

Abstract
d-amino acids are useful building blocks for de novo peptide design and they play a role in aging-related diseases associated with gradual protein racemization. For amino acids with achiral side chains, one should be able to presume that the conformational propensities of l- and d-amino acids are a reflection of one another due to the straightforward geometric inversion at the Cα atom. However, this presumption does not account for the directionality of the backbone dipole and the inverted propensities have never been definitively confirmed in this context. Furthermore, there is little known of how alternative side chain chirality affects the backbone conformations of isoleucine and threonine. Using a GGXGG host–guest pentapeptide system, we have completed exhaustive sampling of the conformational propensities of the d-amino acids, including d-allo-isoleucine and d-allo-threonine, using atomistic molecular dynamics simulations. Comparison of these simulations with the same systems hosting the cognate l-amino acids verifies that the intrinsic backbone conformational propensities of the d-amino acids are the inverse of their cognate l-enantiomers. Where amino acids have a chiral center in their side chain (Thr, Ile) the β-configuration affects the backbone sampling, which in turn can confer different biological properties.
Keywords: conformational sampling, d-amino acids, epimerization, MD simulation, racemization
Introduction
d-amino acids began to be isolated from natural products in 1935, first from alkaloids and then antibiotics produced by Bacillus brevis (Lipmann et al., 1941). It is for this prevalence in prokaryotic cyclic peptide antibiotics that d-amino acids are well-known (Bodanszky and Perlman, 1968; Mor et al., 1992; Skaugen et al., 1994; Heck et al., 1996; Sahl et al., 2008). Lesser known is the extent to which d-amino acids appear across the various kingdoms of life, including both invertebrates and vertebrates (Bodanszky and Perlman, 1968; Corrigan, 1969; Mor et al., 1992; Skaugen et al., 1994; Heck et al., 1996; Andreu and Rivas, 1998; Iida et al., 2001; Bozzi et al., 2008; Sahl et al., 2008; Ohide et al., 2011). Recent interest in d-amino acids has stemmed from mounting evidence of their role in human physiology. Within humans, d-amino acids exist as both free residues (Bodanszky and Perlman, 1968; Corrigan, 1969; Mor et al., 1992; Skaugen et al., 1994; Heck et al., 1996; Fuchs et al., 2005; D'Aniello, 2007; Sahl et al., 2008; Wolosker et al., 2008) and within peptides and proteins (Fisher et al., 1986; Shapira and Chou, 1987; D'Aniello et al., 1992; Fujii et al., 2011). A gradual racemization of proteins has been observed in aging populations, now implicated in many aging-related diseases (Mor et al., 1992; Fuchs et al., 2005; Fujii et al., 2011). Earlier, and more widespread, interest in d-amino acids originates from their utility in drug design (Pritsker et al., 1998; Das et al., 2003; Wilkemeyer et al., 2004; Funke and Willbold, 2009; Figueiredo et al., 2012). Proteins and peptides composed of d-amino acids are more resistant to proteolysis by endogenous l-enzymes, prolonging their biological half-lives (Milton et al., 1992; Pritsker et al., 1998; Ben-Yedidia et al., 2002), and they can still illicit an immune response (Sela and Zisman, 1997; Ben-Yedidia et al., 2002). Hence, d-amino acid containing peptides make attractive drug and vaccine targets. With increased accessibility to chemically synthesized peptides and proteins (Schnölzer et al., 1992; Cupido et al., 2007), including retro-inverso methods that can generate topochemically equivalent reverse sequence peptides (C- to N-terminal) (Wade et al., 1990), d-amino acids are increasingly being utilized in redesigned and de novo designed peptides and proteins (Cochran et al., 2001; Funke and Willbold, 2009; Sievers et al., 2011; Kumar and Sim, 2014).
Knowledge of the intrinsic propensities of l-amino acids has long been believed to be crucial in understanding nascent structure and initiation of folding (Wright et al., 1988). In contrast, knowing the d-amino acid conformational propensities will aid understanding of the disruption of regular secondary structure, for example in disease states (Fujii et al., 2011), or their predilection for certain advantageous secondary structure in the case of cyclic peptides (Yongye et al., 2009). In organic chemistry, there is unquestionable evidence that l- and d-enantiomers are structural mirrors of one another; hence, it seems logical that the behaviors of the l- and d-amino acids should also mirror one another. However, there is a possibility that, because of the asymmetry and directionality of the backbone dipole in proteins and peptides (Gunner et al., 2000; Ripoll et al., 2005), the conformational propensities of l- and d-amino acids might not mirror one another as closely as one would presume. Except for surveys (Mitchell and Smith, 2003; Annavarapu and Nanda, 2009) of the Protein Data Bank (PDB) (Berman et al., 2000) where the propensities of d-amino acids are under-represented, no one to our knowledge has rigorously studied the intrinsic propensities of d-amino acids outside of specific contextual systems (Imperiali et al., 1992; Chalmers and Marshall, 1995; Krause et al., 2000).
The presence of d-amino acids in peptides or proteins is usually the result of post-translational modification (Mor et al., 1992; Kreil, 1994; Soyez et al., 2000), although in some prokaryotes they are also incorporated through non-ribosomal biosynthesis involving multi-enzyme complexes (Kleinkauf and Döhren, 1996; Luo et al., 2002). In both ribosomal and non-ribosomal biosynthesis, isomerization to the d-configuration involves a mechanism that rearranges the Hα and HN atoms. This mechanism, α-epimerization, only inverts the structure at a single point in the peptide backbone with the chirality of the side chains retained. Such a simple inversion at the Cα position results in the expectation that the geometry, secondary structure preferences and conformational behavior of d-amino acids and proteins will mirror their l-amino acid counterparts. It has already been shown that mirror images of a number of proteins can be obtained when composed of d-amino acids (Das et al., 2003; Wiesehan et al., 2003; Pentelute, 2008; Funke and Willbold, 2009), as first confirmed by the Kent lab's production of a mirror image of the wild-type HIV-1 protease through assembly of the same amino acid sequence using the cognate d-amino acids (Milton et al., 1992). Hence, it is expected that the intrinsic conformational propensities of the d-amino acids will also mirror that of their l-counterparts.
Two exceptions that warrant special attention are isoleucine and threonine; both these residues have chiral side chains resulting in four possible optically active antipodes (l, l-allo, d and d-allo). For the l-amino acids only one enantiomer is naturally dominant (Ile: 2S, 3S; Thr: 2S, 3R); l-allo forms are rarely seen (Meyer and Rose, 1936; Rabinovitz et al., 1955). As mentioned above, in vivo mechanisms for incorporating d-amino acids into proteins or peptides are consistent with chirality being modified only at the Cα position in the backbone, the original chirality of the side chains at Cβ being retained in both l- and d-forms (d-allo-isoleucine and d-allo-threonine) (Bodanszky and Perlman, 1968). Consequently, it is expected we will always be presented with the d-allo antipodes of Ile and Thr in nature (Bodanszky and Perlman, 1968; Bada et al., 1970; Kreil, 1994), which may have propensities that do not mirror those of the naturally dominant l-forms.
