 of 70 unrelated AS patients, while about 1/3 of AS patients are caused by the P253R mutation, and both of these mutations are the most common mutations of AS (22). A study by Oldridge et al. demonstrated a new ser252phe mutation in FGFR2 with CG-to-TT in the cDNA (23). In 2002, Kan et al. reported the M186T (c.557C>T) mutation of FGFR2 in AS through genomic screening of fibroblast growth factor receptor 2 (24). Next, several rare shear mutations were identified: 940-2A_G, 940-3_-4insAlu and 1041_1042insAlu (25). Recently, a novel E731K (c.2191G>A) mutation of exon18 of FGFR2 in a Korean AS patient was reported (26). A brief outline of FGFR2 mutations in AS patients is presented below (Table 1).
Table 1. The FGFR2 mutations for Apert syndrome.
| Disease | Gene | Nucleotide mutation | Amino acid change | Reference |
| Apert syndrome | FGFR2 | c.557C>T | p.Met186Thr | (26) |
| c.755C>G* | p.Ser252Trp | (22) | ||
| c.755CG>TT | p.Ser252Phe | (23) | ||
| c.758C>G* | p.Pro253Arg | (22) | ||
| c.940-2A G | shear mutation | (25) | ||
| c.940-3_-4insAlu | shear mutation | (25) | ||
| c.1041_1042insAlu | shear mutation | (25) | ||
| c.2191G>A | p.Glu731Lys | (26) |
common mutation type.
4. The changes of cell activities and signal pathways by mutated FGFR2
FGFs/FGFRs signaling plays a vital role in regulating the balance of cell proliferation, differentiation and apoptosis, which is essential to the normal formation of cranial bones. The gain of function mutation of FGFR2 may disrupt the balance, which may further lead to AS.
4.1. The effect of mutated FGFR2 on cell proliferation
FGFR2 has a regulatory role on cell reproduction, whose mutation may impair cell proliferation. In 1998, a study of the calvarial cells from fetuses and infants with AS caused by the FGFR2 S252W mutation showed normal cell growth (27). PCNA immunolocation of the AS fetuses cranial coronal sutures demonstrated no difference between the AS patients and age-matched controls (28). However, AS skull osteoblasts demonstrated a decreased growth rate compared with normal cells in vitro (29). Comparative analysis of the proliferation of wild-type (WT) (from 3 individuals) and mutated-type (MT) (from 3 AS patients) fibroblasts and mesenchymal stem cells showed increased cell growth in MT fibroblasts and decreased in MT MSCs (30).
AS mouse models with the FGFR2 S252W or P253R mutation, which represented clinical features of AS patients, provided a good model for the study of the AS mechanism. In 2003, Chen et al. introduced the FGFR2 S252W mutant mouse and analysis of several sutures of different developmental stages E16.5, E18.5, P5 and P8 of the mutant mouse by both BrdU and (3H) thymidine incorporation assays, and demonstrated no significant difference of cell proliferation between mutant and control cells (31). Cell proliferation assay of coronal sutures during embryonic calvaria development between E12.5 and E16.5 showed that until E16.5 calvarial cell growth rate of the FGFR2S252W/+ mouse appeared to be slower than that of WT controls in vivo, while compared the P1 calvarial osteoblasts isolated from the FGFR2S252W/+ mouse and WT littermates in vitro, the BrdU incorporation assay showed that the FGFR2S252W/+ cells have a higher rate of DNA synthesis than the WT controls (32). Osteoblasts from limbs of the P0 FGFR2S252W/+ mouse also proliferated faster than that of normal mice (33).
Until now, the influence of mutated FGFR2 on cell proliferation is controversial, partly due to different subjects and analysis methods applied in different studies.
4.2. The effect of mutated FGFR2 on differentiation of cells
Premature fusion of cranial sutures, the main characteristic of AS, may be partly ascribed to the altered differentiation of calvarial cells. Lomri et al. analyzed differentiation of calvarial cells by histological analysis, which revealed an increased extent of subperiosteal bone formation and alkaline phosphatase-positive preosteoblastic cells in Apert (S252W) fetal calvaria compared with age-matched controls in vivo, and both the expression of alkaline phosphatase and type 1 collagen, and production of the mineralized matrix of Apert cells were higher than controls in vitro (27). Fragale et al. demonstrated that Apert (P253R) calvarial osteoblasts represented higher alkaline phosphatase activity, increased mineralization and expression of noncollagenous matrix proteins than control osteoblasts (29). Another study by Lemonnier et al. stated that immunohistochemical analysis of the Apert calvaria suture showed higher type 1 collagen, osteocalcin and osteopontin expression in preosteoblasts compared with controls and the expression of the markers in cultured Apert calvaria osteoblasts was also higher than those of normal calvarial osteoblasts (28). Long-term cultured human fetal calvarial osteoblasts from two Apert (S252W) fetuses increased expression of osteoblast markers alkaline phosphatase (ALP), type 1 collagen (COLIA1) and osteocalcin (OC) compared to normal osteoblasts (34). Tanimoto et al. compared differentiation of osteoblasts from the digital bone of two Apert patients with the FGFR2 S252W mutation and two independent non-syndromic polydactyly patients, and revealed that the Apert osteoblasts showed more prominent ALP, OC and osteopontin mRNA expression and mineralized nodule formation (35). In 2012, Yeh et al. found both that the coronal suture periosteal fibroblasts and MSCs from three unrelated AS patients showed enhanced osteogenic differentiation compared to cells from age- and sex-matched control subjects in vivo and vitro(30).
