

Summary
Alzheimer's disease (AD) is a devastating neurodegenerative disease with progressive loss of memory and cognitive function, pathologically hallmarked by aggregates of the amyloid-beta (Aβ) peptide and hyperphosphorylated tau in the brain. Aggregation of Aβ under the form of amyloid fibrils has long been considered central to the pathogenesis of AD. However, recent evidence has indicated that soluble Aβ oligomers, rather than insoluble fibrils, are the main neurotoxic species in AD. The cellular prion protein (PrPC) has newly been identified as a cell surface receptor for Aβ oligomers. PrPC is a cell surface glycoprotein that plays a key role in the propagation of prions, proteinaceous infectious agents that replicate by imposing their abnormal conformation to PrPC molecules. In AD, PrPC acts to transduce the neurotoxic signals arising from Aβ oligomers, leading to synaptic failure and cognitive impairment. Interestingly, accumulating evidence has also shown that aggregated Aβ or tau possesses prion-like activity, a property that would allow them to spread throughout the brain. In this article, we review recent findings regarding the function of PrPC and its role in AD, and discuss potential therapeutic implications of PrPC-based approaches in the treatment of AD.
Keywords: PRNP gene, protein misfolding, N1 fragment, Fyn kinase, long-term potentiation (LTP)
1. Introduction
Alzheimer's disease (AD) is one of the most common neurodegenerative diseases, characterized clinically by progressive loss of memory and decline in cognitive function and pathologically by cerebral accumulation of amyloid-beta (Aβ) peptides in extracellular senile plaques and formation of intracellular neurofibrillary tangles constituted by hyperphosphorylated tau protein. The two pathological events are thought to be sequentially associated (1,2). AD is named after the German psychiatrist and neuropathologist Alois Alzheimer who first described the disease in 1907 (3–5). It is the most common form of dementia, while being a leading cause of death or disability. AD occurs most often in people over 65 years of age, although a less-prevalent early-onset type can occur much earlier. Around 35 million people are estimated to be afflicted with AD worldwide, and the incidence rises exponentially with advancing age, posing a huge challenge for society and health care (6). There are no treatments so far to cure, delay or stop the disease progression. Although the etiology of AD is not fully understood, accumulation of amyloid-beta (Aβ) peptides in the brain is considered the causative component of AD pathogenesis (amyloid hypothesis) (7–10). Aβ peptides of varying length are produced by sequential cleavage of amyloid precursor protein (APP) by β-secretase (mostly β-site APP-cleaving enzyme 1, BACE1) and gammasecretase (11–14). Compared to Aβ 40 (peptide with 40 amino acid residues), the longer form Aβ 42 (peptide with 42 amino acid residues) has an increased propensity to oligomerize and aggregate to form fibrillar amyloid plaques in the brain, and is widely regarded as the main pathogenic species causing AD. Genetic mutations in APP or preselinin-1 (PS-1) or PS-2 (catalytic subunits of gamma-secretase) lead to overproduction of Aβ 42, and cause early onset AD (15).
Upon failure of all the Aβ-centric approaches that reached Phase III clinical trials, scientists began to question the pathogenic role of amyloid aggregates (senile plaques) that comprise Aβ fibrils, which is the main theme of the amyloid hypothesis (10), and speculate that the soluble pre-fibrillar Aβ oligomers are most likely the principal toxic forms of Aβ peptide (16–20). Soluble Aβ oligomers are found to be elevated in AD brains, and their levels are strongly correlated with disease onset and severity (21–23). The cellular mechanisms of Aβ oligomer-mediated neurotoxicity are poorly understood. Recent evidence indicate that the Aβ oligomers, also referred to as amyloid-derived diffusible ligands (ADDLs) (22,24,25), may bind to a surface receptor on neurons, thereby initiating signaling transduction pathways that lead to synaptic dysfunction and neuronal death (26–29). One interesting receptor for Aβ oligomers so far identified is the cellular prion protein (PrPC) (30), which is a cell membrane glycoprotein ubiquitously expressed but enriched in the brain.
The discovery of PrPC as a cell surface receptor for Aβ oligomers has sparked a major interest in research focusing on identification of downstream effectors that mediate the neuronal toxicity and synaptic dysfunction in AD. This subject will be discussed in details later.
2. Cellular prion protein (PrPC)
Prion diseases are a group of fatal infectious neurodegenerative diseases comprising Creutzfeldt-Jacob disease (CJD), variant Creutzfeldt-Jacob disease (vCJD), Gerstmann-Straussler-Scheinker disease (GSS), fatal familial insomnia (FFI), and Kuru in humans, as well as bovine spongiform encephalopathy (BSE) (otherwise known as mad cow disease) and scrapie in animals (31,32). In prion diseases, the normal PrPC is converted into the β-sheet rich, protease-resistant pathogenic form — scrapie prion protein (PrPSc), which is infectious and spreads throughout the brain (33). Stanley Prusiner at the University of California San Francisco (UCSF), USA, first described in 1982 that novel proteinaceous infectious particles (prions) could replicate and propagate without nucleic acids and cause scrapie (32).
PrPC is encoded by the PRNP gene (PRioN Protein) on chromosome 20 in human and the corresponding chromosome 2 in mouse (34,35). PrPC is synthesized in the endoplasmic reticulum (ER) and transits the Golgi on its way to the cell surface. The structure of mouse PrPC is illustrated in Figure 1 (36). Post-translational modifications of PrPC include removal of the N-terminal signal peptide (residues 1–22), N-linked glycosylation at Asn-180 and Asn-196, formation of a disulfide bond between residues 178 and 231, and attachment of a glycosylphosphatidylinositol (GPI) anchor following removal of the C-terminal hydrophobic peptide (residues 231-254) (36–39), rendering a mature PrPC with about 210 amino acid residues and a molecular weight of 33-35 kDa. PrPC is almost ubiquitously expressed across tissues, with an enrichment in synaptic membranes and astrocytes in the brain.