Previously, we used molecular dynamics (MD) simulations of a Gly-based host–guest system (GGXGG) with each of the l-amino acids placed in the central X position to determine their intrinsic backbone conformational propensities (Beck et al., 2008). The Gly-based system was chosen to confer the greatest freedom of movement for the central guest residues and allow sampling reflective of the natural conformational tendencies of the central amino acids. This study revealed that the proteogenic l-amino acids do indeed have intrinsic conformational propensities. Here, to determine the corresponding intrinsic conformational propensities of the d-amino acids, we performed MD simulations using the same GGXGG pentapeptides with the central position substituted with the d-enantiomers of the naturally occurring amino acids, including various protonation states and alternative side chain chirality for Ile and Thr. All simulation trajectories were assessed to have converged using methods published previously (Beck et al., 2008; Towse et al., submitted). Comparisons of the conformational sampling of multiple trajectories of the same guest residue and of the first and latter portions of individual trajectories showed a strong correlation (Supplementary Tables SI and SII). Simulations of these particular host–guest systems for the l-amino acids have previously been experimentally validated against chemical shifts from nuclear magnetic resonance experiments resulting in a Pearson correlation coefficient of >0.9 between the simulated and experimental shifts (Beck et al., 2008). Here, we take a first step in understanding the structural and dynamic roles of the d-amino acids and present the results for the d-amino acid counterparts and compare them with our previous l-amino acid containing GGXGG simulations. Furthermore, we investigate the impact of the β-configuration on the conformational sampling of the two d-antipodes of the Ile and Thr residues. The resulting d-amino acid Ramachandran maps have been used to create conformational libraries that can be used for protein and peptide design. For example, we are using them to design inhibitors of amyloidosis (Hopping et al., 2014).
Materials and methods
MD simulations of GGXGG pentapeptides
Gly-based host–guest pentapeptides were built with each of the 20 ‘guest’ amino acids as the central X residue and explicitly modeled with N-acetylated and C-amidated blocked termini to prohibit artificial interaction with the central residues. All pentapeptides were built with extended conformations, with the ψ and ϕ angles set to −180° and +180°, respectively. Two sets of pentapeptides were simulated, one set with l-isomer guest residues and a second with d-isomer guest residues. Ile and Thr are residues that have a second chiral center in the side chain; the naturally dominant l-enantiomers (Ile: 2S, 3S; Thr: 2S, 3R) and both of the possible d-enantiomers were simulated. Where applicable, pentapeptides with alternative protonation states for the central guest residue were generated. For Asp and Glu, both neutral and acidic protonation states were used (Asp, Ash, Glu, Glh) and, for histidine, three individual simulations were performed for the two neutral tautomers, Hid (δH), Hie (ϵH), and the acidic protonation state, Hip (δH and ϵH). Cysteine was modeled in the reduced state with the side chain protonated (–CH2–SH, named Cyh in our force field).
The simulation protocol was as reported previously (Beck and Daggett, 2004; Beck et al., 2008) using our in lucem molecular mechanics (ilmm) package (Beck et al., 2000–2014) with the Levitt et al. force field (Levitt et al., 1995; Beck and Daggett, 2004). The microcanonical NVE ensemble (constant number of particles, volume and energy) was used with non-bonded interactions treated with an 8 Å force-shifted cutoff (Beck et al., 2005) and the pentapeptides solvated by flexible F3C explicit water models (Levitt et al., 1997). All systems were simulated with a box size that reproduced the experimental density of 0.9970 g/ml at 298 K. To assess convergence, multiple simulations were performed for the pentapeptides containing both the l- and d-isomer Ala. For the l-isomer dataset, duplicate simulations of the Arg, Ash, Asn, Asp, Cys, Gln, Glu, Gly, Hid, Ile, Leu and Lys systems were also acquired. All simulations were performed for 101 ns to assess convergence; due to system equilibration, the first nanosecond of each trajectory was discarded and the remaining 100 ns used for analysis. The conformational propensities of the duplicate simulations were determined, but only the multiple simulations of Ala are presented in detail here; the other amino acids showed comparable behavior. The total simulation time for the simulations used for this analysis was 53 μs.
Calculation of conformational propensities
To assess the conformational propensities of the central ‘guest’ residues, populations were calculated first for the four quadrants of the conformational ψ/ϕ space and then for specific conformational regions within these quadrants. For the l-amino acids, these conformational regions were defined as: αR: −100° ≤ ϕ ≤ −30°, −80° ≤ ψ ≤ −5°; near-αR: −175° ≤ ϕ ≤ −100°, −55° ≤ ψ ≤ −5°; αL: 5° ≤ ϕ ≤ 75°, 25° ≤ ψ ≤ 120°; β: −180° ≤ ϕ ≤ −50°, 80° ≤ ψ ≤ −170°; PIIL: −110° ≤ ϕ ≤ −50°, 120° ≤ ψ ≤ 180°; PIR: −180° ≤ ϕ ≤ −115°, 50° ≤ ψ ≤ 100°. For the d-amino acids, the corresponding conformational regions are defined as: DαR: 30° ≤ ϕ ≤ 100°, 5° ≤ ψ ≤ 80°; near-DαR: 100° ≤ ϕ ≤ 175°, 5° ≤ ψ ≤ 55°; DαL: −75° ≤ ϕ ≤ −5°, −120° ≤ ψ ≤ −25°; Dβ: 50° ≤ ϕ ≤ 180°, −80° ≤ ψ ≤ 170°; DPIIL: 50° ≤ ϕ ≤ 110°, −180° ≤ ψ ≤ −120°; DPIR: 115° ≤ ϕ ≤ 180°, −100° ≤ ψ ≤ −50°. These regions are illustrated in Figs. 1A and B. For both l- and d-amino acids, the actual β-region percentages used in this paper cover the listed bounds above, minus the overlapping PIIL and PIR regions; this was referred to as the non-polyproline β-region (nPβ).
Fig. 1.
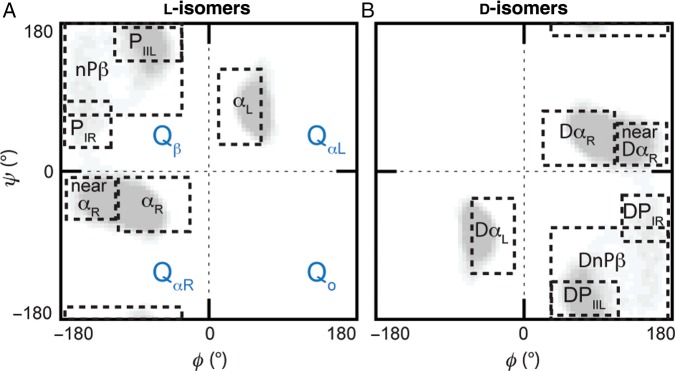
Defined quadrants and conformational regions for the l- and d-amino acids. (A) Labeling convention used for the four quadrants, QαR Qβ, QαL and Qo, and the regions within the quadrants pertaining to specific conformations: right-handed α-helix (αR), left-handed α-helix (αL), polyproline type II left-handed helix (PIIL), polyproline type I right-handed helix (PIR) and remainder of non-polyproline β-region (nPβ). (B) Corresponding conformational regions for the d-amino acids: right-handed α-helix (DαR), left-handed α-helix (DαL), polyproline type II left-handed helix (DPIIL), polyproline type I right-handed helix (DPIR) and remainder of non-polyproline β-region (DnPβ).