Due to the limitation of AS patient samples, animal and cell models of AS have been created. Mansukhani et al. compared the differentiation of the mouse calvarial osteoblasts stably transfected with S252W mutated and normal FGFR2, and showed that osteoblasts expressing mutated FGFR2 had reduced alkaline phosphatase (ALP) and mineralization (36). Another study stated that there was no significant difference in mineralization between chicken calvarial osteoblasts transfected with P253R mutated and WT FGFR2 (37). At the same time, Chen et al. also detected no significant difference of expression of osteoblast differentiation markers between FGFR2S252W and control mice by in situ hybridization (31). However, Holmes et al. checked osteoblast marker gene expression in E15.5 calvaria, which showed only minor increases in FGFR2S252W/+ mice compared with WT controls, and ALP activity was higher in FGFR2S252W/+ osteoblasts in culture than the controls (32). A study by Miraoui et al. also demonstrated that in mesenchymal C3H10T1/2 cells and calvarial preosteoblast MC3T3-E1 cells stably transfected with WT and S252W mutated FGFR2 respectively, both cell types expressing MT FGFR2 presented enhanced osteodifferentitation ability by analysis of mineralization, ALP activity and expression of the osteoblast differentiation markers compared with those expressing WT FGFR2 (38). A recent study stated primary calvarial osteoblasts derived from FGFR2IIIcS252W transgenic mice showed enhanced mineralization, higher ALP activity and greater expression of the differentiation markers than cells from WT mice (8).
In most of the above studies, the FGFR2 mutation which caused the AS enhanced osteoblast differentiation in vivo and vitro. While up to now, the molecular basis of the altered differentiation of the cell is not well understood. The activation of the signal pathways induced by the mutated FGFR2 may play a vital role in this process.
4.2.1. ERK1/2 pathway
The study by Miraoui et al. demonstrated that ERK1/2 played a great role in differentiation of C3H10T1/2 cells expressing WT FGFR2 rather than MT cells (38). However, in FGFR2P253R Apert mouse models, phosphorylation of ERK was increased less than 1.5-fold in the neurocranium compared to WT controls (39). In another study activation of ERK1/2 was higher in primary calvarial osteoblasts from the FGFR2S252W mouse than normal osteoblasts (8). The FGFR2 E731K mutation which was found in a Korean AS patient enhanced phosphorylation and activation of ERK1/2 (26). Furthermore, the study by Yin et al. revealed inhibition of ERK1/2 activity partly prevented premature closure of coronal sutures (40). Another study also verified that the FGFR2S252W AS mice treated with U1206, a pharmacological inhibitor of MEK1/2 that blocks phosphorylation and activation of ERK1/2 during pregnancy and early postnatal stages, significantly repressed craniosynostosis and improved skeletal abnormalities. It also showed that a small hairpin RNA targeting the dominant mutant form of FGFR2 (FGFR2S252W) without affecting wild-type mRNA levels completely prevents the Apert-like syndrome in mice, during which the alterations of ERK1/2 activity and FGF-FGFR modulators and downstream genes were observed (41). These data clearly demonstrate that ERK1/2 plays a vital role in osteoblast differentiation and AS occurrence.
4.2.2. AKT pathway
AKT, a downstream target of phosphphatidylinositol-3-kinase (PI3-K), is an important mediator of cell proliferation and survival via phosphorylation of a variety of targets leading to activation or inhibition of their functions (42–44). AKT was also reported to promote osteoblast differentiation (45). In calvaria tissue and cultured osteoblasts isolated from the FGFR2S252W Apert mouse models, Holmes et al. detected a significant increase of AKT phosphorylation compared to normal controls (32). This indicated that AS osteoblasts enhanced differentiation via activation of the AKT pathway. However, in another AS mouse model with the FGFR2 P253R mutation, phosphorylated AKT was not obviously different compared with that of WT controls (39). The different mutations in FGFR2 may alter osteoblast differentiation via different pathways.
