

Abstract
Background
BCR-ABL kinase domain (KD) mutations are detected in approximately 45% of imatinib-resistant CML. Patterns of KD mutations in Philadelphia chromosome (Ph)+ acute lymphoblastic leukemia (ALL) are less well-studied.
Methods
We assessed KD mutations in relapsed Ph+ ALL following treatments that included one or more kinase inhibitors (n = 24) or no prior KI therapy (n = 12).
Results
ABL KD mutations were detected by direct sequencing in 15 of 17 (88%) relapsed Ph+ ALL with prior imatinib (n = 16) or dasatinib (n = 1) treatment, and in 6 of 7 (86%) resistant/relapsed tumors treated with 2 or more KIs, compared with 0 of 12 relapsed Ph+ ALL never treated with KI. A restricted set of mutations was seen, mostly Y253H and T315I, detected on average 13 months following KI initiation, and mutations were not detected in the initial tumor samples prior to KI therapy in 12 patients assessed. Using a more sensitive pyrosequencing method, we did not detect mutations at codons 315 and 253 in the diagnostic samples from these 12 patients or in 30 Ph+ ALL patients who never relapsed.
Conclusions
ABL KD mutations, especially at codons 315 and 253, emerge upon relapse in the vast majority of patients with Ph+ ALL receiving maintenance KI therapy. Ongoing KI exposure may thus alter the patterns of relapse and favor outgrowth of clones with KI-resistant mutations.
Keywords: kinase inhibitor, therapy resistance, biological selection, lymphoid leukemia, chronic myeloid leukemia
Introduction
BCR-ABL kinase domain (KD) point mutations are detected in the dominant tumor clone(s) in approximately 45% of CML at the time of disease resistance, developing after an average of 20-35 months of imatinib (Gleevec®) therapy.1-3 Second and third generation kinase inhibitors (KIs), including dasatinib,4 nilotinib5, bosutinib6, INNO-4066, and MK-04577 are now used for imatinib-resistant CML and are each associated with a somewhat different spectrum of KD mutations likely related to differing sensitivity of mutant clones to these agents.8, 9
The algorithms for monitoring following imatinib resistance in CML are becoming well-established.10, 11 The clinical scenario in which BCR-ABL KD mutations are detected in Philadelphia chromosome (Ph)+ acute lymphoblastic leukemia (ALL) is less clear but has mostly been associated with relapsed disease following treatment with chemotherapy followed by maintenance therapy with tyrosine kinase inhibitors.12 Since current therapy for Ph+ ALL includes chemotherapy combined with imatinib therapy,13-16 the timing of emergence and overall pattern of BCR-ABL KD mutations is highly relevant. Furthermore, the differences between the biology of lymphoid blast crisis versus de novo Ph+ ALL remains unclear.17 To determine the pattern of KD mutation in Ph+ ALL, we assessed the patterns of BCR-ABL KD mutations at diagnosis, upon relapse and following salvage therapy with second KIs.
Materials and Methods
Patient population
Patients with Ph+ ALL seen at our institution over the last 5 years that had adequate available RNA from an involved sample (>10% blasts) were analyzed. All but two patients were adults over 19 years old. Diagnostic criteria were those of the World Health Organization International Histological Classification of Tumours.18 All but two ALL cases were of B-cell lineage as determined by an extensive flow cytometric antigen panel; the remaining two cases were myeloid/B-cell biphenotypic lineage. Cases presenting as lymphoid blast crisis of CML were excluded. Therapy for Ph+ ALL, in most cases, was hyper-fractionated cyclophosphamide, vincristine, doxorubicin and dexamethasone (CVAD) chemotherapy alternating with high-dose ara-C and methotrexate along with imatinib at 400 to 800 mg/day, with imatinib with or without vincristine or POMP as maintenance (individual treatments are summarized in Table I).13 Patients who presented outside of our institution were initially treated with a variety of intensive multiagent chemotherapy regimens with 400-800 mg/day of imatinib. Twelve relapsed Ph+ ALL who had received Hyper-CVAD (+/- rituximab) but no imatinib therapy were also studied for comparison. Therapy for relapsed disease was variable and included dasatinib (with or without additional chemotherapy) in 9,4, 5 others TKIs in 4, and MK-0457 in 3.7 Eight patients underwent stem cell transplant in 1st or 2nd remission.19
Table I.
Summary of clinical data in BCR-ABL kinase domain mutational analysis of relapsed Ph+ ALL.