Figure 1.
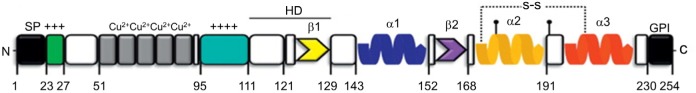
Scheme of PrPC primary structure. The N-terminal part includes (from left to right): a signal peptide (SP, residues 1–22) (removed during PrP biosynthesis in the endoplasmic reticulum), a polybasic region (residues 23–27, green), five histidine-containing octapeptide repeats (residues 51–90, gray) (bind Cu2+ and other bivalent metal ions), a central region (CR) (residues 95–111, cyan, positively charged), and a hydrophobic domain (HD, residues 111–130, highly conserved region). The C-terminal part includes (from left to right): two short β-strands (residues 127–129, yellow; and 166–168, purple), three α-helices (residues 143–152, blue; 171–191, orange; and 199–221, red), and a C-terminal peptide (residues 231–254, black), which is removed during biosynthesis, followed by covalent attachment of a glycosylphosphatidylinositol (GPI) anchor, which attaches the protein to the outer leaflet of the plasma membrane. PrPC also contains two N-linked oligosaccharide chains (at Asn-180 and Asn-196, black lollipops) and a disulfide bond between residues 178 and 231 (indicated by a dashed line). Residues correspond to the mouse sequence. This figure is adapted from Biasini et al. Trends in Neurosciences (2012) with permission.
The precise physiological function of PrPC is still unknown. Several pieces of evidence have shown that PrPC plays a role in metal ion trafficking (40,41), cell adhesion (42–44), cell survival (36,45), immune regulation (46,47) and signal transduction (26,48,49). PRNP gene knockout mice are developmentally normal and have no signs of neurodegeneration (50,51), indicating that the prion pathology is unlikely to be the result of a loss of PrPC function. On the contrary, depletion of neuronal PrPC is protective and reverses the disease pathology in scrapie-infected mice (52), presumably due to depletion of the substrate for generation of PrPSc (53). Scrapie infection of transgenic mice expressing PrPC lacking the GPI anchor causes efficient prion replication, but no pathology (54). Therefore, normal GPI-anchored PrPC is required for the neurotoxicity of PrPSc (55,56).
3. PrPC: a receptor to mediate Aβ toxicity
It is now widely accepted that the soluble Aβ oligomers are the toxic species that leads to synaptic and cognitive dysfunction as well as neurodegeneration in AD (17,25). This concept is supported by studies showing the strong correlation of the synaptic loss with cortical levels of soluble Aβ species rather than with plaque distribution in AD patients (20,21,24), and the inhibitory effect of soluble Aβ oligomers on long-term potentiation (LTP) (19,22,57–59).
The mechanism of Aβ oligomer toxicity remained largely unknown until the Strittmatter's group at Yale University identified PrPC as a receptor capable of mediating the neurotoxic effect of Aβ oligomers (30). To identify candidates for this receptor, they conducted an unbiased expression-cloning screen of a brain cDNA library for binding sites with an oligomeric preparation of synthetic biotinylated Aβ 42 peptides. From more than 225,000 expressed clones, two independent positive clones were isolated and both were found to encode full-length mouse PrP. They found that Aβ 42 oligomers bound to PrP with high affinity and specificity. They then tested the function of this interaction in cultured hippocampal slices, and found that nanomolar concentration of Aβ 42 oligomers potently suppressed CA1 hippocampal LTP and this suppression was not observed in slices generically lacking PrPC or in presence of an anti-PrP antibody that blocks the binding of Aβ 42 oligomers to PrPC, indicating that the suppression of LTP is specifically mediated by binding of Aβ 42 oligomers to PrPC (30). Therefore, they have provided compelling evidence that PrP is a specific binding partner for Aβ 42 oligomers and mediates the inhibitory effect of Aβ 42 on synaptic plasticity.
The Strittmatter's group then sought to test in vivo whether PrPC is essential for the ability of brain-derived Aβ to suppress cognitive function. They crossed familial AD transgenes encoding APPswe and PSen1DeltaE9 into Prnp−/− mice, and found that mice lacking PrPC, but containing Aβ plaques derived from APPswe/PSen1DeltaE9 transgenes, showed no detectable impairment of spatial learning and memory, while the AD transgenic mice with intact PrPC exhibited dramatic deficits in spatial learning and memory, indicating that PrPC is selectively required for the toxicity of the naturally occurring Aβ in the brain that leads to the cognitive phenotypes in these AD transgenic mice (60), which is in consistency with previous reports that Aβ oligomers isolated from the brain of Alzheimer's patients (20,23,59,61) requires PrPC to suppress LTP (62,63).
The Strittmatter's group further found that soluble Aβ assemblies derived from the brains of individuals with Alzheimer's disease interacted with PrPC at the postsynaptic density to activate the Src kinase Fyn, which phosphorylates the NR2B subunit of NMDA receptor and causes transient increase of NR2B on the cell surface with consequent excitotoxicity, while rendering destabilization of dendritic spines. Both NR2B phosphorylation and spine destabilization incurred by Aβ oligomers were eliminated in Prnp−/−and Fyn−/− neurons, indicating a specific association of Aβ-PrPC-Fyn-mediated toxic signaling (26,64). This study sheds new light on the molecular mechanism of PrPC-mediated Aβ toxicity, while indicating a prion connection of Aβ and Fyn (22,49). Another group further demonstrated that soluble Aβ binds to PrPC at neuronal dendritic spines, where it forms a complex with Fyn, and results in the activation of the kinase and subsequent Fyn-dependent tau hyperphosphorylation in a PRNP gene dose-dependent manner (2), making another prion connection that links together the two hallmark pathological events in AD — amyloid accumulation and tau hyperphosphorylation (22,65,66). However, how binding of Aβ oligomers to PrPC activates Fyn still remains enigmatic. PrPC is an extracellular protein attached to the outer surface of the cell membrane by a GPI anchor, and Fyn is located in the cytoplasm. Current evidence indicates that factors like caveolin-1 or the neural cell adhesion molecule (NCAM) could potentially connect PrPC and Fyn from the two opposite sides of the cell membrane (48,67–69).