4.2.3. PKC pathway
Protein kinase C (PKC) is a serine/threonine protein kinase and mediates various cellular functions such as cell proliferation and differentiation (21). PKC signaling also regulates osteoblast differentiation and is necessary for bFGF-induced bone formation (46). A study by Lemonnier et al. demonstrated calvarial osteoblasts isolated from two Apert fetuses with the FGFR2 S252W mutation represented higher PKC activity than normal osteoblasts. In the following experiments, inhibition of PKC by calphostin C or the PKCα-specific inhibitor, obviously repressed the osteodifferentiation of the mutant osteoblasts (34). This may demonstrate PKC pathways are involved in the enhanced differentiation derived from S252W mutated FGFR2. In another study, murine mesenchymal C3H10T1/2 cells stably expressing S252W FGFR2 showed enhanced differentiation and increased PKC activity compared with cells expressing WT FGFR2. Pharmacologic inhibition of PKCα slightly reduced matrix calcification in the WT C3H10T1/2 cell, but completely inhibited mineralization induced by MT FGFR2 (38). These experiments confirm that PKC plays a predominant role in mutated FGFR2 induced differentiation in osteoblasts and mesenchymal stem cells.
4.2.4. p38 pathway
The p38 MAPKs belong to the MAPK superfamily and have been shown to be implicated in proliferation, differentiation, apoptosis, senescence and cytokine production (47,48). Osteoblasts lacking p38α showed reduced osteodifferentation marker expressions and defective mineralization in vitro, which indicated that p38α was an essential positive regulator of osteoblast differentiation (49). Holmes et al. detected an obvious increase of p38 phosphorylation in the calvarial tissues of FGFR2S252W AS mouse models compared to WT controls (32). In calvaria tissues from FGFR2P253R AS mouse models, Wang et al. also detected higher p38 phosphorylation than normal mice (39). Another study stated that Apert calvarial osteoblasts showed enhanced differentiation and increased p38 phosphorylation compared to normal cells. Mutant osteoblasts treated with SB203580, a specific p38 inhibitor, significantly inhibited the expression of differentiation markers and obviously reduced mineralization (8). These studies show that the p38 pathway plays a vital role in enhanced differentiation of Apert mutant osteoblasts.
4.2.5. PLCγ pathway
Phospholipase C (PLC) converts phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) and regulates production of important second messengers which determines cell behavior (50). It was reported that sustained Platelet-derived growth factor receptor α signaling in osteoblasts resulted in craniosynostosis by overactivating the Phospholipase C pathway (51). Another study stated that human Apert mutant osteoblasts expressed more PLCγ than control cells. Suzuki et al. detected a significant increase of PLCγ phosphorylation in calvarial osteoblasts from AS mouse models with the FGFR2 S252W mutation compared to osteoblasts expressing sFGFR2IIIc-Ap. However, Apert mutant osteoblasts treated with a PLCγ inhibitor (U73122) didn't obviously reduce its mineralization (8). It shows the involvement of the PLC pathways in accelerating osteogenesis in the mutant cells, although it seems to have a weak effect.
4.2.6. Other signal pathways
In fibroblasts isolated from AS patients (S252W), JNK phosphorylation is significantly higher than that of the WT osteoblasts. Apert fibroblasts treated with SP600125, a JNK phosphorylation inhibitor reduced its ALP activity and mineralization (30). S252W mutated FGFR2 enhanced EGFR and PDGFRα mRNA expression via activation of PKCα-dependent AP-1 transcriptional activity. Inhibitation of EGFR and PDGFRα by a specific EGFR inhibitor or the PDGFR inhibitor repressed expression of differentiation markers and reduced mineralization in Apert mutant osteoblasts (52). Another study revealed that PDGFR influenced neural crest derived osteogenesis by stimulating the PLC pathway (51). These studies confirm that JNK, EGFR and PDGFR pathways are involved in the enhancement of osteoblast differentiation caused by the activated FGFR2.
The mutated FGFR2 which caused AS drives cells osteodifferentation via ERK1/2, AKT, PKC, p38, PLCγ, JNK, EGFR, PDGFR and other signal pathways. Inhibitors specific to the pathways may be helpful for the treatment of AS patients.
4.3. The effect of mutated FGFR2 on cell apoptosis
Apoptosis, also called programmed cell death, is a widespread phenomenon which plays a crucial role in a variety of physiological and pathological processes (53). A study also indicated that apoptosis was involved in normal and pathological osteogenesis (53). In 2000, Mansukhani et al. first reported S252W mutated FGFR2 induced apoptosis in mouse calvarial osteoblasts (36). Next, Lemonnier et al. also detected significantly increased apoptosis in Apert coronal sutures in vivo and cultured Apert calvarial osteoblasts in vitro. In addition, they found increased apoptosis induced by S252W mutated FGFR2 was PKC-dependent via overexpression and activation of its downstream signals IL-1 and Fas (54). Chen et al. demonstrated that the MT FGFR2 increased Bax expression and apoptosis of osteogenic cells in the mutant coronal suture of the FGFR2S252W AS mouse models (31). In AS mouse models with the same type mutation in FGFR2, it was reported that apoptosis accompanied fusion, but was restricted to bone fronts in contact with one another, while no apoptosis was detected in WT mouse sutures (32). However, in another Apert mouse model with P253R mutated FGFR2, there was no obvious difference in apoptosis in the coronal sutures between the MT and WT mouse models (39). A recent study demonstrated only the inter-premaxillary suture exhibited significantly increased apoptosis in bone bordering the suture mesenchyme in AS mouse models with S252W mutated FGFR2 compared with normal mice (55).