Convergence analysis
Convergence of the simulated trajectories was assessed by determining the populations of the quadrants for the central three residues of each pentapeptide. The terminal residues are able to sample conformational space more extensively than those in the center of the pentapeptide; hence, these three central residues alone were used to assess convergence. As each pair of backbone ϕ/ψ angles for the each residue can populate one of the four quadrants, this results in 34 conformational combinations for the three central residues (Supplementary Fig. S1A). Hence, assessing convergence requires consideration of 64 conformational states. The population of these states was compared between simulations as well as within the first and latter halves of an individual trajectory.
Effect size analysis
With datasets of the size analyzed here, even negligible variations will be determined to be statistically significant when using traditional hypothesis testing methods. Hence, to detect if the change of a central residue from an l- to d-configuration influenced the ϕ/ψ distributions, effect size analysis was performed (Hedges and Olkin, 1985; Fritz et al., 2012). Circular statistics were first used to calculate the mean and standard deviation of the ϕ and ψ angles for the central guest residues within defined conformational regions. The effect size, E, between the two distributions was then calculated using Equation (1), where is the mean, N is the number of data points and δ is the standard deviations of the respective l- and d-amino acid simulations.
| (1) |
To determine effect sizes between the l- and d-pairs, the average angles for the d-amino acids were first multiplied by −1 (point reflection) to coincide with the same geometry of the l-amino acids.
Results
Simulation convergence
For each individual trajectory, the conformational sampling within the first and latter portions was compared in order to assess whether it had converged. The first nanosecond of each trajectory was discarded as the equilibration period. Hence, for each trajectory the sampling in the 1–51 ns portion was compared with that in the 51–101 ns portion. This analysis was performed for pentapeptide systems containing both l-amino acids (Supplementary Fig. S1B) and d-amino acids (Supplementary Fig. S1C). Strong correlations (>0.8) were observed for all pentapeptides (Supplementary Table SI).
Multiple simulations were performed for some of the pentapeptides to further test for convergence of sampling. For these analyses the full trajectories, excluding the first nanosecond equilibration period, were used, i.e. 1000–100 999 ps. As the convergence of sampling for individual simulations was observed to be comparable (Supplementary Table SI), triplicate simulations were only performed on the Ala-containing pentapeptides for the d-amino acid dataset and compared against that of the l-Ala systems (Supplementary Fig. S2). Duplicate simulations were acquired for a number of pentapeptides containing l-isomer guest residues to verify convergence across different amino acid types. Comparison of duplicated simulations for both l- and d-amino acids demonstrated a strong correlation between the conformational sampling in different trajectories. Hence, the simulations converged (Supplementary Table SII).
Conformational sampling of ϕ/ψ space
A comparison of the intrinsic propensities for the l- and d-amino acids was made by examination of the sampling across the four quadrants of the Ramachandran plot and within smaller specified conformational regions (Fig. 1A). The defined conformational regions in ϕ/ψ space for the l-amino acids were as reported previously (Beck et al., 2008; Towse et al., submitted); to assess similarity with the d-amino acids, we defined inverted conformational regions that took into account the inverted geometry at the Cα atom by reflecting through the origin ϕ = 0, ψ = 0 (Fig. 1B). Two things were assessed to conclude whether the pairs of l- and d-residues unequivocally mirrored one another. First, we examined both the bias towards and population of the conformational regions. Second, we used effect size analysis to establish if the underlying ϕ/ψ distributions within each of these individual regions were equivalent. Based on replicate simulations, a population change >8% in any region, or an effect size larger than 0.5 standard deviations, was deemed to be indicative of a substantive difference (Supplementary Tables SV and SVI).
Across all simulations, the backbone ϕ/ψ angle sampling of the d-amino acids mirrored those of the l-amino acids (Fig. 2). Except for Pro and Thr, all the amino acids demonstrated the ability to sample all quadrants, although not to the same extent (Supplementary Table SIII, Fig. 2). The l-amino acids preferentially sampled the QαR and Qβ quadrants with the largest population in QαR, whereas the d-amino acids preferred the inverted counterpart quadrants, QαL and Qo (Fig. 3). The largest populations for the l- and d-amino acids were in the QαR and QαL quadrants, respectively, and were of equivalent magnitude consistent with the geometrical inversion at the Cα position (Fig. 3). The preferences of the d-amino acids for specific conformational regions within a quadrant also mimicked that of the cognate l-amino acids with equivalent populations concentrated in the inverted counterpart regions (Fig. 4). Biases were evident, as reported previously, with l- and d-amino acids preferring the αR and DαR regions, respectively. The β-branched residues Ile and Val showed a preference shifted towards near-αR and near-DαR. Proline exhibited the most bias, with an overwhelming preference for the PIIL and DPIIL regions.
Fig. 2.

Sampling of conformational space by the guest l- and d-amino acids. Ramachandran plots of the conformational populations of the guest residues in all #1 simulation runs. The conformational regions are colored by increasing percentage population and normalized on a scale of 0–1; legend is inset.
Fig. 3.
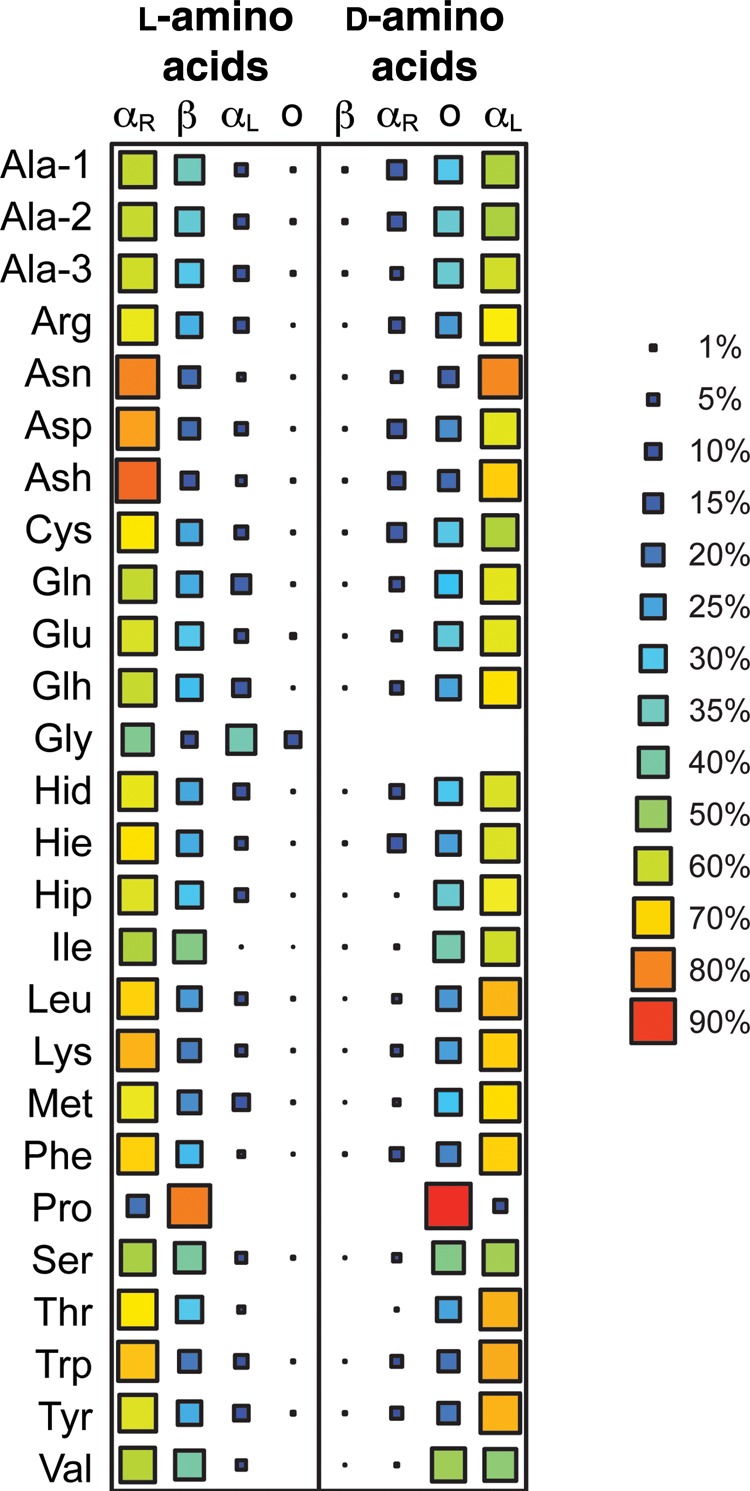
Population of the four quadrants of the Ramachandran plot highlighting the mirrored sampling of conformational space. The l-amino acids show preference for the QαR and Qβ regions and the d-amino acids preferentially sample the opposite regions, QαL and Qo.
