

Abstract
The extensive study of genetic alterations in colorectal cancer (CRC) has led to molecular diagnostics playing an increasingly important role in CRC diagnosis and treatment. Currently, it is believed that CRC is a consequence of the accumulation of both genetic and epigenetic genomic alterations. It is known that there are at least 3 major pathways that lead to colorectal carcinogenesis: (1) the chromosomal instability pathway, (2) the microsatellite instability pathway, and (3) the cytosine-phospho-guanine island methylator phenotype pathway. With recent advances in CRC genetics, the identification of specific molecular alterations responsible for CRC pathogenesis has directly influences clinical care. Patients at high risk for developing CRC can be identified by genetic testing for specific molecular alterations, and the use of molecular biomarkers for predictive and prognostic purposes is also increasing. This is clearly supported by the recent advances in genetic testing for CRC whereby specific molecular alterations are identified for the purpose of guiding treatment with targeting therapies such as anti-endothelial growth factor receptor monoclonal antibodies.
Keywords: Colorectal neoplasms, Chromosomal instability, Microsatellite instability, Epigenetic instability, Biological markers
INTRODUCTION
Despite recent advances in detection and treatment, colorectal cancer (CRC) represents a major global public health problem. In Korea, the incidence of CRC has been rapidly increasing over the last few years. According to the National Cancer Registry, CRC is the second most common cancer in males and the third most common cancer in females, and the fourth leading cause of cancer death.1
The original theory of the multi-step process leading to CRC, which suggested that the disease result from the accumulation of mutations in oncogenes and tumor suppressor genes in colonic mucosa cells, has been largely revised following the observation that epigenetic modifications of several genes occur in the average CRC genome.2 It is believed that CRC is the consequence of the accumulation of both genetic and epigenetic genomic alterations.3
The 5-year survival rates are approximately 90% in early CRC patients but decrease to less than 10% in patients with distant metastases.3 Recurrence is observed in 10%-20% of patients with stage II CRC and 30%-40% of those with stage III CRC.4 The risk of recurrence and subsequent death due to CRC is closely related to the stage of the disease at the time of first diagnosis.5 Therefore, considerable efforts have been directed a identifying biomarker that enables early diagnosis and assists in selecting the most suitable therapeutic methods. Over the past few years, a growing body of evidence has been implying that the genetic features of the tumor determine the prognosis and response to targeted treatment.6
In this review, we will discuss the current knowledge on colorectal carcinogenesis and its clinical implications for the early detection and personalized treatment of CRC.
COLORECTAL CARCINOGENESIS
The mechanism underlying CRC pathogenesis continues to require extensive investigation in the field of cancer biology. There are at least 3 major pathways that lead to colorectal carcinogenesis: (1) the chromosomal instability (CIN) pathway, (2) the microsatellite instability (MSI) pathway, and (3) the cytosine-phospho-guanine (CpG) island methylator phenotype (CIMP) pathway.
Genomic stability is strictly controlled to maintain cell homeostasis. Any defect in the mechanisms governing this phenomenon will promote mutational processes and the selection, and clonal expansion of mutated cells, contributing to cancer progression.7 Two types of genetic instability have been identified in CRC. CIN is the most common genomic instability encompassing approximately 85% of all sporadic CRCs.5,8 CIN refers to an accelerated rate of gains or losses of whole or large portions of chromosomes, resulting in karyotypic variability from cell to cell.9,10,11 The consequence of CIN is an imbalance in chromosome number (aneuploidy), subchromosomal genomic amplifications, and a high frequency of loss of heterozygosity.10 The accumulation of a characteristic set of mutations in specific tumor suppressor genes and oncogenes is coupled with the karyotypic abnormalities observed in CIN tumors which leads to the activation of pathways critical for CRC initiation and progression.10,12,13 CRC caused by CIN is more commonly observed in distal than in proximal CRC, and usually has a poor prognosis regardless of stage and therapy.14,15,16