Based on the above studies, we conclude that mutated FGFR2 induces apoptosis, which has been confirmed by analysis of human bone samples and mouse models in vivo and vitro. At the same time, we face another question “Whether enhanced apoptosis leads to premature suture closure or is secondary to the craniosynostosis”. Chen et al. suggested that accelerated cell death possibly reduced the space between osteogenic fronts of flat bones and resulted in physical contact of these bones (31). On the other hand, Holmes et al. thought apoptosis appeared to be a consequence rather than a cause of sutural fusion based on their study which showed craniosynostosis was an early onset during embryo development while E16.5 apoptosis began to appear in the FGFR2S252W coronal sutures and were strictly limited to sites of osteoid contact between the frontal and parietal bones (32). The answer to this question needs more experiments in the future to confirm.
4.4. Other cell activities altered by mutated FGFR2
It was reported that mutated FGFR2 increased cell-cell aggregation and N- and E-cadherin expression in human calvarial osteoblasts. Neutralizing anti-N-cadherin antibody or N-cadherin antisense oligonucleotides suppressed increased cell-cell aggregation and reduced osteoblast differentiation markers overexpression in mutant osteoblasts (34). A study by Holmes et al. suggested that the critical event of Apert craniosynostosis was to increase the recruitment or advancement of osteoprogenitor cells at the sites where sutures should normally form (32). Both of the above studies suggested that cell adhesion and recruitment play important roles in the pathogenesis of AS.
5. The altered chondrogenesis by mutated FGFR2
Bone formation is formed through intramembranous and endochondral bone formation. Two studies detected FGFR2 expression in a chondrocyte lineage, which suggested FGFR2 may play an important role in development of chondrocytes (56,57). Wang et al. found ectopic cartilage at the midline sagittal suture, and cartilage abnormalities in the basicranium, nasal turbinates and trachea in FGFR2S252W mice (58). It indicated that altered chondrogenesis was involved in the occurrence of AS. In another study, chondrocytes with S252W mutated FGFR2 in hydrogel culture also exhibited strong staining of the cartilage specific marker: collagen type II, while only minimal staining was seen in the WT control (33). In another Apert mouse model with P253R mutated FGFR2, it presented shortened synchondroses, short trabecular bones and a delayed secondary ossification center in the tibia; which stated that the FGFR2 P253R mutation in mice resulted in retarded endochondral ossification (40). Nagata et al. also confirmed that P253R mutated FGFR2 accelerated maturation and hypertrophy of cranial base chondrocytes, which resulted in disturbance of the cranial base growth with precocious endochondral ossification in mice with the mutation (59). Based on the studies, we can conclude that altered chondrogenesis caused by activated FGFR2 mutation plays a vital role in the occurrence of AS.
6. Acne in As
Acne is a chronic inflammatory disease and also a clinical manifestation of AS (60). Major elements contributed to the acne pathogenesis include abnormal follicular differentiation with hyperproliferation, increased sebaceous gland activity with increased sebum, as well as increased bacterial colonization, inflammation and immunological mechanisms. Androgens play an important role in the stimulation and growth of sebocytes, sebum production, and keratinocyte proliferation in the ductus seboglandularis and the acroinfundibulum (61,62). Studies also demonstrated that FGFR2 plays an essential role in homeostasis of the epidermis and sebaceous gland development (63–65). FGFR2 generates two splice variants by alternative splicing: FGFR2b and FGFR2c which are expressed in epithelial and mesenchymal cells respectivly. The FGFR2 mutation in AS altered FGFR2b-mediated downstream signal pathways is involved in pathogenesis of AS (66). The mutated FGFR2b altered cell proliferation and MMP expression via the MAPK pathway, induced lipogenesis and terminal sebocyte differentiation via the PI3K/AKT and Shh/MC5R pathways and induced IL-1a, and inflammatory reactions via the phospholipase Cγ/protein kinase C pathway (66). Melnik stated that known anti-acne agents which attenuated FGFR2-signaling pathways was a common mode of action (67). So, the above studies demonstrated that increased fibroblast growth factor receptor 2 (FGFR2)-signaling caused by the mutated FGFR2 contributed to the occurrence of acne in AS.