Fig. 4.

Population of specific regions of conformational space demonstrating similarities in the conformational propensities of l- and d-amino acids. The conformational regions are as defined in Figs. 1A and B and declared in the Supplementary Information. Inverted conformational regions are used to quantify the propensities of the d-amino acids and labeled as for the complementary l-amino acid regions from which they have been transposed (i.e. αR for the d-amino acids is DαR, as shown in Fig. 1 and discussed in the text; the same is true for the other conformational states).
The largest differences detected by both changes in population of the conformational regions and the underlying ϕ/ψ distributions were for the Ile and Thr simulations, which we discuss separately. Ignoring the special cases of Ile and Thr, the largest differences between l and d populations were observed for Pro. The population of DPIIL was 11% greater than that of the PIIL population obtained for l-Pro and the population of DαR was moderately reduced (−13%) compared with that in the corresponding αR region. Such variations between the Pro l/d pair were anticipated given the cyclized nature of the side chain and possible stronger interaction with the local backbone dipole than for other residues. Other notable differences between the sampling of the l and d counterpart regions were observed for Ash (αR/DαR, 9%), Glh ((αR/DαR, 9%), Met (αL/DαL, 9%) and Val (PIIL/DPIIL, 9%). Another interesting discrepancy was that l-Hip sampled αL 5.4% of the time, but at no point was DαL sampled by d-Hip. Although the difference in population of these mirrored regions are marginally larger than the average differences observed between replicates, effect size analysis showed there to be no substantive difference in the underlying ϕ/ψ distributions within these regions (Supplementary Table SVII). One exception was the αL region sampled by d-Hip where there was no population with which to perform such analysis. Interestingly, despite αL and DαL conformations having relatively low populations, some of the largest differences between l/d residue pairs were observed in this region. Although the changes themselves were small and comparable with the observed variability between replicate simulations, for nearly all residues the population in αL/DαL was more than twice that in the corresponding region sampled by the residue of opposite chirality. Approximately half of the residues exhibiting this behavior was as a result of increased sampling of the DαL region by the d-amino acid.
Allo-forms of isoleucine and threonine
As Ile and Thr have a second chiral center in their side chains, four enantiomers exist: l, l-allo, d and d-allo (Fig. 5). The sampling of the naturally dominant l-forms of Ile and Thr and the corresponding mirror-image reflections (d-Ile and d-Thr) demonstrate that the propensity for certain conformational regions is retained (Fig. 5). In both cases, there is a preference for near-αR/near-DαR and αR/DαR in Ile and Thr, respectively. Except for some variation in these sampled regions (≤11%), there was little difference in the size of the populations within the corresponding conformational regions. For the l/d-Thr pair, this parity was confirmed by effect size analysis, which confirmed there to be no substantive difference in the ϕ/ψ distributions of the l- and d-amino acids (Table I). However, the slightly greater population differences between the l/d-Ile pair were accompanied by moderate shifts in the underlying distributions, a likely consequence of the longer Ile side chain.
Fig. 5.
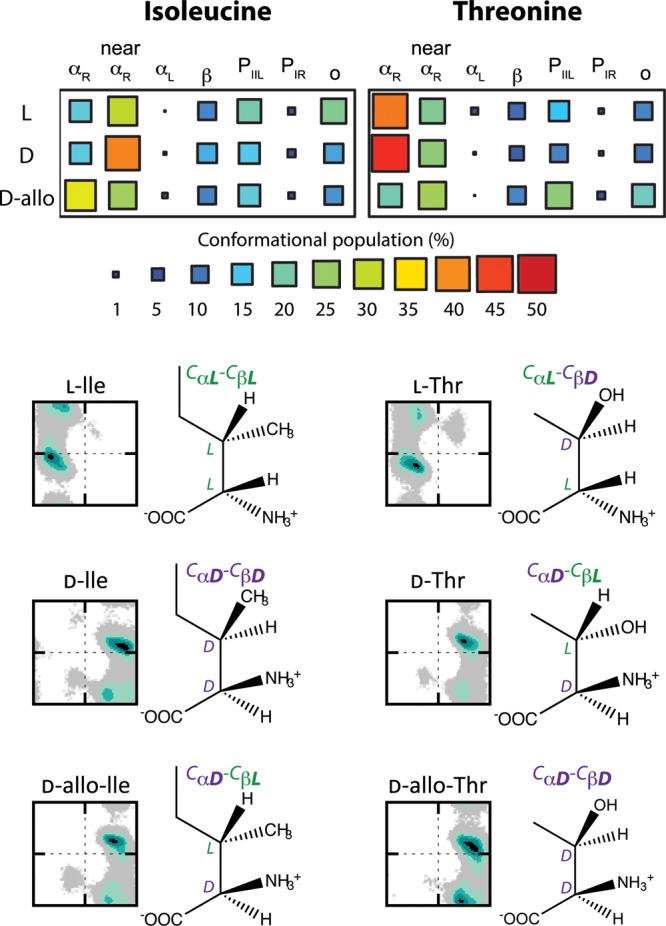
Differences in the conformational propensities of three enantiomers of isoleucine and threonine possible due to the additional chiral center in the side chains. d-Ile and d-Thr are true reflections of the naturally occurring l-Ile and l-Thr enantiomers. The allo-forms, d-allo-isoleucine and d-allo-threonine, have been observed from post-translational modifications involving epimerization at the Cα position; only the stereochemistry at the backbone is affected with that of the side chains remaining the same. The percentage population of specified conformational regions is shown along with the corresponding Ramachandran plots demonstrating the impact of the alternative stereochemistry in the side chain on the sampling of the d-amino acids. For the d-forms, the conformational regions are those inverted across the glide plane to correspond with the same areas specified for the l-forms as shown in Figs. 1A and B.
Table I.