| Clinical setting when KDM detected | Prior KDM status (mo after KI start, method) | KDM mutation at relapse, level/method | Time on KI (mo) | Time from Dx (mo) | KI dose before KDM detected | NC KI | Treatment summary before mutation | Treatment after detection of mutation | Fate of KDM | FU (mo) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| KDM emerging at relapse in patients initially diagnosed at our institution | |||||||||||
| 1 | On IM, 1st relapse | nd | 253H, 91%/py | 15 | 15 | IM 600×14m | N | CVAD>IM/V/Pred | HCVAD/dasat | 253H lost
315I and 450G gain |
DOD
28.3 |
| 2 | On IM, 2nd relapse | None
(2m/py) |
253H, 50%/ds | 10 | 15 | IM 400×2m - off tx- IM 800×3m | Y | CVAD/IM
Relapse: CVAD/IM |
dasat 70 | ni | DOD
16.8 |
| 3 | On IM, 1st relapse | None
(0m/py) |
253H, 50%/ds
P309A, 50%/ds |
12 | 12 | IM 600×12m | N | CVAD/IM>IM | none | ni | DOD
30.4 |
| 4 | On IM, 2nd relapse | None
(0m/ds-py) |
315I, 20%/py
255K, 70%/py |
10 | 91 | IM 600×10m | N | CVAD-R/IDEC>POMP
Relapse: V/Dec/IM |
bosutinib | ni | DOD
94.6 |
| 5 | On dasat, 1st relapse in CNS post-bmt | None
(0m/py) |
315I, 85%/ds-py | 12 | 13 | dasat 100×12m | N | CVAD >V/dasat | no KI, salvage chemotherapy | 315I persist | AWD
15.5 |
| 6 | On IM, 1st relapse in CNS post-bmt | nd | 317L, 24%/ds-py | 16 | 16 | IM 400-300 | Y | CVAD, A/RT (CNS) > bmt > IM | none | ni | AWD
12.2 |
| 7 | On IM, CNS relapse post-bmt | None
(3m/ds) |
355Q, 50%/ds | 6 | 15 | IM 400×3m - off tx-G1400×6m | Y | CVAD>POMP>bmt>IM | dasat 100 | 317L gain, 355Q lost | AWD
37.5 |
| 8 | On IM, 1st relapse | None
(0m/ds) |
359V, 50%/ds | 10 | 10 | IM 400>800×10m | N | CVAD/IM | none | ni | DOD
11.8 |
| 9 | Om IM, 2nd relapse post-bmt | None
(2m/ds) |
396R, 100%/ds | 10 | 29 | IM 400× 5m - off tx- IM 300×10m | Y | I/A>IM/V
Relapse: CVAD/IM>bmt |
bmt | ni | DOD
39.9 |
| 10 | On IM, 1st relapse in CNS | None
(0m/ds) |
459K, 75%/ds | 14 | 14 | IM 400>200>600 ×14m | Y | CVAD/R>IM/V/Pred | dasat>bmt | ni | DOD
31.7 |
| Patients referred to our institution for relapsed disease with KDM mutation | |||||||||||
| 11 | On IM, 1st relapse | nd | 253H, 100%/ds | 13 | 13 | IM 400×12m | ? | Larson A-B-C/IM>IM | nilotinib | 253H persist | DOD
19.3 |
| 12 | On IM, 2nd relapse | None
(12m/py) |
253H, 100%/ds | 16 | 22 | IM 400-600 ×16m | Y | CVAD>IM,
Relapse: CVAD/IM>IM/V |
dasat | ni | DOD
28.6 |
| 13 | On nilot, 2nd relapse | None
(14m/ds-py) |
253H, 100%/ds | 9 | 23 | IM 400× 1m > off 4m > nilot 800×9m | Y | CVAD>CVAD/IM>bmt
Relapse: nilot |
none | ni | DOD
39.6 |
| 14 | On dasat, 3rd relapse | None
(2m/py) |
253H, 100%/ds | 6 | 24 | IM 800×4m > dasat 140×1m | N | V/Pred/Asp>MTX/V/Pred
Relapse:V/Pred/Mito>IM/MT X/A>CVAD/IM>dasat |
decitabine >bmt | ni | DOD
26 |
| 15 | On IM, 1st relapse | nd | 315I, 50%/ds
253H, 50%/ds |
10 | 10 | IM dose/duration unknown | N | CVAD/IM | bosutinib | 315I gain
253H lost |
DOD
15.1 |
| 16 | On dasat, 2nd relapse | nd | 315I, 100%/ds | 5 | 26 | IM 400 > Dasat 140×5m | Y | D/V/Pred/IM>
MTX/Etop/D/VP Persist: > dasat 140 |
MK-0457 | 315I persist | DOD
42.8 |
| 17 | On dasat, 3rd relapse | nd | 315I, 100%/ds | ? | 23 | IM 400-800 ×12m > dasat 100×5m | N | CVAD
Relapse: bmt>IM 400-800 |
MK-0457 | 315I persist | DOD
38.1 |
| 18 | On dasat | nd | 315I, 66%/py | 6 | 6 | IM 800 ×4m > dasat 70×1m | ? | IM>dasat | HCVAD-dasat > bmt | 315I lost
255K gain |
AWD
14.8 |
| 19 | On IM, 3rd relapse | None
(5m/ds-py) |
315I, 100%/ds | 12 | 12 | IM 400×5m | Y | AALL-0232>bmt
Relapse: AALL-0232> CVAD/IM |
dasat and chemotherapy | ni | DOD
18.5 |
| 20 | On dasat, 3rd relapse | nd | 315I, 63%/ds-py
317L, 24%/ds-py 355G, 25%/ds |
? | 32 | IM > dasat dose/duration unknown | ? | CVAD/IM
Relapse: sarafenib >dasat |
MK-0457 | 315I and 355G loss | AWD
50.8 |
| 21 | On IM, CNS relapse post-bmt | nd | 317L, 75%/ds | 24 | 26 | IM 600-400 ×12m | Y | BFCI-ALL>IM-bmt
Relapse: CVAD/IM |
dasat | 317L persist | DOD
57 |
| Patients with relapsed disease with no KDM mutation detected | |||||||||||
| 22 | nr | None
(0m/ds) |
None | nr | nr | IM 400-600 ×9m | N | CVAD/R/IM | INNO-406 | nr | DOD
26.4 |
| 23 | nr | None
(0m/ds) |
None | nr | nr | IM 800×3m> off 5m> IM 800×2m | Y | D-V-Asp > bmt>IM | ni | nr | AWD
11.2 |
| 24 | nr | nd | None | nr | nr | IM 400×24m BMS×1m | Y | HCVAD > IM/V/Pred >
HCVAD/dasat |
nr | nr | AWD
34m |
All cases were e1a2/p190 BCR-ABL transcript type except for case 1 (b2a2/210), case 2 (b3a2/p210) and case 24 (b3a2 & b2a2/p210).
Abbreviations: AWD: alive with disease, bmt: stem cell transplantation, CNS: central nervous system, DOD: died of disease, ds: direct sequencing method (20% sensitivity, semiquantitative), DX: diagnosis, FU: followup, KDM: BCR-ABL kinase domain mutations, KI inhibitor, m: months, NC KI: non-continuous/breaks in KI maintenance therapy, nd: not done, ni: no information, nr: not relevant py: pyrosequencing method (1-5% sensitivity, quantitative +/- 5%). Doses of KI are listed as milligrams/day.