Two distinct binding sites for Aβ oligomers have been identified on PrPC by deletion analysis, antibody binding (30), and biophysical techniques such as site-directed spin labeling and surface plasmon resonance (70). Both sites are rich in positively charged basic residues: one is immediately adjacent to the central region (residues 95–110) and the other is at the extreme N-terminus (residues 23–27) (Refer to Figure 1). It is very likely that the two sites act in concert to render high affinity binding for Aβ oligomers, and deletion of either region results in a major loss of the binding capacity (36,70).
4. PrPC: a tale of an “evil angel”
As we know, PrPC is converted into an aggregated, β-sheet-rich neurotoxic isoform called PrPSc in prion diseases (33,71). PrPC serves not only as the substrate for PrPSc conversion and propagation, but also as a transducer of PrPSc-associated neuronal death (52,56,72). Another noxious function of PrPC is to serve as a cell surface receptor for Aβ oligomers to mediate signal transduction leading to neuronal toxicity (26,30,60), which we have already discussed above.
However, this “evil” protein has been found to have numerous beneficial “angel” functions. One notable function is that PrPC suppresses glutamate-mediated neuronal excitatoxicity by inhibiting NMDA receptor (36,73). Another function is that PrPC physically interacts with the APP cleaving enzyme BACE1 through its N-terminal polybasic domain (residues 23-26) and inhibits its enzyme activity, resulting in a reduction of Aβ production (74–76), which indicates a preventive role against AD. In both cell and animal models, PrPC has been shown to lower Aβ production by inhibiting BACE1 (75,76).This function is thought to be modulated by PRNP polymorphism at codon 129 (M129V), which may be associated with increased risk of AD (77–81). Interestingly, binding of Aβ oligomers to PrPC impairs the inhibitory effect of PrPC on BACE1 activity (64), which may indicate another mechanism of Aβ oligomer toxicity.
A physiological process that makes PrPC a “double-faced gem” is that PrPC undergoes constitutive proteolytic cleavage between residues 111/112, yielding a soluble N-terminal fragment (N1) and a membrane-bound C-terminal fragment (C1), which have a protective role in AD and prion disease, respectively. N1 binds to Aβ oligomers with high affinity, and blocks the neurotoxicity of Aβ oligomers through neutralizing toxic assemblies of Aβ. Therefore, N1 may serve as a potent inhibitor of Aβ oligomer toxicity and represent an entirely new class of therapeutic agents for the treatment of AD (82). N1 is a naturally occurring soluble fragment that is generated by endogenous proteolytic processing of PrPC at the α-site (residues 111 and 112) (83), presumably by ADAM (a disintegrin and metalloprotease) proteases (84–87). Blocking ADAM10 synaptic trafficking has been shown to be able to generate a model of sporadic Alzheimer's disease (88). Agents that could stimulate α-cleavage of PrPC should be good drug candidates. On the other hand, the C1 fragment of PrPC can inhibit PrPSc formation and accumulation of neurotoxic forms of PrP. The C1 transgenic mice inoculated with PrPSc were found healthy and did not exhibit PrPSc accumulation, indicating that C1 is not a substrate for conversion to PrPSc. Manipulating C1 fragment may thus have therapeutic value for prion diseases (89).
PrPC homodimerization has been found to be an important regulator of PrPC α-cleavage and stimulate the production of N1 and C1 fragments. The increase of N1 is protective against the toxicity of Aβ oligomers. Thus, manipulation of PrPC homodimerization may represent a potential therapeutic avenue against Aβ toxicity in Alzheimer's disease (90). Interestingly, the APP processing enzyme α-secretase (belongs to ADAM family of zinc metalloproteases), which precludes Aβ production by cleaving APP within the Aβ domain (91), also cleaves PrPC at the α-site (residues 111 and 112), releasing N1 from the membrane. Therefore, enhancing the activity of α-secretase may represent a novel therapeutic strategy by reducing the toxic Aβ production and increasing the protective N1 production (92).
5. AD: a story of two prions
The misfolding and aggregation of Aβ and tau proteins were traditionally thought to contribute in parallel to pathogenesis of AD. Accumulating evidence indicated that misfolded, toxic oligomers of Aβ and tau spread through the brain in a way much like misfolded PrP (93–95). The misfolded forms of Aβ or tau have a seeding effect, and can induce normal Aβ or tau in the cells to misfold, spread and become toxic (96–102). Therefore, AD can be regarded as a disease that harbors two proteins with prion-like behavior: Aβ and tau (103,104). The prion-like propagation of additional proteins whose misfolding into β-sheet-rich structures underlies other well-known neurodegenerative diseases has also been indicated (105–107). Thus, a prion-based mechanism is proposed to unite a wide array of neurodegenerative diseases, all of which may stem from misfolded proteins self-propagating through the brain (103,108). Local injection of misfolded Aβ in the brains of AD transgenic mice has been found to trigger the misfolding and spreading of otherwise normal Aβ throughout the brain, indicating the prion-like activity of Aβ (108–114). Injection of AD brain extracts into the hippocampus of mice expressing human wild-type APP induces Aβ deposition, which progressively increases over time after inoculation and spreads to brain areas far from the injection site, where other Aβ-related pathology is also observed (114). It is believed that certain Aβ conformations tend to self-propagate, and targeting those specific Aβ assemblies may interrupt the spread of Aβ deposition, hence exerting therapeutic effect on AD (109).