Effect size analysis and percentage change in population of conformational regions sampled by the l-, d- and d-allo-isoleucine and threonine simulations
| Simulation #1 | Simulation #2 | Region |
θL − θD (°) |
Pooled δ (°) |
Effect size |
|% Δ| | |||
|---|---|---|---|---|---|---|---|---|---|
| ϕ | ψ | ϕ | ψ | ϕ | ψ | ||||
| l-Ile | d-Ile | αR | 0.1 | 2.4 | 11.0 | 15.1 | 0.01 | 0.16 | 0.5 |
| nαR | 6.9 | 3.1 | 15.3 | 12.0 | 0.45 | 0.26 | 11.2 | ||
| αL | 8.3 | 9.4 | 14.3 | 14.8 | 0.58 | 0.63 | 0.2 | ||
| β | 11.3 | 19.4 | 25.2 | 24.8 | 0.45 | 0.78 | 2.1 | ||
| PIIL | 3.0 | 9.4 | 15.0 | 14.1 | 0.20 | 0.67 | 4.3 | ||
| PIR | 12.1 | 8.6 | 12.9 | 14.4 | 0.94 | 0.60 | 0.4 | ||
| l-Ile | d-allo-Ile | αR | 2.1 | 8.6 | 11.3 | 14.3 | 0.19 | 0.60 | 16.4 |
| nαR | 4.0 | 11.0 | 12.4 | 12.7 | 0.32 | 0.86 | 3.3 | ||
| αL | 23.2 | 3.0 | 12.4 | 15.9 | 1.87 | 0.19 | 0.9 | ||
| β | 8.3 | 20.4 | 22.7 | 28.1 | 0.37 | 0.73 | 0.3 | ||
| PIIL | 1.0 | 4.8 | 15.0 | 14.8 | 0.07 | 0.32 | 3.2 | ||
| PIR | 1.3 | 7.6 | 10.7 | 14.5 | 0.12 | 0.52 | 0.3 | ||
| d-Ile | d-allo-Ile | αR | 2.1 | 11.0 | 11.4 | 13.3 | 0.18 | 0.83 | 16.9 |
| nαR | 10.9 | 7.9 | 15.1 | 12.3 | 0.72 | 0.64 | 14.6 | ||
| αL | 14.9 | 6.4 | 13.0 | 15.6 | 1.15 | 0.41 | 0.7 | ||
| β | 19.6 | 1.0 | 26.0 | 26.0 | 0.75 | 0.04 | 2.3 | ||
| PIIL | 2.0 | 4.6 | 15.0 | 15.0 | 0.13 | 0.31 | 1.1 | ||
| PIR | 13.4 | 1.0 | 13.0 | 14.8 | 1.03 | 0.07 | 0.8 | ||
| l-Thr | d-Thr | αR | 1.3 | 2.2 | 12.5 | 14.3 | 0.11 | 0.15 | 5.6 |
| nαR | 0.8 | 0.4 | 12.9 | 12.3 | 0.06 | 0.03 | 2.3 | ||
| αL | 1.8 | 3.0 | 11.2 | 16.3 | 0.16 | 0.18 | 1.4 | ||
| β | 2.3 | 4.4 | 23.7 | 25.3 | 0.10 | 0.18 | 2.2 | ||
| PIIL | 0.3 | 4.0 | 14.3 | 15.3 | 0.02 | 0.26 | 3.4 | ||
| PIR | 0.7 | 0.7 | 10.2 | 14.7 | 0.07 | 0.05 | 0.3 | ||
| l-Thr | d-allo-Thr | αR | 4.4 | 3.5 | 12.5 | 14.8 | 0.35 | 0.23 | 22.5 |
| nαR | 3.1 | 2.7 | 13.3 | 12.9 | 0.24 | 0.21 | 4.5 | ||
| αL | 23.8 | 16.0 | 11.1 | 16.5 | 2.14 | 0.97 | 1.6 | ||
| β | 16.4 | 28.1 | 25.5 | 28.9 | 0.64 | 0.97 | 1.9 | ||
| PIIL | 0.8 | 9.3 | 14.6 | 15.1 | 0.05 | 0.62 | 9.8 | ||
| PIR | 7.6 | 3.4 | 12.0 | 14.5 | 0.64 | 0.23 | 1.1 | ||
| d-Thr | d-allo-Thr | αR | 3.1 | 1.3 | 12.1 | 14.5 | 0.26 | 0.09 | 28.1 |
| nαR | 4.0 | 2.3 | 13.3 | 12.7 | 0.30 | 0.18 | 2.2 | ||
| αL | 22.0 | 13.0 | 12.6 | 14.4 | 1.75 | 0.90 | 0.2 | ||
| β | 18.7 | 32.6 | 25.3 | 27.4 | 0.74 | 1.19 | 4.1 | ||
| PIIL | 1.1 | 13.3 | 14.5 | 14.7 | 0.08 | 0.91 | 13.1 | ||
| PIR | 6.9 | 2.7 | 12.3 | 14.4 | 0.56 | 0.19 | 1.4 | ||
Larger sampling differences were observed between the other d-antipodes and their cognate l-amino acids (Table I). For the d-antipodes of Ile and Thr, the sampling of the Ramachandran quadrants was most similar for d-Ile and d-allo-Ile. Hence, the difference between the Ile antipodes was not immediately apparent across the quadrants with only a 10% spread in the populations of QαR and QαL, indicative of the antipodes sharing similar helical propensities (Supplementary Table SV). The effect of the β-configuration on Thr sampling was more pronounced across the four quadrants with a difference of ∼20% between the QαL and Qo populations of d-Thr and d-allo-Thr (Supplementary Table SV).
The difference between the antipodes became more apparent when focusing on conformational regions related to specific secondary structure (Fig. 5). Although l-Thr and d-Thr propensities mirrored each other well, d-allo-Thr showed a preference for more extended structures, with nearly equivalent populations of PIIL and αR (Fig. 5, Supplementary Table SV). Conversely, it was l-Ile that exhibited a similar balance of populations across the conformational regions and instead it was both the d- and d-allo forms that demonstrated a greater preference for the DnαR and DαR regions, respectively. For both Ile and Thr, of the two most sampled conformational regions, αR and near-αR, the slight preference of one by the allo-forms is opposite to that observed for the mirror-image l/d pair. For example, d-allo-Ile has a larger population in DαR whereas the l- and d-Ile forms preferentially sample the related near-αR and near-DαR regions (Fig. 5). Effect size analysis showed that, in addition to the distinguishable populations in given conformational regions, there were substantial shifts in some of the ϕ/ψ distributions (Table I).
The conformational propensities of these two residues suggest a relationship to the configurations at the two chiral centers. Where the configuration at the two chiral centers is the same, e.g. CαL-CβL, a similar preference for certain regions of conformational space is seen (Fig. 5). For example, d-Ile and d-allo-Thr and l-Ile all have the same configuration at the Cα and Cβ positions and exhibit comparable conformational propensities. Analogous to this, the antipodes of Ile and Thr that have mixed configurations at the two chiral centers (i.e. CαL-CβD or CαD-CβL) also have similar (although inverted) propensities that are distinct from those observed where the two configurations are the same (Fig. 5).