Treatments: “>” indicates sequential treatment, “/” indicates concurrent treatments, A: ara-C, Asp: asparaginase, BFCI-ALL: daunorubicin, vincristine, high-dose methotrexate, L-asparaginase, prednisone, bmt: stem cell transplant, D: daunorubicin, dasat: dasatinib (Sprycel), Dex: dexamethasone, etop: etoposide, HCVAD: hyper-fractionated cyclophosphamide, vincristine, doxorubicin and dexamethasone alternating with high-dose ara-C and methotrexate, I: idarubicin, HU: hydroxyurea, IM: imatinib mesylate (Gleevec®), Larson A: vincristine, prednisone, l-asparaginase, and doxorubicin, Mito: mitoxantrone, MTX: intrathecal methotrexate, nilot: nilotinib (Tasigna, AMN107), Pred: prednisone, POMP: 6-MP and methotrexate, with or without vincristine and steroids, R: rituximab, V: vincristine.
For monitoring of treatment response, BCR-ABL PCR studies were done every 1 to 3 months on peripheral blood during the induction phase. Standard cytogenetic analysis of Giemsa (G)-banded metaphases was done on bone marrow aspirates, and fluorescence in situ hybridization using BCR-ABL fusion probes was done along with the PCR studies on blood every 3-6 months during the maintenance phase and during remission and at relapse.
RNA isolation and quantitative BCR-ABL PCR
White blood cells from bone marrow aspirate or peripheral blood specimens were isolated by centrifugation following red blood cell lysis. Total RNA was extracted using Trizol reagent (Gibco-BRL, Gaithersburg, MD) and RNA quality assessed by agarose gel electrophoresis prior to cDNA synthesis. Real-time quantitative PCR for BCR-ABL was performed in a one-tube reaction with normalization to total ABL levels, and post-PCR sizing of fusion transcript products, as previously described.20, 21
KD mutation detection
To avoid interference from the normal ABL allele, a nested PCR sequencing approach was used with a 1st round amplification of the BCR-ABL transcript followed by two separate PCR reactions that cover exons 221 to 380 and codons 350 to 500 of the ABL kinase domain.22 Standard dideoxy chain-termination DNA sequencing was performed using Big Dye chain terminator reagents on an ABI3700 analyzer and analyzed using Sequence Analysis software V3.3 and the SeqScape software V2 (ABI, Foster City, CA).1 All mutations were confirmed by sequencing of forward and reverse strands, with a sensitivity of 10% mutation-bearing transcripts in the analyzed population. Mutation level for direct sequencing (ds) was reported semiquantitatively in Table I, as only mutant (100%), mostly mutant (75%), mixed (50%), or mostly unmutated (25%).
For mutation analysis of codons 311-317 and codons 250-255, pyrosequencing was performed following first round PCR, as above and different 2nd-round PCR reactions. Second round PCR was performed using one biotin-tagged primer and single-stranded product isolated by avidin-sepharose beads (GE Life Sciences, Piscataway, NJ) and sequenced using a HSQ96 Pyrosequencer (Biotage, Uppsala, Sweden). The sensitivity of the pyrosequencing was 1-5% mutation-bearing transcripts in the total RNA pool. Mutation level for pyrosequencing (py) was reported as % mutated product in Table I, with an established precision of 5% for the assay.
Results
BCR-ABL KD Mutations are detectable at relapse in KI-treated but not KI-naïve ALL
Overall, 63 samples from 36 Ph+ patients with relapsed Ph+ ALL were studied, including 24 with kinase inhibitors (KI) as part of their therapy, and 12 patients who had never received KI therapy prior to relapse as a comparison group. There were 34 adults and 2 children (21M 15F, median 45 years old at diagnosis, range 3-83 years), with median followup of 28 months, range 11-96 months). 30 of 36 (83%) tumors had e1a2 BCR-ABL fusion transcripts associated with the p190 BCR-ABL protein, with the other tumors having b2a2 or b3a2 BCR-ABL fusion transcripts producing the p210 protein
Using a nested PCR-based assay to directly sequence the entire ABL kinase domain (KD) of the BCR-ABL fusion transcript, no KD mutations were noted at relapse in the 12 patients with Ph+ ALL who had received no prior KI therapy. The frequency and pattern of BCR-ABL KD mutation development among the 24 patients who had received KI therapy prior to relapse is summarized in Table I. BCR-ABL KD mutations were noted by direct sequencing in 15 of 17 (88%) patients with morphological evidence of relapse after only imatinib (n = 16) or dasatinib (n = 1) therapy. The sites of KD mutation were Y253H in 6 (c.1121T>C), T315I in 4 (c.1308C>T), F317L in 2 (c.1315C>A), and E255K (c.1127G>A), P309A (c.1288C>G), E355Q (c.1427G>C), F359V (c.1439T>G), H396R (c.1551A>G), E459K (c.1739G>A) in 1 each, with 3 patients having 2 detectable mutations (Figure 1). All mutations were detected in peripheral blood or bone marrow aspirate sample, although concurrent relapse in the CNS was commonly noted. The median time from diagnosis to mutation detection was 10 months (range 6-24 months), with mutations in codons 253 and 315 most frequently developing in those receiving continuous KI therapy in the preceding months, versus those patients with some breaks in imatinib maintenance therapy. The two patients on imatinib maintenance who relapsed without detectable KD mutations also had intermittent KI therapy.
Figure 1. Summary of location of mutations in the kinase domain of BCR-ABL in relapsed Ph+ ALL.