7. Conclusion
After decades of investigation, the changes of proliferation, differentiation and apoptosis in cells have been shown to play a prominent role in the occurrence and development of AS. One purpose of this review is to demonstrate that the pathways regulating the cell behavior changes caused by the FGFR2 mutation in AS patients has been established (Figure 1). Though these pathways are mostly incomplete, they provide a basis for future advances. In vivo and vitro studies have shown that these pathways play a vital role in AS and pharmacological inhibitors that specificly repress the activation of these pathways could significantly improve the AS clinical manifestation. The mutated FGFR2b pathway has further lead to the acne occurence in AS. Notwithstanding our extensive knowledge of cell behavior changes by the FGFR2 mutation through these pathways, different types and differentiation stages of cells display distinct responses to the activated FGFR2 mutation in AS, which needs further investigation in the future.
Figure 1.
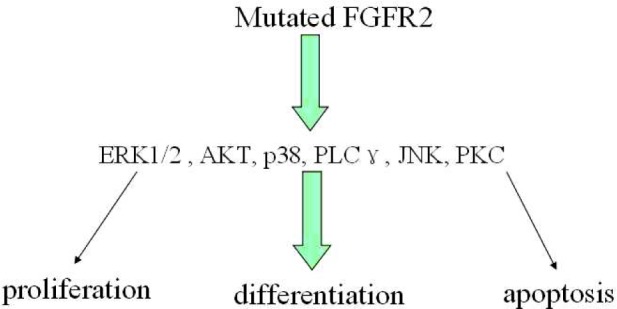
Altered pathways in AS. The mutated FGFR2 which caused AS disrupts the cell balance of proliferation, differentiation and apoptosis of via changing the activation of its downstream signal pathways: ERK1/2, AKT, P38, PLCγ, JNK and PKC.
References
- 1. Park WJ, Theda C, Maestri NE, Meyers GA, Fryburg JS, Dufresne C, Cohen MM, Jr, Jabs EW. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet. 1995; 57:321-328 [PMC free article] [PubMed] [Google Scholar]
- 2. Cohen MM, Jr, Kreiborg S, Lammer EJ, Cordero JF, Mastroiacovo P, Erikson JD, Roeper P, Martínez-Frías ML. Birth prevalence study of Apert syndrome. Am J Med Genet. 1992; 42:655-659 [DOI] [PubMed] [Google Scholar]
- 3. De D, Narang T, Kanwar AJ, Dogra S. Brachycephaly and syndactyly: Apert's syndrome. Indian J Dermatol Venereol Leprol. 2008; 74:395-396 [DOI] [PubMed] [Google Scholar]
- 4. Coomaralingam S, Roth P. Apert syndrome in a newborn infant without craniosynostosis. J Craniofac Surg. 2012; 23:e209-211 [DOI] [PubMed] [Google Scholar]
- 5. Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995; 9:165-172 [DOI] [PubMed] [Google Scholar]
- 6. Ibrahimi OA, Chiu ES, Mccarthy JG, Mohammadi M. Understanding the molecular basis of Apert syndrome. Plast Reconstr Surg. 2005; 115:264-270 [PubMed] [Google Scholar]
- 7. Marie PJ, Debiais F, Hay E. Regulation of human cranial osteoblast phenotype by FGF-2, FGFR-2 and BMP-2 signaling. Histol Histopathol. 2002; 17:877-885 [DOI] [PubMed] [Google Scholar]
- 8. Suzuki H, Suda N, Shiga M, Kobayashi Y, Nakamura M, Iseki S, Moriyama K. Apert syndrome mutant FGFR2 and its soluble form reciprocally alter osteogenesis of primary calvarial osteoblasts. J Cell Physiol. 2012; 227:3267-3277 [DOI] [PubMed] [Google Scholar]
- 9. Cohen MM, Jr, Kreiborg S. A clinical study of the craniofacial features in Apert syndrome. Int J Oral Maxillofac Surg. 1996; 25:45-53 [DOI] [PubMed] [Google Scholar]
- 10. Kreiborg S, Cohen MM, Jr. The oral manifestations of Apert syndrome. J Craniofac Genet Dev Biol. 1992; 12:41-48 [PubMed] [Google Scholar]
- 11. Tiwari A, Agrawal A, Pratap A, Lakshmi R, Narad R. Apert syndrome with septum pellucidum agenesis. Singapore Med J. 2007; 48:e62-65 [PubMed] [Google Scholar]
- 12. Soanca A, Dudea D, Gocan H, Roman A, Culic B. Oral manifestations in Apert syndrome: Case presentation and a brief review of the literature. Rom J Morphol Embryol. 2010; 51:581-584 [PubMed] [Google Scholar]
- 13. Cohen MM, Jr, Kreiborg S. Skeletal abnormalities in the Apert syndrome. Am J Med Genet. 1993; 47:624-632 [DOI] [PubMed] [Google Scholar]
- 14. Cohen MM, Jr, Kreiborg S. Cutaneous manifestations of Apert syndrome. Am J Med Genet. 1995; 58:94-96 [DOI] [PubMed] [Google Scholar]
- 15. Freiman A, Tessler O, Barankin B. Apert syndrome. Int J Dermatol. 2006; 45:1341-1343 [DOI] [PubMed] [Google Scholar]