Discussion
Although it was expected that the intrinsic conformational propensities of the d-amino acids would mirror those of their l-amino counterparts, there are scant confirmatory data. For those amino acids with achiral side chains, our simulations confirmed the expected mirroring of conformational behaviors (Figs. 2, 3 and 4). However, a larger problem concerns Ile and Thr. These two residues have chiral side chains and the incorporation of these d-amino acids through in vivo α-epimerization, whether via ribosomal or non-ribosomal pathways, results in d-allo-Ile and d-allo-Thr where the side chain chirality is the same as the naturally dominant l-amino acid counterpart (Bodanszky and Perlman, 1968; Bada et al., 1970; Kreil, 1994). Unfortunately, when Ile and Thr are used in peptide design, the details of the side chain chiralities are often unspecified (or unknown). Presumably, in many studies where mirror-image structures were obtained of proteins or peptides containing Ile and Thr, d-Ile and d-Thr were used with both chiral centers inverted (Milton et al., 1992; Hung et al., 1998; Das et al., 2003; Wiesehan et al., 2003; Pentelute, 2008; Wei et al., 2009). However, the ambiguity of the forms of Ile and Thr used makes assessing the impact of the β-configuration on conformational propensities, and any related biological activity, difficult. Here, we summarize the data that existed previously regarding the conformational propensities of the d-amino acids, confirm the sampling behaviors of the amino acids and discuss the impact of the side chain chirality in the cases of Ile and Thr.
An initial survey of the PDB for conformations of d-amino acids made it difficult to conclude if they mirror those of l-amino acids, as not all the expected ϕ/ψ regions were sampled (Mitchell and Smith, 2003). For example, there was a lack of d-amino acid conformations in the inverted αL region, which was expected for any d-amino acids present in α-helices (Mitchell and Smith, 2003). The dissonance between what was expected and actually observed is a simple reflection of the low occurrence of d-amino acids present in the PDB and the limited contextual representation. In total, only 148 entries were identified that contained non-artifactual d-amino acids and only one natural protein contained a d-amino acid with the remainder appearing in backbone cyclic peptides, such as gramicidin-S. Hence, the results of the PDB survey are somewhat expected given the known proclivity of d-amino acids for turn structures (Imperiali et al., 1992; Struthers et al., 1996; Davies, 2003; Mitchell and Smith, 2003). Another PDB survey considered l- and d-amino acids specifically within turn structures to predict propensities of d-amino acids for αR-helix formation (Annavarapu and Nanda, 2009). Outside of these contexts, little is known of the d-amino acids structural propensities and the true conformational behavior of d-amino acids is under-represented in the available structural databases.
Our simulations confirmed the expected mirroring of l- and d-amino acid conformational propensities for those residues with achiral side chains. Furthermore, as our peptides were not as conformationally restricted as those in the PDB, our simulations demonstrated that d-amino acids do indeed sample conformations within the inverted DαL region. Some small differences in the populations of the quadrants and specified conformational regions between the d- and l- forms of a number of the residues with achiral side chains were observed (Figs. 3 and 4). This suggests that it is possible that modest differences in d-amino acid propensities may be seen in some cases and could be a result of backbone dipole and side chain interactions. However, the variability observed between multiple simulations of a given pentapeptide ranged from 2% (Asn) to as high as 10% (Gly) (Supplementary Table SV). As the variation between simulations of the l- and d-enantiomers of a given residue were of comparable magnitude to the simulation-to-simulation variability (Supplementary Tables SIII and SIV), we concluded that there were no significant differences. An exception was the unmistakable differences in the conformational sampling by the antipodes of Ile and Thr. We determined that d-allo-Thr preferred more extended structures than d-Thr; d-Thr sampled QαL 75% of the time with a clear preference for αR conformations. Ile antipodes also showed slight preferences towards helical regions. Although d-allo-Ile showed a shift to αR from the near-αR preferred by l-Ile, unlike Thr the propensities were less distinct and both d-allo-Ile and d-Ile exhibited similar propensities for helical structures. From collective examination of the antipodes, it appeared that the conformational propensities of Ile and Thr were tied to the configurations at the two chiral centers. The preference for a conformational region, naturally inverted for the d-amino acids, appears dependent on whether the configuration of the two chiral centers match, e.g. CαL-CβL, with l-Ile and d-allo-Thr showing very similar conformational propensities (Fig. 5). Where antipodes have the same configuration at the Cα and Cβ positions, they exhibit comparable conformational propensities.
Due to the inverted stereochemistry at the chiral centers and corresponding preferences of d-amino acids for opposing regions of conformational space to that of the l-counterparts, the introduction of d-amino acids into l-peptide or proteins often results in destabilization of secondary structure (Krause et al., 1995, 2000; McInnes et al., 2000; Mitchell and Smith, 2003; Lee et al., 2004). For example, where l-amino acids preferentially form αR-helices, it is possible that the corresponding d-amino acid more favorably forms αL structure, such that this d-amino acid in an l-helix disrupts the structure (Krause et al., 2000). Our results show agreement with these experimentally derived rankings of helix destabilization. Previously, a link between the destabilization of α-helices by d-amino acids and a low propensity of the l-counterparts for αL turns was suggested, these low propensities were believed to reflect favorable αR-helix formation by the d-amino acid (Annavarapu and Nanda, 2009). Hence, the extent of sampling of the DαL region, which marginally overlaps with the αR region, should also be an indication of potential d-amino acid propensities for αR-helix formation; sparse sampling of DαL should be consistent with the extent to which a given d-amino acid can destabilize a αR-helix. Consequently, we inspected the sampling of the DαL region by the d-amino acids and observed similar patterns in helix destabilizing propensity to those determined experimentally (Krause et al., 2000; Annavarapu and Nanda, 2009). Interestingly, nearly all d-residues that had a DαL population twice that of αL for the corresponding l-amino acid correlated with those previously determined to be weak helix destabilizers (Krause et al., 2000). Based on the DαL populations, our simulations suggest that the β-branched residues and Pro would be strong destabilizers, with d-His being the weakest αR-helix destabilizer, in agreement with experiment (Annavarapu and Nanda, 2009). This agreement with experiment lends more support to validation of our simulations in addition to the extensive convergence analysis performed.
As mentioned above, the ambiguity in declaring the side chain chiralities of Ile and Thr in peptide design studies means that there is very little experimental data for the impact of the β-configuration on the conformational preferences of these residues. However, there are reports that the β-configuration has an impact on biological properties (Rabinovitz et al., 1955; Gullino et al., 1956; Winitz et al., 1956). For example, enantiomers of Ile and Thr, where the absolute configuration is different at the two chiral centers, i.e. CαLCβD or CαDCβL, are less susceptible to oxidase enzymes than their counterparts with the same stereochemistry at both chiral centers (Winitz et al., 1956). Thus, d-Thr and d-allo-Ile, with mixed stereochemistry, are both less susceptible to oxidase than their corresponding d-antipodes. Examination of the conformational sampling of these residues reveals patterns that are consistent with these biological observations. Where the chirality is the same at both chiral centers, the propensities, although inverted, are similar. However, where the chirality is different, there is a shift in the conformational propensities. The d-allo-Ile (CαL-CβD) propensities match that of the l-Thr (inverted) and d-Thr residues all showing a preference towards αR regions (Fig. 5). Where the stereochemistry is the same at both centers (l-Ile and d-Ile and d-allo-Thr), the sampling is biased towards the near-αR regions.
From other studies, we know that only structures analogous to l-Ile, and not l-allo-Ile, are able to inhibit incorporation of radioactive l-Ile into proteins (Rabinovitz et al., 1955), and d-allo forms of Ile and Thr exhibit lower toxicities than the respective d- and l-non-allo forms (Gullino et al., 1956). Although not frequently found in d-amino acids containing natural products, the relevance of the β-configuration may not appear compelling at this time, but it should be noted that d-Ile was shown to be crucial for biological activity in two peptide ethanol antagonists and no mention was made of its side chain chirality or indeed that there are two possibilities (Wilkemeyer et al., 2004). Moreover, it should be kept in mind that the d-allo forms may become a salient point in future studies of the structural effects of in vivo racemization related to aging and disease. Thus, gaining greater insight into the conformational behaviors of allo and non-allo forms has relevance to explaining observed biological differences.