Illustrated is a schematic representation of the kinase domain, including ATP binding pocket (P-loop), catalytic domain (C), and activation loop (A). Mutations which developed on imatinib are shown as open circles, those developing on dasatinib as black circles or triangles, and those on nilotinib and bosutinib as hatched circles. The hatched lines indicate the area covered by the pyrosequencing (pyro) mutation assay. Amino acid designation for the mutations are those of the ABL type 1a protein, with the following approximate KD subdomains: P-loop 248–256, C-helix/catalytic domain 276–290, SH3 contact 294–304, additional imatinib binding pocket 311-318, SH2 contact 331–353, and activation loop 381–402.32
BCR-ABL KD mutations were also noted in 6 of 7 patients (86%) who presented to our institution with relapsed/persistent Ph+ ALL, after having received two or more KIs. The sites of KD mutation were T315I in 4 (c.1308C>T), Y253H (c.1121T>C), F317L (c.1315C>A) and E355G (c.1428A>G) in 1 for those receiving dasatinib after imatinib, and Y253H (c.1121T>C) in 1 patient receiving nilotinib after imatinib. One patient who had previously received imatinib, nilotinib and dasatinib (in that order) had 3 KD mutations, including T315I, F317L and E355G, all present at mixed levels suggesting 3 distinct clones. The median duration of KI therapy at mutation detection was 10 months, and the median time on second KI therapy was 4 months.
Median followup among the cohort of 24 KI-treated patients who relapsed was 29 months (range 11-95 months), with 17 of 24 patients dead of disease at last followup (overall survival, range 12-95 months).
Table II summarizes the cytogenetic findings seen by conventional G-banded karyotype among the 24 KI-treated relapsed tumors. Only 2 cases had t(9;22) as the sole abnormality at diagnosis, with extra copies of the Ph chromosome, as indicated by derivative (der) chromosome 22 or more complex derivative “marker” (mar) chromosomes, seen in 8 cases (all amplifications were confirmed by BCR-ABL fusion FISH study). In cases with complex karyotypes there was usually reemergence of a similar or related clone at time of relapse; however 6 cases at 1st or 2nd relapse had clonal progression or emergence of an apparently distinct (sub)clone from that seen at diagnosis.
Table II.
Karyotypic findings at diagnosis and relapse.
| Case # | Initial karyotype summary | Karyotype at relapse | Comment |
|---|---|---|---|
| 1 | PQ, 46,XX,t(10;19)(q24;q13.3) | 45,XX,-9,t(10;19)(q23;q13.3),t(16;22)(p13.3;q12),del(20)(q11.2q13.3) | Cryptic BCR-ABL, t(10;19) likely constitutional change |
| 2 | 41-46,XY,-9,add(21)(p11),der(22)t(9;22)(q34;q11.2) | 46,XY,add(7)(q36),del(9)(q22),t(9;22)(q34;q11.2),del(13)(q14q22), add(21)(p11) | Related complex clones with gain of add7q at relapse |
| 3 | 46,XX,-9,t(9;22)(q34;q11.2),+der(22)t(9;22) | 47,XX,-9,t(9;22)(q34;q11.2),+der(22)t(9;22)×2 | Several related subclones seen at diagnosis/relapse |
| 4 | 46,XX,t(9;22)(q34;q11.2) | 46,XX,t(9;22)(q34;q11.2) | No change at relapse |
|
|
|||
| 5 | 46,XY,t(9;22)(q34;q11.2) | 46,XY,del(6)(q13q23),t(9;22)(q34;q11.2) | Gain of del6q at relapse |
| 6 | PQ, BCR-ABL FISH+ | 46,XY/ BCR-ABL FISH+ | |
| 7 | NA, BCR-ABL FISH+ | 1st relapse: 46,XX,t(9;22)(q34;q11.2) [1 of 20 metaphases] | Subclonal diversity evident that correlates with shifts in KDM upon dasatinib introduction |
| 46,XX,t(9;22)(q34;q11.2),del(20)(q12) [1 of 20 metaphases] | |||
| 46,XX,t(3;21)(q21;q22),t(9;22)(q34;q11.2) [1 of 20 metaphases] | |||
| 2nd relapse: 45,XX,-8,t(9;22)(q34;q11.2),t(11;15)(q13;q26), add(12)(q24.3),add(16)(q24) | |||
| 8 | 48,XY,add(3)(q29),t(9;22)(q34;q11.2),+der(22)t(9;22) | 49,XY,+4,t(9;22)(q34;q11.2),+der(22)t(9;22),+mar | Several related clones at diagnosis, Clonal shift at relapse |
| 9 | NA, BCR-ABL FISH+ | 45,XX,del(1)(q32q42),-7,t(9;22)(q34;q11.2),-13,-16,+19,+1-3mar | |
| 10 | NA, BCR-ABL FISH+ | 48,XX,der(2)t(1;2)(q11;q37),t(9;22)(q34;q11.2),del(19)(q13.2), +der(22)t(9;22)×2 | |
| Karyotype at relapse | Karyotype at subsequent relapse/persistence | ||
| 11 | 45,XY,del(3)(p13),del(4)(q31.1q35),add(4)(q35),-7,t(9;22)(q34;q11.2),-16,+mar[2] | 45,XY,del(3)(p13),del(4)(q31.1q35),add(4)(q35),-7, t(9;22)(q34;q11.2),-10,-16,+2mar | Similar complex clones with TKI switch |
| 12 | NA, BCR-ABL FISH+ | 46,XY,del(5)(q21q31),t(9;22)(q34;q11.2) | |
| 13 | 46,XX,del(1)(p32),t(2;2)(q31;q35),inv(5)(q11.2q23), der(9)del(9)(p21)t(9;22)(q34;q11.2),t(17;22)(q11.2;q13), der(22)t(9;22) | 46,XX,del(1)(p32),t(2;2)(q37;q35),der(5)inv(5)(q11.2q23) dup(5)(q23q31),der(9)del(9)(p21)t(9;22)(q34;q11.2), t(17;22)(q11.2;q13),der(22)t(9;22) | Similar complex clones at relapses |
| 14 | 44-46,XX,add(3)(q29),-7,der(9)del(9) del(9)(p23)t(9;22)(q34;q11.2),+1-3mar | 45,XX,add(3)(q29),-7,der(9)del(9)(p21)t(9;22)(q34;q11.2), der(22)t(9;22) | Related but distinct complex clones at multiple relapses |