Intracerebral injection of aggregation-prone mutant tau in mice has also been demonstrated to induce wild-type tau to form neurofibrillary tangles and spread throughout the brain (99). Accumulating evidence indicates that Aβ works upstream of tau in AD pathogenesis (65,115–121). Aβ can bind to tau and induce formation of tau oligomers, which can then self-propagate without additional Aβ, indicating a cross talk between the two prions (104).
6. PrPC: a novel therapeutic target for AD
PrPC has been identified as a major player in mediating the toxicity of Aβ oligomers that leads to synaptic loss and cognitive impairment in AD. Therefore, targeting PrPC, its interaction with Aβ oligomers, or downstream mediators can be considered the new line of choice for therapeutic development for treatment of Alzheimer's disease.
Genetic ablation of PrPC in mice rescues the neurotoxic phenotypes of Aβ oligomers (30,60). It might be reasonable to speculate that using shRNA or siRNA to knock down the expression of PrPC may represent a therapeutic approach for AD, though little has been done in this regard. Nevertheless, knocking down PrPC will also affect other functions of PrPC, causing various complications. For example, PrPC reduces Aβ production by inhibiting BACE1 activity, and has a protective role in AD (74,75). This can be jeopardized by PrPC knockdown.
Attempts have been made to seek antibodies that could efficiently bind to PrPC and block the binding site(s) of Aβ oligomers, which may have therapeutic effect on AD by preventing the Aβ oligomer/PrPC-initiated noxious signaling. Michael Rowan group at University College Dublin in the U.K. found that antibodies against the epitopes at the PrP principal Aβ-binding site and helix-1 were able to block Aβ binding and block the Aβ-mediated disruption of synaptic plasticity (63). Chung et al. from New York University School of Medicine intraperitoneally injected the monoclonal anti-PrP antibody, 6D11, in APP/PS1 transgenic mice, and found that the treatment with 6D11 antibody completely rescue the cognitive and behavioral deficits of the transgenic animals (122). The 6D11 antibody is directed against the epitope (residues 93–109), which is the region suggested to be involved in Aβ oligomer binding.
Screening for small molecules that could efficiently target either the Aβ oligomer/PrPC interaction or the downstream mediators may represent a promising avenue for therapeutic development.
The Fyn kinase has been found to be activated upon binding of Aβ oligomers with PrPC, which then initiate downstream signaling to mediate Aβ toxicity, for example, activation of Fyn kinase lead to hyperphosphorylation of tau (2,66). Targeting Fyn kinase or other Aβ/PrPC downstream mediators, for example by genetic engineering, RNAi, or small molecule modulators, may also be of therapeutic value.
Synthetic N1 fragment, equivalent of that released by α-cleavage of endogenous PrPC, has been found to bind Aβ oligomers with high affinity, sequester Aβ oligomers in the extracellular space, and hence block the Aβ oligomer-mediated synaptic toxicity (82). Therefore, exogenous administration of N1 or enhancement of endogenous α-cleavage of PrPC represents a brand-new class of therapeutic approaches for AD. Among others, seeking modulators that prevent Aβ oligomerization or inhibit the prion-like activity of Aβ or tau may represent another category of therapeutic development strategies for the treatment of AD.
The ‘PrPC axis’ of therapeutic development strategies for AD is illustrated in Figure 2.
Figure 2.
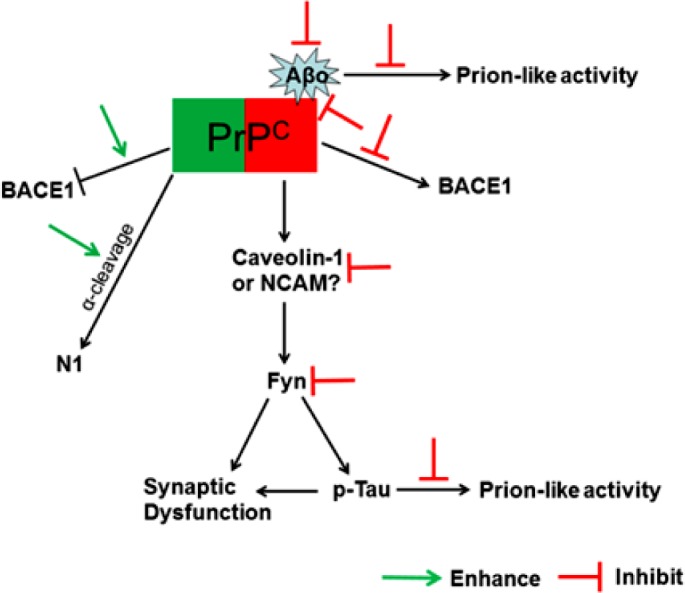
Therapeutic development strategies for AD (The ‘PrPC axis’). The green color portion of PrPC indicates the protective function of PrPC (left-hand side), and the red color portion of PrPC indicates PrPC as the receptor of Aβ oligomers (righthand side) to mediate the Aβ toxicity. The left-hand side of the figure indicates therapeutic strategies by enhancing the normal function of PrPC (enhancing the inhibitory effect on BACE1, which reduces Aβ production, and enhancing the α-cleavage, which increases production of the protective N1 fragment of PrPC). The right-hand side of the figure indicates therapeutic strategies by targeting the Aβ oligomer/PrPC-mediated toxic signaling pathway, which encompass measures to inhibit Aβ oligomerization, Aβ prion-like activity, interaction of Aβ oligomers with PrPC, Aβ oligomer/PrPC-mediated disinhibition of BACE1, intermediate mediators (such as caveolin-1 and NCAM), Fyn kinase, and prion-like activity of hyperphosphorylated Tau. (Abbreviations: Aβo: Aβ oligomers; N1: N1 fragment of PrPC; p-Tau: hyperphosphorylated Tau.)
7. Discrepancies
The Strittmatter group's discovery of PrPC as an Aβ oligomer receptor to mediate synaptotoxicity has created an exciting hot spot, which has greatly stimulated research in the field. However, studies from different groups around the world showed discrepancies and some groups came to a completely opposite conclusion to the Strittmatter group's, which made scientists begin to question the role of PrPC in mediating the toxic effects of Aβ oligomeric assemblies.