In summary, through exhaustive sampling, we have confirmed that the intrinsic conformational propensities of the l- and d-amino acids mirror one another. The bias observed by l-amino acids for certain regions of conformational space is adhered to by d-amino acids in the corresponding inverted regions. This library of d-amino acid propensities now forms part of our Structural Library of Intrinsic Residue Propensities available at http://dynameomics.org. More importantly, we have investigated for the first time the impact of β-configurations on the conformational propensities of the Ile and Thr backbones. We show an interesting contrast between the d- and d-allo-series of the Thr and Ile residues that suggests that this should be taken into account in designing synthetic peptides to mimic those d-containing peptides found in nature. This comprehensive view of the conformational propensities of the l- and d-amino acids provides a foundation to assist in future peptide design work and to better understand the conformational changes associated with incorporation of d-amino acids in proteins.
Supplementary data
Funding
This work was supported by the National Institutes of Health (grant numbers GM 50789 and GM 95836). We are grateful to the National Energy Research Scientific Computing Center, supported by the Office of Science and US Department of Energy under contract number DE-AC02-05CH11231, for providing computing time.
Supplementary Material
Acknowledgements
We thank Jiri Vymetal for supplying a python script used to create Figs. 3–5.
References
- Andreu D., Rivas L. Biopolymers. 1998;47:415–433. doi: 10.1002/(SICI)1097-0282(1998)47:6<415::AID-BIP2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Annavarapu S., Nanda V. BMC Struct. Biol. 2009;9:61. doi: 10.1186/1472-6807-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bada J.L., Luyendyk B.P., Maynard J.B. Science. 1970;170:730–732. doi: 10.1126/science.170.3959.730. [DOI] [PubMed] [Google Scholar]
- Beck D.A.C., Alonso D.O.V., Inoyama D., Daggett V. Proc. Natl Acad. Sci. USA. 2008;105:12259–12264. doi: 10.1073/pnas.0706527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck D.A.C., Armen R.S., Daggett V. Biochemistry. 2005;44:609–616. doi: 10.1021/bi0486381. [DOI] [PubMed] [Google Scholar]
- Beck D.A.C., Daggett V. Methods. 2004;34:112–120. doi: 10.1016/j.ymeth.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Beck D.A.C., McCully M.E., Alonso D.O.V. In lucem Molecular Mechanics (ilmm) Seattle: University ofWashington; 2000–2014. [Google Scholar]
- Ben-Yedidia T., Beignon A.-S., Partidos C.D., Muller S., Arnon R. Mol. Immunol. 2002;39:323–331. doi: 10.1016/s0161-5890(02)00095-0. [DOI] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodanszky M., Perlman D. Nature. 1968;218:291–292. doi: 10.1038/218291a0. [DOI] [PubMed] [Google Scholar]
- Bozzi A., Mangoni M.L., Rinaldi A.C., Mignogna G., Aschi M. Biopolymers. 2008;89:769–778. doi: 10.1002/bip.21006. [DOI] [PubMed] [Google Scholar]
- Chalmers D.K., Marshall G.R. J. Am. Chem. Soc. 1995;117:5927–5937. [Google Scholar]
- Cochran A.G., Skelton J.J., Staravasnik M.A. Proc. Natl Acad. Sci. USA. 2001;98:5578–5583. doi: 10.1073/pnas.091100898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan J.J. Science. 1969;164:142–149. doi: 10.1126/science.164.3876.142. [DOI] [PubMed] [Google Scholar]
- Cupido T., Tulla-Puche J., Spengler J., Albericio F. Curr. Opin. Drug Discov. Devel. 2007;10:768–783. [PubMed] [Google Scholar]
- D'Aniello A. Brain Res. Rev. 2007;53:215–234. doi: 10.1016/j.brainresrev.2006.08.005. [DOI] [PubMed] [Google Scholar]
- D'Aniello A., Vetere A., Fisher G.H., Cusano G., Chavez M., Petrucelli L. Brain Res. 1992;592:44–48. doi: 10.1016/0006-8993(92)91656-y. [DOI] [PubMed] [Google Scholar]
- Das C., Berezovska O., Diehl T.S., Genet C., Buldyrev I., Tsai J.-Y., Hyman B.T., Wolfe M.S. J. Am. Chem. Soc. 2003;125:11794–11795. doi: 10.1021/ja037131v. [DOI] [PubMed] [Google Scholar]
- Davies J.S. J. Pept. Sci. 2003;9:471–501. doi: 10.1002/psc.491. [DOI] [PubMed] [Google Scholar]
- Figueiredo A.C., Clement C.C., Zakia S., Gingold J., Philipp M., Pereira P.J.B. PLoS ONE. 2012;7:e34354. doi: 10.1371/journal.pone.0034354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher G.H., Garcia N.M., Payan I.L., Cadilla-Perezrios R., Sheremata W.A., Man E.H. Biochem. Biophys. Res. Commun. 1986;135:683–687. doi: 10.1016/0006-291x(86)90047-1. [DOI] [PubMed] [Google Scholar]
- Fritz C.O., Morris P.E., Richler J.J. J. Exp. Psychol. Gen. 2012;141:2–18. doi: 10.1037/a0024338. [DOI] [PubMed] [Google Scholar]
- Fuchs S.A., Berger R., Klomp L.W.J., deKoning T.J. Mol. Genet. Metab. 2005;85:168–180. doi: 10.1016/j.ymgme.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Fujii N., Kaji Y., Fujii N. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011;879:3141–3147. doi: 10.1016/j.jchromb.2011.05.051. [DOI] [PubMed] [Google Scholar]
- Funke S.A., Willbold D. Mol. BioSyst. 2009;5:783–786. doi: 10.1039/b904138a. [DOI] [PubMed] [Google Scholar]
- Gullino P., Winitz M., Birnbaum S.M., Cornfield J., Otey M.C., Greenstein J.P. Arch. Biochem. Biophys. 1956;64:319–332. doi: 10.1016/0003-9861(56)90276-4. [DOI] [PubMed] [Google Scholar]
- Gunner M.R., Saleh M.A., Cross E., ud-Doula A., Wise M. Biophys. J. 2000;78:1126–1144. doi: 10.1016/S0006-3495(00)76671-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck S.D., Faraci W.S., Kelbaugh P.R., Saccomano N.A., Thadeio P.F., Volkmann R.A. Proc. Natl Acad. Sci. USA. 1996;93:4036–4039. doi: 10.1073/pnas.93.9.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges L.V., Olkin I. Statistical Methods for Meta-Analysis. London: Academic Press; 1985. [Google Scholar]