| 15 | 46,XY,t(2;21)(p13;q22),add(9)(p22),t(9;22)(q34;q11.2),der(20)t(1;20)(q23;q13.1) | 39-44,XY,t(2;21)(p13;q22),del(5)(q31q35),add(8)(p23), del(9)(p22p24),t(9;22)(q34;q11.2),der(10)t(1;10)(q21;q26),add(11)(p15), add(12)(p13),-17,-19,del(20)(q11.2) | Related but distinct complex clones at multiple relapses |
| 16 | 45,XY,-7,t(9;22)(q34;q11.2),der(12)t(7;12)(q11.2;p11.2) | 45,XY,-7,t(9;22)(q34;q11.2),der(12)t(7;12)(q11.2;p11.2) | Related complex clones with disease persistence |
| 17 | PQ, BCR-ABL FISH+ | 41-45,X,-X,t(9;22)(q34;q11.2),ins(10)(p15q25q26), der(19)t(1;19)(q23;p13.3) | |
| 18 | 46,XY,t(9;22)(q34;q11.2) | 6,XY,i(8)(q10),t(9;22)(q34;q11.2) | Gain of i8q associated with KDM shift with relapse after SCT |
| 19 | 46,XY,t(1;4)(q22;p15.3),del(2)(p14),add(2)(q37),add(7)(q36), der(9)add(9)(p24)t(9;22)(q34;q11.2),+10,add(11)(p14),-14, del(14)(q21),add(18)(q22),der(22)t(9;22) | ||
| 20 | 46,XX,add(7)(p11),t(9;22)(q34;q11.2),-14, der(16)t(1;16)(q23;q22),+der(22)t(9;22) | 46,XX,-7,t(9;22)(q34;q11.2),der(14)t(7;14)(q11.2;q22), der(16)t(1;16)(q23;q13),+der(22)t(9;22) | Related but distinct complex clones at multiple relapses |
| 21 | 44-45,XY,-2,-2,del(7)(p13p21), t(9;22)(q34;q11.2),del(12)(p11.2),add(16)(q24),+2mar | 45-46,XY,inv(3)(q21q26),-7,t(9;22)(q34;q11.2), del(10)(q11),add(11)(p15),del(12)(p11.2),add(16)(q24),-18,+1-2mar | Distinct clones at 1st/2nd relapse |
| Initial karyotype | Karyotype at relapse | ||
| 22 | 45,XX,der(9)del(9)(p13)t(9;22)(q34;q11.2),-20,-21,der(22)t(9;22),+mar | 46,XX,der(9)del(9)(p13)t(9;22)(q34;q11.2), -19,add(20)(q13.3),der(22)t(9;22),+mar | Related complex clones seen at diagnosis/relapse |
| 23 | NA, BCR-ABL FISH+ | 46,XX,t(9;13)(q32;q12),t(9;22)(q34;q11.2) | |
| 24 | NA | 44,XX,der(9)add(9)(p11.2)t(9;22)(q34;q11.2),-13,-13 |
Case numbers and groupings as in Table I.
Abbreviations: add: additional/altered chromosomal material present, del: deletion of chromosome material, der: derivative chromosome, mar: marker (unassigned) chromosomal material, i: isochromosome, inv: chromosomal inversion, na: karyotype not available, pq: poor quality metaphases.
Shifts in KD mutation can occur following switch to a different KI
Table I also summarizes the outcome of KD mutations who switched from one KI to another. Complete regression of one mutation and emergence of another was seen in 3 patients (#1, 7, 15), such as the complete shift from E355Q to F317L (c.1315C>A) at 5 months following switch from imatinib to dasatinib. Using quantitative pyrosequencing, we noted that persistence and level of particular mutations sometimes paralleled their predicted sensitivity to particular KIs from in vitro studies,8, 23-25 For example in 3 patients, F317L and T315I exhibited no regression with dasatinib treatment (#5, 18, 21) but T315I did regress with MK-0457 therapy in 1 of 3 patients (#20) and with stem cell transplant (#18). These data suggest that although mutations can develop rapidly in Ph+ ALL, exposure to a relevant KI is essential for their full emergence and persistence. Several cases that showed shifts to a new KD mutation with introduction of a 2nd TKI had evidence of emergence of a distinct clone (case #7) or clonal progression (cases #1, 15 and 20) by cytogenetic analysis.
BCL-ABL KD mutations are not detectable at diagnosis in Ph+ ALL
In order to assess whether significant levels of BCR-ABL transcripts with KD mutations were present prior to KI use, we studied the diagnostic sample from 12 patients who ultimately developed a mutation at relapse and did not detect any KD mutations. Using a more sensitive quantitative pyrosequencing assay, we also did not any detect mutations in Y253H T315I and F317L in these samples or in the initial diagnostic samples from 25 additional cases of Ph+ ALL who did not relapse (not shown). KD mutations were also rare (2 of 12 patients) in persistent ALL during the first months of treatment, and in those cases mutations were not seen at the relapsed mutational hotspots (Table III).
Table III.
Detection of BCR-ABL kinase domain mutations by direct sequencing during disease course of Ph+ ALL.
| At time of diagnosis | Residual disease during therapy including KI | At relapse with no prior KI treatment | At relapse with one prior KI treatment | At relapse after two or three KIs |
|---|---|---|---|---|
| 0/121 | 2/122 | 0/123 | 15/174 | 6/7 |
Direct sequencing refers to full ABL kinase domain Sanger sequencing performed following a nested PCR reaction (See methods).
Tested were the diagnostic or pre-KI-treated samples from 12 patients with Ph+ ALL that eventually relapsed with a KD mutation.
Tested were samples with residual Ph+ ALL in the first 6 months of treatments with chemotherapy and KI. Low levels of S438C (c.1674C>G) and Q252H (c.1120G>T) mutation detected by direct sequencing in 1 sample each.
Tested were 12 relapse samples from patients with Ph+ ALL who received chemotherapy regimens with no KI.