The work published in Nature a year later from Roberto Malinow's group at the University of California at San Diego in the USA reported that PrPC is not required for Aβ-induced synaptic toxicity, having raised a conflicting concern that Aβ-mediated synaptic defects do not require PrPC. The Aβ-induced depression of synaptic transmission was observed in both wild-type and Prnp−/− mouse slices (123).
Gianluigi Forloni and his team at the Mario Negri Institute for Pharmacological Research in Italy injected Aβ 42 oligomers into the lateral ventricle of C56BL/6 mice and found that PrP-expressing and PrP knock-out mice were equally susceptible to cognitive impairment, suggesting that PrPC is not required for Aβ 42 oligomermediated cognitive impairment, although they in the meantime confirmed that Aβ 42 oligomers do interact with PrPC with nanomolar affinity (124).
Andriano Aguzzi and his group at the University Hospital Zurich in Switzerland have also reported findings that challenge the role of PrPC as an Aβ toxicity mediator. They found that deletion or overexpression of PrPC had no effect on the impairment of hippocampal synaptic plasticity, while having also confirmed the efficient binding of Aβ 42 oligomers to PrPC (125), once again showing contradictory results to those of Strittmatter and his team (30,60).
However, those conflicting reports do not necessarily negate the findings of Strittmatter and his team. They might arise from differences in animal models, experimental settings, and preparations of Aβ oligomers (method of preparation, material source, and size and conformation of Aβ oligomeric assemblies may all matter) (126).
A recent study from Michael Rowan group at University College Dublin in the U.K., which we have mentioned previously, clearly shows that PrP is required for the plasticity-impairing effects of toxic Aβ species from human AD brain and that standardized ADDL preparations disrupt hippocampal synaptic plasticity in a PrP-dependent manner (63). They further found that antibodies that block Aβ binding to PrPC block the toxic effect on synaptic plasticity.
Sylvain Lesne group at the University of Minnesota in the USA has recently demonstrated that soluble Aβ binds to PrPC at neuronal dendritic spines, where PrPC is enriched, and causes hyperphosphorylation of tau by activation of the Fyn kinase. The PrPC antibody 6D11 prevents Aβ oligomers from binding to PrPC, and abolishes subsequent Fyn activation and Fyn-dependent tau hyperphosphorylation.
There are also other studies that have indicated a role of PrPC in mediating the toxicity of Aβ oligomers, supporting the findings of the Strittmatter group (64,127). Therefore, the argument is far from conclusive.
8. Concluding remarks and perspectives
Although there are conflicting reports regarding the function of PrPC as a cell surface receptor to mediate the deleterious effects of Aβ oligomers in AD, there are no ambiguities for two end points: high affinity binding of Aβ oligomers to PrPC (30,70,124,125), and high synaptic toxicity of Aβ oligomers (23,59–61). The challenge remaining for scientists is to ‘make the two ends meet’.
Strittmatter group's finding that PrPC acts as the receptor for mediating Aβ oligomer neurotoxicity (30) has at least opened a new direction towards understanding the molecular mechanism that connect Aβ oligomers and their toxic effects. The research discoveries of his team have apparently been supported by multiple studies from other groups, though there have been conflicting results reported. Sophisticated studies using advanced animal models and optimized experimental conditions are needed to elucidate the precise role of PrPC in mediating Aβ oligomer neurotoxicity, or to identify other potential cell surface receptors and signaling networks that make the two ends — toxin and toxicity — meet.
If the role of PrPC in Aβ oligomer-mediated pathogenic process turns out to be substantial, it would be of interest to seek potential co-receptors or to examine whether other Aβ-mediated signaling pathways are also PrPC-dependent, making a complete prion connection network of AD.
To conclude, endeavors to gain precise understanding of the Aβ oligomer-mediated neurotoxic signaling pathways will greatly facilitate the development of novel therapies that would be able to target specific Aβ oligomeric assemblies and their downstream associates, and offer new hope to AD patients and families. Identification of the biophysical features of naturally occurring toxic Aβ species in human AD brain would be of critical reference to clinically relevant translational research.
Acknowledgments
We are grateful to Dr. Emiliano Biasini for critical reading of the manuscript and helpful advice.