- Hopping G., Kellock J., Barnwal R.P., Law P., Bryers J.D., Varani G., Caughey B., Daggett V. eLIFE. 2014;3:e01681. doi: 10.7554/eLife.01681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung L.-W., Kohmura M., Ariyoshi Y., Kim S.-H. Acta Crystallogr. D Biol. Crystallogr. 1998;54:494–500. doi: 10.1107/s0907444997012225. [DOI] [PubMed] [Google Scholar]
- Iida T., Santa T., Toriba A., Imai K. Biomed. Chromatogr. 2001;15:319–327. doi: 10.1002/bmc.80. [DOI] [PubMed] [Google Scholar]
- Imperiali B., Moats R.A., Fisher S.L., Prins T.J. J. Am. Chem. Soc. 1992;114:3182–3188. [Google Scholar]
- Kleinkauf H., Döhren,Von H. Eur. J. Biochem. 1996;236:335–351. doi: 10.1111/j.1432-1033.1996.00335.x. [DOI] [PubMed] [Google Scholar]
- Krause E., Beinert M., Schmieder P., Wenschuh H. J. Am. Chem. Soc. 2000;122:4865–4870. [Google Scholar]
- Krause E., Beyermann M., Dathe M., Rothemund S., Bienert M. Anal. Chem. 1995;67:252–258. doi: 10.1021/ac00098a003. [DOI] [PubMed] [Google Scholar]
- Kreil G. Science. 1994;266:996–997. doi: 10.1126/science.7973683. [DOI] [PubMed] [Google Scholar]
- Kumar J., Sim V. Prion. 2014;8:119–124. doi: 10.4161/pri.28220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.L., Powers J.P.S., Pflegerl K., Vasil M.L., Hancock R.E.W., Hodges R.S. J. Pept. Res. 2004;63:69–84. doi: 10.1046/j.1399-3011.2003.00106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M., Hirshberg M., Sharon R., Daggett V. Comput. Phys. Commun. 1995;91:215–231. [Google Scholar]
- Levitt M., Hirshberg M., Sharon R., Laidig K.E., Daggett V. J. Phys. Chem. B. 1997;101:5051–5061. [Google Scholar]
- Lipmann F., Hotchkiss R.D., Dubos R.J. J. Biol. Chem. 1941;141:163–169. [Google Scholar]
- Luo L., Kohli R.M., Onishi M., Linne U., Marahiel M.A., Walsh C.T. Biochemistry. 2002;41:9184–9196. doi: 10.1021/bi026047+. [DOI] [PubMed] [Google Scholar]
- McInnes C., Kondejewski L.H., Hodges R.S., Sykes B.D. J. Biol. Chem. 2000;275:14287–14294. doi: 10.1074/jbc.275.19.14287. [DOI] [PubMed] [Google Scholar]
- Meyer C.E., Rose W.C. J. Biol. Chem. 1936;115:721–729. [Google Scholar]
- Milton R.C., Milton S.C., Kent S.B. Science. 1992;256:1445–1448. doi: 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- Mitchell J.B., Smith J. Proteins Struct. Funct. Bioinform. 2003;50:563–571. [Google Scholar]
- Mor A., Amiche M., Nicolas P. Trends Biochem. Sci. 1992;17:481–485. doi: 10.1016/0968-0004(92)90333-5. [DOI] [PubMed] [Google Scholar]
- Ohide H., Miyoshi Y., Maruyama R., Hamase K., Konno R. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011;879:3162–3168. doi: 10.1016/j.jchromb.2011.06.028. [DOI] [PubMed] [Google Scholar]
- Pentelute B.L. J. Am. Chem. Soc. 2008;130:9702–9707. doi: 10.1021/ja801352j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritsker M., Jones P., Blumenthal R., Shai Y. Proc. Natl Acad. Sci. USA. 1998;95:7287–7292. doi: 10.1073/pnas.95.13.7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitz M., Olson M.E., Greenberg D.M. J. Am. Chem. Soc. 1955;77:3109–3111. [Google Scholar]
- Ripoll D.R., Vila J.A., Scheraga H.A. Proc. Natl Acad. Sci. USA. 2005;102:7559–7564. doi: 10.1073/pnas.0502754102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahl H.-G., Jack R.W., Bierbaum G. Eur. J. Biochem. 2008;230:827–853. doi: 10.1111/j.1432-1033.1995.tb20627.x. [DOI] [PubMed] [Google Scholar]
- Schnölzer M., Alewood P.F., Jones A., Alewood D., Kent S.B.H. Int. J. Pept. Protein Res. 1992;40:180–193. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- Sela M., Zisman E. FASEB J. 1997;11:449–456. doi: 10.1096/fasebj.11.6.9194525. [DOI] [PubMed] [Google Scholar]
- Shapira R., Chou C.H. Biochem. Biophys. Res. Commun. 1987;146:1342–1349. doi: 10.1016/0006-291x(87)90797-2. [DOI] [PubMed] [Google Scholar]
- Sievers S.A., Karanicolas J., Chang H.W., Zhao A., Jiang L., Zirafi O., Stevens J.T., Munch J., Baker D., Eisenberg D. Nature. 2011;475:96–100. doi: 10.1038/nature10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaugen M., Nissen-Meyer J., Jung G., Stevanovic S., Sletten K., Inger C., Abildgaard M., Nes I.F. J. Biol. Chem. 1994;269:27183–27185. [PubMed] [Google Scholar]
- Soyez D., Toullec J.Y., Ollivaux C., Géraud G. J. Biol. Chem. 2000;275:37870–37875. doi: 10.1074/jbc.M007302200. [DOI] [PubMed] [Google Scholar]
- Struthers M.D., Cheng R.P., Imperiali B. J. Am. Chem. Soc. 1996;118:3073–3081. [Google Scholar]
- Towse C.-L., Vymetal J., Vondrasek J., Daggett V. submitted. [Google Scholar]
- Wade D., Boman A., Wahlin B., Drain C.M., Andreu D., Boman H.G., Merrifield R.B. Proc. Natl Acad. Sci. USA. 1990;87:4761–4765. doi: 10.1073/pnas.87.12.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G., de Leeuw E., Pazgier M., Yuan W., Zou G., Wang J., Ericksen B., Lu W.-Y., Lehrer R.I., Lu W. J. Biol. Chem. 2009;284:29180–29192. doi: 10.1074/jbc.M109.018085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesehan K., Buder K., Linke R.P., Patt S., Stoldt M., Unger E., Schmitt B., Bucci E., Willbold D. ChemBioChem. 2003;4:748–753. doi: 10.1002/cbic.200300631. [DOI] [PubMed] [Google Scholar]
- Wilkemeyer M.F., Chen S.-Y., Menkari C.E., Sulik K.K., Charness M.E. J. Pharmacol. Exp. Ther. 2004;309:1183–1189. doi: 10.1124/jpet.103.063818. [DOI] [PubMed] [Google Scholar]
- Winitz M., Bloch-Frankenthal L., Izumiya N., Birnbaum S.M., Baker C.G., Greenstein J.P. J. Am. Chem. Soc. 1956;78:2423–2430. [Google Scholar]
- Wolosker H., Dumin E., Balan L., Foltyn V.N. FEBS J. 2008;275:3514–3526. doi: 10.1111/j.1742-4658.2008.06515.x. [DOI] [PubMed] [Google Scholar]
- Wright P.E., Dyson H.J., Lerner R.A. Biochemistry. 1988;27:7167–7175. doi: 10.1021/bi00419a001. [DOI] [PubMed] [Google Scholar]
- Yongye A.B., Li Y., Giulianotti M.A., Yu Y., Houghten R.A., Martinez-Mayorga K. J. Comput. Aided Mol. Des. 2009;23:677–689. doi: 10.1007/s10822-009-9295-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.