16 pts had been treated with IM only, 1 with dasatinib only prior to KDM detection. 10 of these pts were diagnosed at our institution (summarized in top portion of Table I), with the remainder being referred for relapsed ALL treated elsewhere (summarized below in Table I).
Abbreviations: KI: kinase inhibitor, KDM: ABL kinase domain mutation.
Discussion
We note that BCR-ABL KD mutation in Ph+ ALL occurs predominantly in the setting of relapse following KI maintenance therapy and involves a restricted subset of mutations that are among the most resistant in vitro to imatinib and most of the newer KIs, namely T315I and Y253H.26 Mutations were not observed at relapse in those patients not receiving KI as part of their treatments.
The higher incidence, rapid emergence and restricted group of KD mutations seen here in relapsed Ph+ ALL as compared to KI-resistant CML11, 22, 27 is similar to another series recently reported,28 but our results differ in some respects. By comparing KD mutations present at low levels at the time of diagnosis, Pfeifer et al concluded that acquired therapy resistance in Ph+ ALL is due in part to mutated BCR-ABL clones that exist prior to KI therapy.28 They concluded that this supported the use of KIs as initial therapy to eradicate these pre-existing KD-mutated populations. Given the absence of a substantial population of any given KD-mutated clone detectable prior to mutation growth, our results would favor selection following initiation of KI therapy as the primary determinant of mutation emergence. In fact, many of these patients who developed KD mutations at relapse were on continuous KI therapy from the time of diagnosis. The rapidity of KD mutation shifts that we observed in some patients following introduction of a different KI (median 4 months) also supports rapid KD mutation outgrowth in response to KI-driven selective pressure.
The selective pressure reflected in the high rate of KD mutation also provides strong evidence that BCR-ABL expression remains critical to growth of Ph+ ALL at relapse. The more restricted sites of BCR-ABL mutations in Ph+ ALL as compared to KI-resistant CML may be related to need for more active BCR-ABL kinase for growth/transformation of lymphoid cells versus myeloid cells. This hypothesis would also be consistent with the frequent presence in Ph+ ALL of BCR-ABL gene amplification, as evidenced by extra Philadelphia chromosomes and the frequent finding of higher BCR-ABL transcript levels. Effects due to differences in the signaling pathways for the p190 versus p230 BCR-ABL protein following KI therapy in lymphoid versus biphenotypic versus myeloid stem cell populations also cannot be excluded. Finally differences in the entry, binding and metabolism of KIs in different leukocyte populations may influence the selection of KD mutated clones in ALL versus CML.
KIs clearly have an important role in therapy of relapsed Ph+ ALL. Dasatinib, in particular, has also been shown to be effective for imatinib-treated resistant/relapsed Ph+ ALL.29, 30 However, the high rate of KD mutation development at relapse suggests that KI-selected mutations can promote disease progression and has implications for the timing and duration of induction and maintenance therapy with KIs in Ph+ ALL.13 Our data suggests that sequential or combination therapy with both imatinib and dasatinib may preferentially select for tumor clones with mutations that mediate resistance to both agents. The most common mutation that develops in this setting, T315I, is cross-resistant to most KIs. Whether other chemotherapeutic agents in maintenance regimens beside KIs plays a role in mediating outgrowth of mutated clones deserves further study. In that regard, strategies to alternate KIs with other therapies during the maintenance phase31 or replacement of KIs with other cytostatic chemotherapies once mutations are detected to allow clonal regression may be strategies worth exploring to minimize biological selection of resistant disease.
Figure 2. Mapping of BCR-ABL KD mutations seen in relapsed Ph+ ALL.
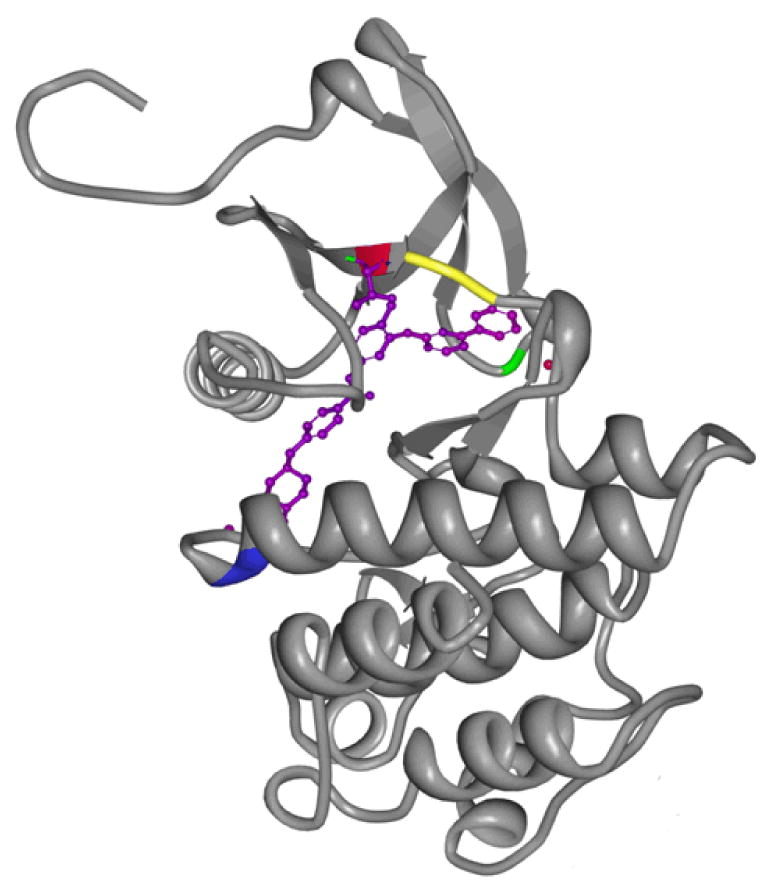
Human Abl kinase domain in complex with imatinib (accession 2HYY,33 accessed from the RCSB Protein Data Bank), was rendered with MBT protein workshop software.34 One ABL chain is shown, with imatinib in its binding pocket highlighted in purple; ABL codon 253 is labeled green, codon 315 is red, codon 317 is yellow, and codon 355 is blue.