References
- 1. Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010; 362:329-344 [DOI] [PubMed] [Google Scholar]
- 2. Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, Aguzzi A, Lesne SE. The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer's disease. J Neurosci. 2012; 32:16857-16871a [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3. Berchtold NC, Cotman CW. Evolution in the conceptualization of dementia and Alzheimer's disease: Greco-Roman period to the 1960s. Neurobiol Aging. 1998; 19:173-189 [DOI] [PubMed] [Google Scholar]
- 4. Maurer K, Volk S, Gerbaldo H. Auguste D and Alzheimer's disease. Lancet. 1997; 349:1546-1549 [DOI] [PubMed] [Google Scholar]
- 5. Alzheimer A. About a peculiar disease of the cerebral cortex. Centralblatt fur Nervenheilkunde Psychiatrie. 1907; 30:177-179 (in German) [Google Scholar]
- 6. Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer's disease. Alzheimers Dement. 2007; 3:186-191 [DOI] [PubMed] [Google Scholar]
- 7. Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991; 12:383-388 [DOI] [PubMed] [Google Scholar]
- 8. Selkoe DJ. Alzheimer's disease results from the cerebral accumulation and cytotoxicity of amyloid β-protein. J Alzheimers Dis. 2001; 3:75-80 [DOI] [PubMed] [Google Scholar]
- 9. Yankner BA, Lu T. Amyloid β-protein toxicity and the pathogenesis of Alzheimer disease. J Biol Chem. 2009; 284:4755-4759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science. 2002; 297:353-356 [DOI] [PubMed] [Google Scholar]
- 11. Vassar R, Bennett BD, Babu-Khan S, et al. β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999; 286:735-741 [DOI] [PubMed] [Google Scholar]
- 12. Ray WJ, Yao M, Mumm J, Schroeter EH, Saftig P, Wolfe M, Selkoe DJ, Kopan R, Goate AM. Cell surface presenilin-1 participates in the gamma-secretase-like proteolysis of Notch. J Biol Chem. 1999; 274:36801-36807 [DOI] [PubMed] [Google Scholar]
- 13. Zhang H, Ma Q, Zhang YW, Xu H. Proteolytic processing of Alzheimer's β-amyloid precursor protein. J Neurochem. 2012; 120(Suppl 1):9-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Selkoe D, Mandelkow E, Holtzman D. Deciphering Alzheimer disease. Cold Spring Harb Perspect Med. 2012; 2:a011460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bertram L, Tanzi RE. The genetics of Alzheimer's disease. Prog Mol Biol Transl Sci. 2012; 107:79-100 [DOI] [PubMed] [Google Scholar]
- 16. Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: An appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011; 10:698-712 [DOI] [PubMed] [Google Scholar]
- 17. Lublin AL, Gandy S. Amyloid-β oligomers: Possible roles as key neurotoxins in Alzheimer's Disease. Mt Sinai J Med. 2010; 77:43-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gandy S, Simon AJ, Steele JW, Lublin AL, Lah JJ, Walker LC, Levey AI, Krafft GA, Levy E, Checler F, Glabe C, Bilker WB, Abel T, Schmeidler J, Ehrlich ME. Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-β oligomers. Ann Neurol. 2010; 68:220-230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011; 31:6627-6638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999; 46:860-866 [DOI] [PubMed] [Google Scholar]
- 21. Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999; 155:853-862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998; 95:6448-6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006; 440:352-357 [DOI] [PubMed] [Google Scholar]
- 24. Walsh DM, Selkoe DJ. A β oligomers — a decade of discovery. J Neurochem. 2007; 101:1172-1184 [DOI] [PubMed] [Google Scholar]
- 25. Krafft GA, Klein WL. ADDLs and the signaling web that leads to Alzheimer's disease. Neuropharmacology. 2010; 59:230-242 [DOI] [PubMed] [Google Scholar]
- 26. Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012; 15:1227-1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verdier Y, Zarandi M, Penke B. Amyloid β-peptide interactions with neuronal and glial cell plasma membrane: Binding sites and implications for Alzheimer's disease. J Pept Sci. 2004; 10:229-248 [DOI] [PubMed] [Google Scholar]
- 28. Origlia N, Capsoni S, Cattaneo A, Fang F, Arancio O, Yan SD, Domenici L. Aβ-dependent Inhibition of LTP in different intracortical circuits of the visual cortex: The role of RAGE. J Alzheimers Dis. 2009; 17:59-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang HY, Stucky A, Liu J, Shen C, Trocme-Thibierge C, Morain P. Dissociating β-amyloid from alpha 7 nicotinic acetylcholine receptor by a novel therapeutic agent, S 24795, normalizes alpha 7 nicotinic acetylcholine and NMDA receptor function in Alzheimer's disease brain. J Neurosci. 2009; 29:10961-10973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature. 2009; 457:1128-1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011; 3:a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982; 216:136-144 [DOI] [PubMed] [Google Scholar]
- 33. Prusiner SB. The prion diseases. Brain Pathol. 1998; 8:499-513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kretzschmar HA, Stowring LE, Westaway D, Stubblebine WH, Prusiner SB, Dearmond SJ. Molecular cloning of a human prion protein cDNA. DNA. 1986; 5:315-324 [DOI] [PubMed] [Google Scholar]
- 35. Sparkes RS, Simon M, Cohn VH, Fournier RE, Lem J, Klisak I, Heinzmann C, Blatt C, Lucero M, Mohandas T, Dearmond SJ, Westaway D, Prusiner SB, Weiner LP. Assignment of the human and mouse prion protein genes to homologous chromosomes. Proc Natl Acad Sci U S A. 1986; 83:7358-7362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Biasini E, Turnbaugh JA, Unterberger U, Harris DA. Prion protein at the crossroads of physiology and disease. Trends Neurosci. 2012; 35:92-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harris DA. Trafficking, turnover and membrane topology of PrP. Br Med Bull. 2003; 66:71-85 [DOI] [PubMed] [Google Scholar]
- 38. Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987; 51:229-240 [DOI] [PubMed] [Google Scholar]
- 39. Haraguchi T, Fisher S, Olofsson S, et al. Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch Biochem Biophys. 1989; 274:1-13 [DOI] [PubMed] [Google Scholar]
- 40. Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. J Biol Chem. 1998; 273:33107-33110 [DOI] [PubMed] [Google Scholar]
- 41. Watt NT, Taylor DR, Kerrigan TL, Griffiths HH, Rushworth JV, Whitehouse IJ, Hooper NM. Prion protein facilitates uptake of zinc into neuronal cells. Nat Commun. 2012; 3:1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chiesa R, Harris DA. Fishing for prion protein function. PLoS Biol. 2009; 7:e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mangé A, Milhavet O, Umlauf D, Harris D, Lehmann S. PrP-dependent cell adhesion in N2a neuroblastoma cells. FEBS Lett. 2002; 514:159-162 [DOI] [PubMed] [Google Scholar]