- 16. Elwood T, Sarathy PV, Geiduschek JM, Ulma GA, Karl HW. Respiratory complications during anaesthesia in Apert syndrome. Paediatr Anaesth. 2001; 11:701-703 [DOI] [PubMed] [Google Scholar]
- 17. Cohen MM, Jr, Kreiborg S. Visceral anomalies in the Apert syndrome. Am J Med Genet. 1993; 45:758-760 [DOI] [PubMed] [Google Scholar]
- 18. Yaghoobi R, Bagherani N, Tajalli M, Paziar N. Apert syndrome. Indian J Dermatol Venereol Leprol. 2010; 76:724. [DOI] [PubMed] [Google Scholar]
- 19. Ludwig K, Salmaso R, Manara R, Cosmi E, Baldi M, Rugge M. Apert syndrome with fused thalami. Fetal Pediatr Pathol. 2012; 31:410-414 [DOI] [PubMed] [Google Scholar]
- 20. Carinci F, Pezzetti F, Locci P, Becchetti E, Carls F, Avantaggiato A, Becchetti A, Carinci P, Baroni T, Bodo M. Apert and Crouzon syndromes: Clinical findings, genes and extracellular matrix. J Craniofac Surg. 2005; 16:361-368 [DOI] [PubMed] [Google Scholar]
- 21. Holmes G. Mouse models of Apert syndrome. Childs Nerv Syst. 2012; 28:1505-1510 [DOI] [PubMed] [Google Scholar]
- 22. Slaney SF, Oldridge M, Hurst JA, Moriss-Kay GM, Hall CM, Poole MD, Wilkie AO. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet. 1996; 58:923-932 [PMC free article] [PubMed] [Google Scholar]
- 23. Oldridge M, Lunt PW, Zackai EH, Mcdonald-Mcginn DM, Muenke M, Moloney DM, Twigg SR, Heath JK, Howard TD, Hoganson G, Gagnon DM, Jabs EW, Wilkie AO. Genotype-phenotype correlation for nucleotide substitutions in the IgII-IgIII linker of FGFR2. Hum Mol Genet. 1997; 6:137-143 [DOI] [PubMed] [Google Scholar]
- 24. Kan SH, Elanko N, Johnson D, Cornejo-Roldan L, Cook J, Reich EW, Tomkins S, Verloes A, Twigg SR, Rannan-Eliya S, McDonald-McGinn DM, Zackai EH, Wall SA, Muenke M, Wilkie AO. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet. 2002; 70:472-486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bochukova EG, Roscioli T, Hedges DJ, Taylor IB, Johnson D, David DJ, Deininger PL, Wilkie AO. Rare mutations of FGFR2 causing apert syndrome: Identification of the first partial gene deletion, and an Alu element insertion from a new subfamily. Hum Mutat. 2009; 30:204-211 [DOI] [PubMed] [Google Scholar]
- 26. Park J, Park OJ, Yoon WJ, Kim HJ, Choi KY, Cho TJ, Ryoo HM. Functional characterization of a novel FGFR2 mutation, E731K, in craniosynostosis. J Cell Biochem. 2012; 113:457-464 [DOI] [PubMed] [Google Scholar]
- 27. Lomri A, Lemonnier J, Hott M, De PN, Lajeunie E, Munnich A, Renier D, Marie PJ. Increased calvaria cell differentiation and bone matrix formation induced by fibroblast growth factor receptor 2 mutations in Apert syndrome. J Clin Invest. 1998; 101:1310-1317 [PMC free article] [PubMed] [Google Scholar]
- 28. Lemonnier J, Delannoy P, Hott M, Lomri A, Modrowski D, Marie PJ. The Ser252Trp fibroblast growth factor receptor-2 (FGFR-2) mutation induces PKC-independent downregulation of FGFR-2 associated with premature calvaria osteoblast differentiation. Exp Cell Res. 2000; 256:158-167 [DOI] [PubMed] [Google Scholar]
- 29. Fragale A, Tartaglia M, Bernardini S, Di Stasi AM, Di Rocco C, Velardi F, Teti A, Battaglia PA, Migliaccio S. Decreased proliferation and altered differentiation in osteoblasts from genetically and clinically distinct craniosynostotic disorders. Am J Pathol. 1999; 154:1465-1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yeh E, Atique R, Ishiy FA, Fanganiello RD, Alonso N, Matushita H, Da Rocha KM, Passos-Bueno MR. FGFR2 mutation confers a less drastic gain of function in mesenchymal stem cells than in fibroblasts. Stem Cell Rev. 2012; 8:685-695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen L, Li D, Li C, Engel A, Deng CX. A Ser252Trp (corrected) substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone. 2003; 33:169-178 [DOI] [PubMed] [Google Scholar]