Acknowledgments
This investigation was supported by a developmental grant from the Leukemia SPORE (1P50CA100707-01) awarded by the National Cancer Institute, Department of Health and Human Services. Presented in part at the American Society of Hematology, Orlando, FL, December 9-12, 2006. The Authors would like to thank Nubia Reeves and John Galbincea for sequencing analysis and Drs. Stefan Faderl and Farhad Ravandi for clinical information and feedback.
References
- 1.Jabbour E, Kantarjian H, Jones D, Talpaz M, Bekele N, O'Brien S, et al. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia. 2006;20(10):1767–73. doi: 10.1038/sj.leu.2404318. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16855631. [DOI] [PubMed] [Google Scholar]
- 2.Lahaye T, Riehm B, Berger U, Paschka P, Muller MC, Kreil S, et al. Response and resistance in 300 patients with BCR-ABL-positive leukemias treated with imatinib in a single center: a 4.5-year follow-up. Cancer. 2005;103(8):1659–69. doi: 10.1002/cncr.20922. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15747376. [DOI] [PubMed] [Google Scholar]
- 3.Al-Ali HK, Heinrich MC, Lange T, Krahl R, Mueller M, Muller C, et al. High incidence of BCR-ABL kinase domain mutations and absence of mutations of the PDGFR and KIT activation loops in CML patients with secondary resistance to imatinib. Hematol J. 2004;5(1):55–60. doi: 10.1038/sj.thj.6200319. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14745431. [DOI] [PubMed] [Google Scholar]
- 4.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354(24):2531–41. doi: 10.1056/NEJMoa055229. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16775234. [DOI] [PubMed] [Google Scholar]
- 5.Quintas-Cardama A, Kantarjian H, Jones D, Nicaise C, O'Brien S, Giles F, et al. Dasatinib (BMS-354825) is active in Philadelphia chromosome-positive chronic myelogenous leukemia after imatinib and nilotinib (AMN107) therapy failure. Blood. 2007;109(2):497–9. doi: 10.1182/blood-2006-07-035493. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16990591. [DOI] [PubMed] [Google Scholar]
- 6.Jabbour E, Cortes JE, Ghanem H, O'Brien S, Kantarjian HM. Targeted therapy in chronic myeloid leukemia. Expert Rev Anticancer Ther. 2008;8(1):99–110. doi: 10.1586/14737140.8.1.99. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18095887. [DOI] [PubMed] [Google Scholar]
- 7.Giles FJ, Cortes J, Jones D, Bergstrom D, Kantarjian H, Freedman SJ. MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood. 2007;109(2):500–2. doi: 10.1182/blood-2006-05-025049. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16990603. [DOI] [PubMed] [Google Scholar]
- 8.Burgess MR, Skaggs BJ, Shah NP, Lee FY, Sawyers CL. Comparative analysis of two clinically active BCR-ABL kinase inhibitors reveals the role of conformation-specific binding in resistance. Proc Natl Acad Sci U S A. 2005;102(9):3395–400. doi: 10.1073/pnas.0409770102. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15705718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soverini S, Martinelli G, Colarossi S, Gnani A, Castagnetti F, Rosti G, et al. Presence or the emergence of a F317L BCR-ABL mutation may be associated with resistance to dasatinib in Philadelphia chromosome-positive leukemia. J Clin Oncol. 2006;24(33):e51–2. doi: 10.1200/JCO.2006.08.9128. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17114651. [DOI] [PubMed] [Google Scholar]
- 10.Hochhaus A, Erben P, Ernst T, Mueller MC. Resistance to targeted therapy in chronic myelogenous leukemia. Semin Hematol. 2007;44(1) 1:15–24. doi: 10.1053/j.seminhematol.2006.12.002. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17292737. [DOI] [PubMed] [Google Scholar]
- 11.Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaeda J, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108(1):28–37. doi: 10.1182/blood-2006-01-0092. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16522812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hofmann WK, Komor M, Hoelzer D, Ottmann OG. Mechanisms of resistance to STI571 (Imatinib) in Philadelphia-chromosome positive acute lymphoblastic leukemia. Leuk Lymphoma. 2004;45(4):655–60. doi: 10.1080/10428190310001625755. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15160936. [DOI] [PubMed] [Google Scholar]
- 13.Thomas DA, Faderl S, Cortes J, O'Brien S, Giles FJ, Kornblau SM, et al. Treatment of Philadelphia chromosome-positive acute lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood. 2004;103(12):4396–407. doi: 10.1182/blood-2003-08-2958. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14551133. [DOI] [PubMed] [Google Scholar]
- 14.Ottmann OG, Druker BJ, Sawyers CL, Goldman JM, Reiffers J, Silver RT, et al. A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood. 2002;100(6):1965–71. doi: 10.1182/blood-2001-12-0181. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12200353. [DOI] [PubMed] [Google Scholar]
- 15.Rea D, Legros L, Raffoux E, Thomas X, Turlure P, Maury S, et al. High-dose imatinib mesylate combined with vincristine and dexamethasone (DIV regimen) as induction therapy in patients with resistant Philadelphia-positive acute lymphoblastic leukemia and lymphoid blast crisis of chronic myeloid leukemia. Leukemia. 2006;20(3):400–3. doi: 10.1038/sj.leu.2404115. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16437142. [DOI] [PubMed] [Google Scholar]
- 16.de Labarthe A, Rousselot P, Huguet-Rigal F, Delabesse E, Witz F, Maury S, et al. Imatinib combined with induction or consolidation chemotherapy in patients with de novo Philadelphia chromosome-positive acute lymphoblastic leukemia: results of the GRAAPH-2003 study. Blood. 2007;109(4):1408–13. doi: 10.1182/blood-2006-03-011908. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17062730. [DOI] [PubMed] [Google Scholar]