MSI is another form of genomic instability observed at the nucleotide level. MSI accounts for approximately 15%-20% of sporadic CRCs.5,11,14 Microsatellites are small stretches of repeated DNA sequences of 1-6 bases that are scattered throughout the human genome.12,17,18 These sequence motifs are prone to the accumulation of mutations, mainly because DNA polymerases cannot efficiently bind DNA.12,18 The mismatch repair (MMR) system corrects errors missed by the proofreading function of DNA polymerase and acts as an additional system for preserving genomic integrity.17 Because microsatellite sequences are present in the coding regions of key genes that regulate cell growth and apoptosis, defective MMR can result in frameshift mutations, ultimately creating a favorable environment for cell survival and the carcinogenic process.18 Frameshift mutations in the TGF-βRII gene are found in 90% of CRCs with MSI.19 The inactivation of the DNA MMR system may be due to an epigenetic mechanism or a mutation. Germ-line mutations in one of the MMR genes (MLH1, MSH2, MSH6, and PMS2) are present in Lynch syndrome (hereditary non-polyposis colorectal cancer, HNPCC), whereas sporadic CRCs with MSI are caused by the aberrant epigenetic methylation of MLH1.5,14 MSI tumors are associated with a proximal location, mucinous histology, poor differentiation, and a dense lymphocytic infiltration.17,20,21 Compared to patients diagnosed with CRC without MSI, patients with MSI-associated CRC have a slightly better prognosis at all stages of the disease, despite their resistance to alkylating agents and cisplatin.3,17
Although genomic instability is the most common phenomenon in CRC, epigenetic instability is also an important mechanism in the pathogenesis of CRC.14 The term "epigenetics" is used to describe those mechanisms able to modify the expression levels of selected genes without necessarily altering the primary DNA sequence.2 There are several epigenetic mechanisms that regulate gene expression: DNA methylation, histone modifications, chromatin remodeling and non-coding RNA molecules.2,7,22,23 The most widely studied epigenetic modification in humans is DNA methylation, which refers to the enzymatic addition of a methyl group to the 5' position of cytosine by DNA methyltransferases to produce 5-methyl cytosine predominantly in the CpG dinucleotide.24,25,26 DNA methylation is essential for normal embryonic development and serves an important function in X-chromosome inactivation and genomic imprinting.26,27,28 The non-cancerous mammalian cell genome contains approximately 70%-80% methylated CpGs in the non-promoter region. However, approximately 50% of CpG islands, short regions 0.5-4 kb in length possessing a rich (60%-70%) CpG content, are located in the promoter region and around the transcription start sites and are unmethylated in normal cells.16,29,30 In this unmethylated status, CpG-island containing genes are normally transcribed in the presence of the necessary transcriptional activators.17,25 In cancer cells, the transcriptional silencing of tumor-suppressor genes by CpG-island-promoter hypermethylation is key to the tumorigenic process, contributing to the development of all the typical hallmarks of a cancer cell that result from tumor-suppressor inactivation.17,25 The CIMP pathway refers to widespread promoter CpG island methylation.6,12 CIMP tumors have a distinct clinical, pathological, and molecular profile, including an association with older age, proximal location, poor differentiation, wild type p53, MSI (usually MLH1 methylation), and B-type Raf (BRAF) mutations, and are believed to arise via the serrated pathway.14,16 CIMP is helpful in distinguishing HNPCC from sporadic MSI CRC because HNPCC-related CRCs present with KRAS mutations but not BRAF mutations, whereas sporadic MSI CRCs are associated with BRAF mutation and MLH1 methylation.14,31,32
HEREDITARY COLON CANCER
Genetic mutations can be either inherited or acquired. Any genetic mutation that occurs at or before ovum fertilization is termed a germline mutation and can be transmitted from parent to offspring as an inherited defect. If the mutation occurs spontaneously in the sperm, ovum, or zygote, the affected individual's parents do not manifest the cancer phenotype, but future progeny may inherit the de novo mutation.33
It is presently estimated that 15%-30% of CRCs may have a major hereditary component given the incidence of CRC in first- or second-degree relatives, even though the etiologies are not completely understood.34 Approximately 5% of all CRC cases occur in the setting of an established familial genetic syndrome, demonstrating the profound influence of inheritable genetic mechanisms on the development of this disease (Table 1).12,35,36
Table 1.