- 32. Holmes G, Rothschild G, Roy UB, Deng CX, Mansukhani A, Basilico C. Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology. Dev Biol. 2009; 328:273-284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang F, Wang Y, Zhang Z, Hsu B, Jabs EW, Elisseeff JH. The study of abnormal bone development in the Apert syndrome Fgfr2+/S252W mouse using a 3D hydrogel culture model. Bone. 2008; 43:55-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lemonnier J, Hay E, Delannoy P, Lomri A, Modrowski D, Caverzasio J, Marie PJ. Role of N-cadherin and protein kinase C in osteoblast gene activation induced by the S252W fibroblast growth factor receptor 2 mutation in Apert craniosynostosis. J Bone Miner Res. 2001; 16:832-845 [DOI] [PubMed] [Google Scholar]
- 35. Tanimoto Y, Yokozeki M, Hiura K, Matsumoto K, Nakanishi H, Matsumoto T, Marie PJ, Moriyama K. A soluble form of fibroblast growth factor receptor 2 (FGFR2) with S252W mutation acts as an efficient inhibitor for the enhanced osteoblastic differentiation caused by FGFR2 activation in Apert syndrome. J Biol Chem. 2004; 279:45926-45934 [DOI] [PubMed] [Google Scholar]
- 36. Mansukhani A, Bellosta P, Sahni M, Basilico C. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J Cell Biol. 2000; 149:1297-1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ratisoontorn C, Fan GF, Mcentee K, Nah HD. Activating (P253R, C278F) and dominant negative mutations of FGFR2: Differential effects on calvarial bone cell proliferation, differentiation, and mineralization. Connect Tissue Res. 2003; 44 Suppl 1:292-297 [PubMed] [Google Scholar]
- 38. Miraoui H, Oudina K, Petite H, Tanimoto Y, Moriyama K, Marie PJ. Fibroblast growth factor receptor 2 promotes osteogenic differentiation in mesenchymal cells via ERK1/2 and protein kinase C signaling. J Biol Chem. 2009; 284:4897-4904 [DOI] [PubMed] [Google Scholar]
- 39. Wang Y, Sun M, Uhlhorn VL, Zhou X, Peter I, Martinez-Abadias N, Hill CA, Percival CJ, Richtsmeier JT, Huso DL, Jabs EW. Activation of p38 MAPK pathway in the skull abnormalities of Apert syndrome Fgfr2(+P253R) mice. BMC Dev Biol. 2010; 10:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yin L, Du X, Li C, Xu X, Chen Z, Su N, Zhao L, Qi H, Li F, Xue J, Yang J, Jin M, Deng C, Chen L. A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone. 2008; 42:631-643 [DOI] [PubMed] [Google Scholar]
- 41. Shukla V, Coumoul X, Wang RH, Kim HS, Deng CX. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet. 2007; 39:1145-1150 [DOI] [PubMed] [Google Scholar]
- 42. Datta SR, Brunet A, Greenberg ME. Cellular survival: A play in three Akts. Genes Dev. 1999; 13:2905-2927 [DOI] [PubMed] [Google Scholar]
- 43. Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: A hard Akt to follow. Trends Biochem Sci. 2001; 26:657-664 [DOI] [PubMed] [Google Scholar]
- 44. Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004; 29:233-242 [DOI] [PubMed] [Google Scholar]
- 45. Raucci A, Bellosta P, Grassi R, Basilico C, Mansukhani A. Osteoblast proliferation or differentiation is regulated by relative strengths of opposing signaling pathways. J Cell Physiol. 2008; 215:442-451 [DOI] [PubMed] [Google Scholar]
- 46. Tang CH, Yang RS, Huang TH, Liu SH, Fu WM. Enhancement of fibronectin fibrillogenesis and bone formation by basic fibroblast growth factor via protein kinase C-dependent pathway in rat osteoblasts. Mol Pharmacol. 2004; 66:440-449 [DOI] [PubMed] [Google Scholar]
- 47. Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005; 15:11-18 [DOI] [PubMed] [Google Scholar]
- 48. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010; 429:403-417 [DOI] [PubMed] [Google Scholar]
- 49. Thouverey C, Caverzasio J. The p38alpha MAPK positively regulates osteoblast function and postnatal bone acquisition. Cell Mol Life Sci. 2012; 69:3115-3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kadamur G, Ross EM. Mammalian phospholipase C. Annu Rev Physiol. 2013; 75:127-154 [DOI] [PubMed] [Google Scholar]