- 17.Anastasi J, Feng J, Dickstein JI, Le Beau MM, Rubin CM, Larson RA, et al. Lineage involvement by BCR/ABL in Ph+ lymphoblastic leukemias: chronic myelogenous leukemia presenting in lymphoid blast vs Ph+ acute lymphoblastic leukemia. Leukemia. 1996;10(5):795–802. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8656674. [PubMed] [Google Scholar]
- 18.Jaffe ES, Harris N, Stein H, Vardiman JW. Tumours of Hematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2001. [Google Scholar]
- 19.Jabbour E, Cortes J, Kantarjian HM, Giralt S, Jones D, Jones R, et al. Allogeneic stem cell transplantation for patients with chronic myeloid leukemia and acute lymphocytic leukemia after Bcr-Abl kinase mutation-related imatinib failure. Blood. 2006;108(4):1421–3. doi: 10.1182/blood-2006-02-001933. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16601247. [DOI] [PubMed] [Google Scholar]
- 20.Cortes J, Talpaz M, O'Brien S, Jones D, Luthra R, Shan J, et al. Molecular responses in patients with chronic myelogenous leukemia in chronic phase treated with imatinib mesylate. Clin Cancer Res. 2005;11(9):3425–32. doi: 10.1158/1078-0432.CCR-04-2139. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15867244. [DOI] [PubMed] [Google Scholar]
- 21.Luthra R, Medeiros LJ. TaqMan reverse transcriptase-polymerase chain reaction coupled with capillary electrophoresis for quantification and identification of bcr-abl transcript type. Methods Mol Biol. 2006;335:135–45. doi: 10.1385/1-59745-069-3:135. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16785625. [DOI] [PubMed] [Google Scholar]
- 22.Cortes J, Jabbour E, Kantarjian H, Yin CC, Shan J, O'Brien S, et al. Dynamics of BCR-ABL kinase domain mutations in chronic myeloid leukemia after sequential treatment with multiple tyrosine kinase inhibitors. Blood. 2007;110(12):4005–11. doi: 10.1182/blood-2007-03-080838. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17785585. [DOI] [PubMed] [Google Scholar]
- 23.Bradeen HA, Eide CA, O'Hare T, Johnson KJ, Willis SG, Lee FY, et al. Comparison of imatinib mesylate, dasatinib (BMS-354825), and nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-based mutagenesis screen: high efficacy of drug combinations. Blood. 2006;108(7):2332–8. doi: 10.1182/blood-2006-02-004580. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16772610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.von Bubnoff N, Manley PW, Mestan J, Sanger J, Peschel C, Duyster J. Bcr-Abl resistance screening predicts a limited spectrum of point mutations to be associated with clinical resistance to the Abl kinase inhibitor nilotinib (AMN107) Blood. 2006;108(4):1328–33. doi: 10.1182/blood-2005-12-010132. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16614241. [DOI] [PubMed] [Google Scholar]
- 25.Weisberg E, Catley L, Wright RD, Moreno D, Banerji L, Ray A, et al. Beneficial effects of combining nilotinib and imatinib in preclinical models of BCR-ABL+ leukemias. Blood. 2007;109(5):2112–20. doi: 10.1182/blood-2006-06-026377. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17068153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65(11):4500–5. doi: 10.1158/0008-5472.CAN-05-0259. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15930265. [DOI] [PubMed] [Google Scholar]
- 27.Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003;102(1):276–83. doi: 10.1182/blood-2002-09-2896. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12623848. [DOI] [PubMed] [Google Scholar]
- 28.Pfeifer H, Wassmann B, Pavlova A, Wunderle L, Oldenburg J, Binckebanck A, et al. Kinase domain mutations of BCR-ABL frequently precede imatinib-based therapy and give rise to relapse in patients with de novo Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL) Blood. 2007;110(2):727–34. doi: 10.1182/blood-2006-11-052373. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17405907. [DOI] [PubMed] [Google Scholar]
- 29.Jabbour E, Cortes J, Kantarjian H. Dasatinib for the treatment of Philadelphia chromosome-positive leukaemias. Expert Opin Investig Drugs. 2007;16(5):679–87. doi: 10.1517/13543784.16.5.679. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17461740. [DOI] [PubMed] [Google Scholar]
- 30.Ottmann O, Dombret H, Martinelli G, Simonsson B, Guilhot F, Larson RA, et al. Dasatinib induces rapid hematologic and cytogenetic responses in adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia with resistance or intolerance to imatinib: interim results of a Phase II study. Blood. 2007. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17496201. [DOI] [PubMed]
- 31.Delannoy A, Delabesse E, Lheritier V, Castaigne S, Rigal-Huguet F, Raffoux E, et al. Imatinib and methylprednisolone alternated with chemotherapy improve the outcome of elderly patients with Philadelphia-positive acute lymphoblastic leukemia: results of the GRAALL AFR09 study. Leukemia. 2006;20(9):1526–32. doi: 10.1038/sj.leu.2404320. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16838024. [DOI] [PubMed] [Google Scholar]
- 32.Jiang X, Saw KM, Eaves A, Eaves C. Instability of BCR-ABL gene in primary and cultured chronic myeloid leukemia stem cells. J Natl Cancer Inst. 2007;99(9):680–93. doi: 10.1093/jnci/djk150. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17470736. [DOI] [PubMed] [Google Scholar]
- 33.Cowan-Jacob SW, Fendrich G, Floersheimer A, Furet P, Liebetanz J, Rummel G, et al. Structural biology contributions to the discovery of drugs to treat chronic myelogenous leukaemia. Acta Crystallogr D Biol Crystallogr. 2007;63(Pt 1):80–93. doi: 10.1107/S0907444906047287. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17164530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moreland JL, Gramada A, Buzko OV, Zhang Q, Bourne PE. The Molecular Biology Toolkit (MBT): a modular platform for developing molecular visualization applications. BMC Bioinformatics. 2005;6:21. doi: 10.1186/1471-2105-6-21. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15694009. [DOI] [PMC free article] [PubMed] [Google Scholar]