- 44. Málaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, Stuermer CA. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 2009; 7:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka KV, Nave KA, Weis J, Aguzzi A. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010; 13 :310-318 [DOI] [PubMed] [Google Scholar]
- 46. Cashman NR, Loertscher R, Nalbantoglu J, Shaw I, Kascsak RJ, Bolton DC, Bendheim PE. Cellular isoform of the scrapie agent protein participates in lymphocyte activation. Cell. 1990; 61:185-192 [DOI] [PubMed] [Google Scholar]
- 47. Ballerini C, Gourdain P, Bachy V, Blanchard N, Levavasseur E, Grégoire S, Fontes P, Aucouturier P, Hivroz C, Carnaud C. Functional implication of cellular prion protein in antigen-driven interactions between T cells and dendritic cells. J Immunol. 2006; 176:7254-7262 [DOI] [PubMed] [Google Scholar]
- 48. Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science. 2000; 289:1925-1928 [DOI] [PubMed] [Google Scholar]
- 49. Um JW, Strittmatter SM. Amyloid-β induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion. 2013; 7:37-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992; 356:577-582 [DOI] [PubMed] [Google Scholar]
- 51. Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994; 8:121-127 [DOI] [PubMed] [Google Scholar]
- 52. Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003; 302:871-874 [DOI] [PubMed] [Google Scholar]
- 53. Büeler, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993; 73:1339-1347 [DOI] [PubMed] [Google Scholar]
- 54. Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005; 308:1435-1439 [DOI] [PubMed] [Google Scholar]
- 55. Klingeborn M, Race B, Meade-White KD, Rosenke R, Striebel JF, Chesebro B. Crucial role for prion protein membrane anchoring in the neuroinvasion and neural spread of prion infection. J Virol. 2011; 85:1484-1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996; 379:339-343 [DOI] [PubMed] [Google Scholar]
- 57. Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007; 27:2866-2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of β amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002; 924:133-140 [DOI] [PubMed] [Google Scholar]
- 59. Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008; 14:837-842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Lauren J, Gimbel ZA, Strittmatter SM. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010; 30:6367-6374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002; 416:535-539 [DOI] [PubMed] [Google Scholar]
- 62. Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ. Alzheimer's disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci. 2011; 31:7259-7263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J. Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat Commun. 2011; 2:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rushworth JV, Griffiths HH, Watt NT, Hooper NM. Prion protein-mediated toxicity of amyloid-β oligomers requires lipid rafts and the transmembrane LRP1. J Biol Chem. 2013; 288:8935-8951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model. Science. 2007; 316:750-754 [DOI] [PubMed] [Google Scholar]
- 66. Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011; 31:700-711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Toni M, Spisni E, Griffoni C, Santi S, Riccio M, Lenaz P, Tomasi V. Cellular prion protein and caveolin-1 interaction in a neuronal cell line precedes Fyn/Erk 1/2 signal transduction. J Biomed Biotechnol. 2006; 2006:69469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tomasi V. Signal transduction in neurons: Effects of cellular prion protein on fyn kinase and ERK1/2 kinase. Immun Ageing. 2010; 7(Suppl 1):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol. 2005; 169:341-354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-beta (Aβ) oligomers: Role OF N-terminal residues. J Biol Chem. 2010; 285:26377-26383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998; 95:13363-13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Brandner S, Raeber A, Sailer A, Blattler T, Fischer M, Weissmann C, Aguzzi A. Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc Natl Acad Sci U S A. 1996; 93:13148-13151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, Chen L, Villemaire M, Ali Z, Jirik FR, Zamponi GW. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol. 2008; 181:551-565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Griffiths HH, Whitehouse IJ, Hooper NM. Regulation of amyloid-β production by the prion protein. Prion. 2012; 6:217-222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Parkin ET, Watt NT, Hussain I, Eckman EA, Eckman CB, Manson JC, Baybutt HN, Turner AJ, Hooper NM. Cellular prion protein regulates β-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc Natl Acad Sci U S A. 2007; 104:11062-11067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Griffiths HH, Whitehouse IJ, Baybutt H, Brown D, Kellett KA, Jackson CD, Turner AJ, Piccardo P, Manson JC, Hooper NM. Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J Biol Chem. 2011; 286:33489-33500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dermaut B, Croes EA, Rademakers R, Van den Broeck M, Cruts M, Hofman A, van Duijn CM, Van Broeckhoven C. PRNP Val129 homozygosity increases risk for early-onset Alzheimer's disease. Ann Neurol. 2003; 53:409-412 [DOI] [PubMed] [Google Scholar]
- 78. Gacia M, Safranow K, Styczynska M, Jakubowska K, Peplonska B, Chodakowska-Zebrowska M, Przekop I, Slowik A, Golanska E, Hulas-Bigoszewska K, Chlubek D, Religa D, Zekanowski C, Barcikowska M. Prion protein gene M129 allele is a risk factor for Alzheimer's disease. J Neural Transm. 2006; 113:1747-1751 [DOI] [PubMed] [Google Scholar]
- 79. Rujescu D, Hartmann AM, Gonnermann C, Moller HJ, Giegling I. M129V variation in the prion protein may influence cognitive performance. Mol Psychiatry. 2003; 8:937-941 [DOI] [PubMed] [Google Scholar]
- 80. Del Bo R, Scarlato M, Ghezzi S, Martinelli-Boneschi F, Fenoglio C, Galimberti G, Galbiati S, Virgilio R, Galimberti D, Ferrarese C, Scarpini E, Bresolin N, Comi GP. Is M129V of PRNP gene associated with Alzheimer's disease? A case-control study and a meta-analysis. Neurobiol Aging. 2006; 27:770.e1-770.e5 [DOI] [PubMed] [Google Scholar]
- 81. Choi IG, Woo SI, Kim HJ, Kim DJ, Park BL, Cheong HS, Pasaje CF, Park TJ, Bae JS, Chai YG, Shin HD. Lack of association between PRNP M129V polymorphism and multiple sclerosis, mild cognitive impairment, alcoholism and schizophrenia in a Korean population. Dis Markers. 2010; 28:315-321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fluharty BR, Biasini E, Stravalaci M, Sclip A, Diomede L, Balducci C, La Vitola P, Messa M, Colombo L, Forloni G, Borsello T, Gobbi M, Harris DA. An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J Biol Chem. 2013; 288:7857-7866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem. 1995; 270:19173-19180 [DOI] [PubMed] [Google Scholar]