- 51. Moenning A, Jager R, Egert A, Kress W, Wardelmann E, Schorle H. Sustained platelet-derived growth factor receptor alpha signaling in osteoblasts results in craniosynostosis by overactivating the phospholipase C-gamma pathway. Mol Cell Biol. 2009; 29:881-891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miraoui H, Ringe J, Haupl T, Marie PJ. Increased EGF- and PDGFalpha-receptor signaling by mutant FGF-receptor 2 contributes to osteoblast dysfunction in Apert craniosynostosis. Hum Mol Genet. 2010; 19:1678-1689 [DOI] [PubMed] [Google Scholar]
- 53. Schwartzman RA, Cidlowski JA. Apoptosis: The biochemistry and molecular biology of programmed cell death. Endocr Rev. 1993; 14:133-151 [DOI] [PubMed] [Google Scholar]
- 54. Lemonnier J, Hay E, Delannoy P, Fromigue O, Lomri A, Modrowski D, Marie PJ. Increased osteoblast apoptosis in apert craniosynostosis: Role of protein kinase C and interleukin-1. Am J Pathol. 2001; 158:1833-1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Martinez-Abadias N, Holmes G, Pankratz T, Wang Y, Zhou X, Jabs EW, Richtsmeier JT. From shape to cells: Mouse models reveal mechanisms altering palate development in Apert syndrome. Dis Model Mech. 2013; 6:768-779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lazarus JE, Hegde A, Andrade AC, Nilsson O, Baron J. Fibroblast growth factor expression in the postnatal growth plate. Bone. 2007; 40:577-586 [DOI] [PubMed] [Google Scholar]
- 57. Rice DP, Rice R, Thesleff I. Fgfr mRNA isoforms in craniofacial bone development. Bone. 2003; 33:14-27 [DOI] [PubMed] [Google Scholar]
- 58. Wang Y, Xiao R, Yang F, Karim BO, Iacovelli AJ, Cai J, Lerner CP, Richtsmeier JT, Leszl JM, Hill CA, Yu K, Ornitz DM, Elisseeff J, Huso DL, Jabs EW. Abnormalities in cartilage and bone development in the Apert syndrome FGFR2 (+/S252W) mouse. Development. 2005; 132:3537-3548 [DOI] [PubMed] [Google Scholar]
- 59. Nagata M, Nuckolls GH, Wang X, Shum L, Seki Y, Kawase T, Takahashi K, Nonaka K, Takahashi I, Noman AA, Suzuki K, Slavkin HC. The primary site of the acrocephalic feature in Apert Syndrome is a dwarf cranial base with accelerated chondrocytic differentiation due to aberrant activation of the FGFR2 signaling. Bone. 2011; 48:847-856 [DOI] [PubMed] [Google Scholar]
- 60. Solomon LM, Fretzin D, Pruzansky S. Pilosebaceous abnormalities in Apert's syndrome. Arch Dermatol. 1970; 102:381-385 [PubMed] [Google Scholar]
- 61. Pochi PE, Strauss JS. Sebaceous gland response in man to the administration of testosterone, delta-4-androstenedione, and dehydroisoandrosterone. J Invest Dermatol. 1969; 52:32-36 [DOI] [PubMed] [Google Scholar]
- 62. Thiboutot D, Knaggs H, Gilliland K, Lin G. Activity of 5-alpha-reductase and 17-beta-hydroxysteroid dehydrogenase in the infundibulum of subjects with and without acne vulgaris. Dermatology. 1998; 196:38-42 [DOI] [PubMed] [Google Scholar]
- 63. Danilenko DM, Ring BD, Yanagihara D, Benson W, Wiemann B, Starnes CO, Pierce GF. Keratinocyte growth factor is an important endogenous mediator of hair follicle growth, development, and differentiation. Normalization of the nu/nu follicular differentiation defect and amelioration of chemotherapy-induced alopecia. Am J Pathol. 1995; 147:145-154 [PMC free article] [PubMed] [Google Scholar]
- 64. Kuslak SL, Thielen JL, Marker PC. The mouse seminal vesicle shape mutation is allelic with Fgfr2. Development. 2007; 134:557-565 [DOI] [PubMed] [Google Scholar]
- 65. Lin Y, Liu G, Zhang Y, Hu YP, Yu K, Lin C, Mckeehan K, Xuan JW, Ornitz DM, Shen MM, Greenberg N, Mckeehan WL, Wang F. Fibroblast growth factor receptor 2 tyrosine kinase is required for prostatic morphogenesis and the acquisition of strict androgen dependency for adult tissue homeostasis. Development. 2007; 134:723-734 [DOI] [PubMed] [Google Scholar]
- 66. Melnik BC, Schmitz G, Zouboulis CC. Anti-acne agents attenuate FGFR2 signal transduction in acne. J Invest Dermatol. 2009; 129:1868-1877 [DOI] [PubMed] [Google Scholar]
- 67. Melnik BC. Role of FGFR2-signaling in the pathogenesis of acne. Dermatoendocrinol. 2009; 1:141-156 [DOI] [PMC free article] [PubMed] [Google Scholar]