- 84. Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, De Strooper B, Grassi J, Lopez-Perez E, Checler F. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem. 2001; 276:37743-37746 [DOI] [PubMed] [Google Scholar]
- 85. Liang J, Wang W, Sorensen D, Medina S, Ilchenko S, Kiselar J, Surewicz WK, Booth SA, Kong Q. Cellular prion protein regulates its own α-cleavage through ADAM8 in skeletal muscle. J Biol Chem. 2012; 287:16510-16520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yusa S, Oliveira-Martins JB, Sugita-Konishi Y, Kikuchi Y. Cellular prion protein: From physiology to pathology. Viruses. 2012; 4:3109-3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Alfa Cissé M, Louis K, Braun U, Mari B, Leitges M, Slack BE, Fisher A, Auberger P, Checler F, Vincent B. Isoform-specific contribution of protein kinase C to prion processing. Mol Cell Neurosci. 2008; 39:400-410 [DOI] [PubMed] [Google Scholar]
- 88. Epis R, Marcello E, Gardoni F, et al. Blocking ADAM10 synaptic trafficking generates a model of sporadic Alzheimer's disease. Brain. 2010; 133:3323-3335 [DOI] [PubMed] [Google Scholar]
- 89. Westergard L, Turnbaugh JA, Harris DA. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J Biol Chem. 2011; 286:44234-44242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Beland M, Motard J, Barbarin A, Roucou X. PrPC homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J Neurosci. 2012; 32:13255-13263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromolecular Med. 2010; 12:1-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Vincent B, Cisse MA, Sunyach C, Guillot-Sestier MV, Checler F. Regulation of βAPP and PrPc cleavage by alpha-secretase: Mechanistic and therapeutic perspectives. Curr Alzheimer Res. 2008; 5:202-211 [DOI] [PubMed] [Google Scholar]
- 93. Prusiner SB. Some speculations about prions, amyloid, and Alzheimer's disease. N Engl J Med. 1984; 310:661-663 [DOI] [PubMed] [Google Scholar]
- 94. Kim J, Holtzman DM. Medicine. Prion-like behavior of amyloid-β. Science. 2010; 330:918-919 [DOI] [PubMed] [Google Scholar]
- 95. Novak P, Prcina M, Kontsekova E. Tauons and prions: Infamous cousins? J Alzheimers Dis. 2011; 26:413-430 [DOI] [PubMed] [Google Scholar]
- 96. Nath S, Agholme L, Kurudenkandy FR, Granseth B, Marcusson J, Hallbeck M. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of β-amyloid. J Neurosci. 2012; 32:8767-8777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Harris JA, Devidze N, Verret L, Ho K, Halabisky B, Thwin MT, Kim D, Hamto P, Lo I, Yu GQ, Palop JJ, Masliah E, Mucke L. Transsynaptic progression of amyloid-β-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron. 2010; 68:428-441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Braak H, Del Tredici K. Alzheimer's pathogenesis: Is there neuron-to-neuron propagation? Acta Neuropathol. 2011; 121:589-595 [DOI] [PubMed] [Google Scholar]
- 99. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009; 11:909-913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012; 73:685-697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011; 286:15317-15331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012; 7:e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science. 2012; 336:1511-1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Nussbaum JM, Seward ME, Bloom GS. Alzheimer disease: A tale of two prions. Prion. 2013; 7:14-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011; 70:532-540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010; 33:317-325 [DOI] [PubMed] [Google Scholar]
- 107. Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. 2010; 11:155-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Walker LC, Levine H, 3rd, Mattson MP, Jucker M. Inducible proteopathies. Trends Neurosci. 2006; 29:438-443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Stohr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ, Prusiner SB, Giles K. Purified and synthetic Alzheimer's amyloid beta (Aβ) prions. Proc Natl Acad Sci U S A. 2012; 109:11025-11030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, Roher AE, Walker LC. Evidence for seeding of β-amyloid by intracerebral infusion of Alzheimer brain extracts in β-amyloid precursor protein-transgenic mice. J Neurosci. 2000; 20:3606-3611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Walker LC, Callahan MJ, Bian F, Durham RA, Roher AE, Lipinski WJ. Exogenous induction of cerebral β-amyloidosis in βAPP-transgenic mice. Peptides. 2002; 23:1241-1247 [DOI] [PubMed] [Google Scholar]
- 112. Meyer-Luehmann M, Coomaraswamy J, Bolmont T, et al. Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science. 2006; 313:1781-1784 [DOI] [PubMed] [Google Scholar]
- 113. Eisele YS, Obermuller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science. 2010; 330:980-982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Morales R, Duran-Aniotz C, Castilla J, Estrada LD, Soto C. De novo induction of amyloid-β deposition in vivo. Mol Psychiatry. 2012; 17:1347-1353 [DOI] [PubMed] [Google Scholar]
- 115. King ME, Kan HM, Baas PW, Erisir A, Glabe CG, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar β-amyloid. J Cell Biol. 2006; 175:541-546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Nussbaum JM, Schilling S, Cynis H, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature. 2012; 485:651-655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Aβ 42 fibrils. Science. 2001; 293:1491-1495 [DOI] [PubMed] [Google Scholar]
- 118. Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001; 293:1487-1491 [DOI] [PubMed] [Google Scholar]
- 119. Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to β-amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002; 99:6364-6369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer's disease mouse models. Cell. 2010; 142:387-397 [DOI] [PubMed] [Google Scholar]
- 121. Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Aβ-induced defects in axonal transport. Science. 2010; 330:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci. 2010; 11:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-β. Nature. 2010; 466:E3-4; discussion E4–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010; 107:2295-2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A. Prion protein and Aβ-related synaptic toxicity impairment. EMBO Mol Med. 2010; 2:306-314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008; 283:29639-29643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bate C, Williams A. Amyloid-β-induced synapse damage is mediated via cross-linkage of cellular prion proteins. J Biol Chem. 2011; 286:37955-37963 [DOI] [PMC free article] [PubMed] [Google Scholar]