Characteristic Features of Hereditary Colorectal Cancer Syndrome36
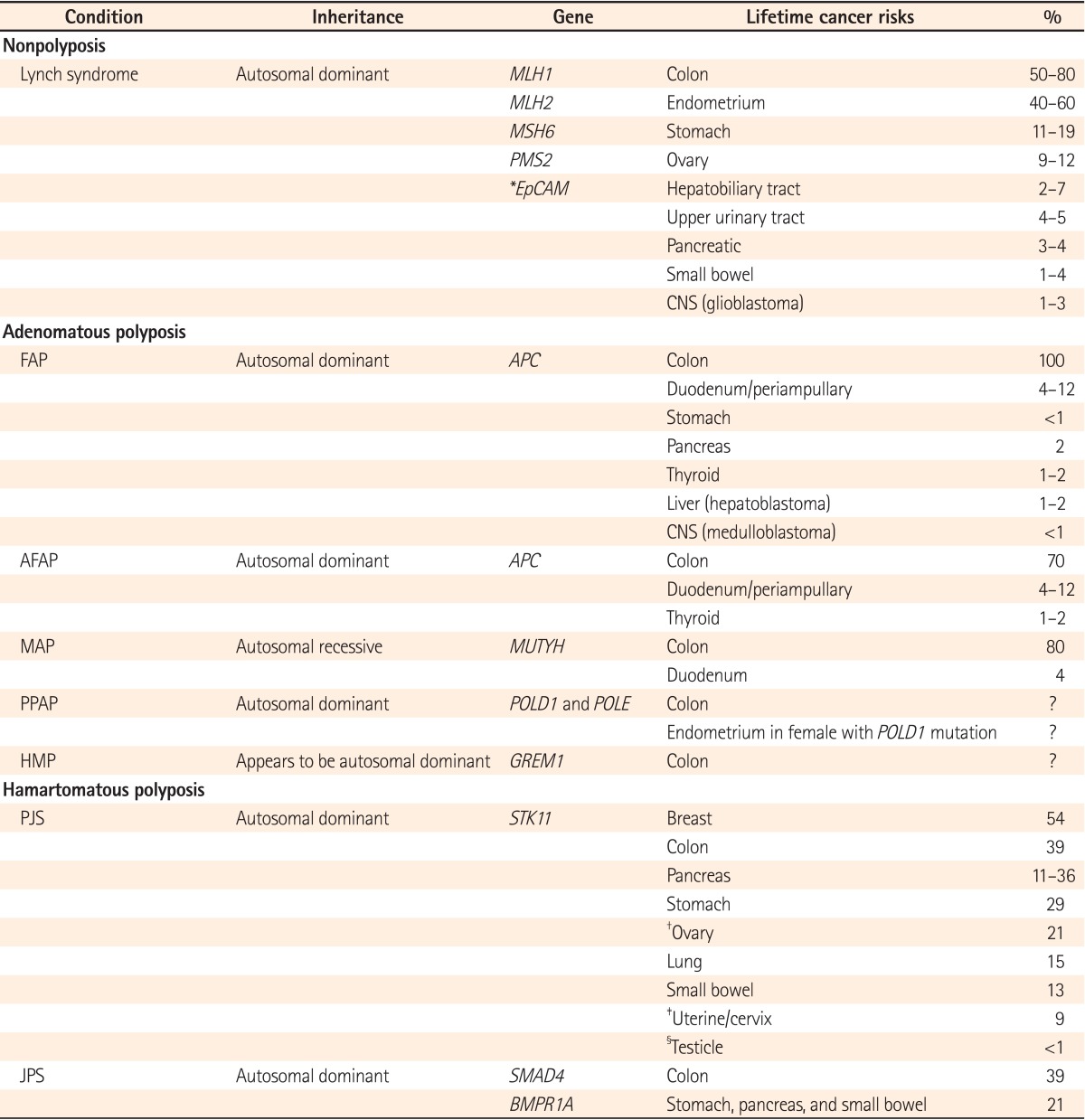
*Risks associated with EpCAM mutations are not yet known.
†Sex cord tumors with annular tubules.
‡Adenoma malignum.
§Sertoli cell tumors.
MLH1, MutL homolog 1; MSH6, MutS homolog 6; PMS2, PostMeiotic Segregation increased 2; EpCAM, epithelial cell adhesion molecule gene; FAP, familial adenomatous polyposis; APC, adenomatous polyposis coli; AFAP, attenuated familial adenomatous polyposis; MAP, MUTYH-associated polyposis; MUTYH, MutY Homolog; PPAP, polymerase proofreading associated polyposis; POLD1, Polymerase (DNA Directed), Delta 1, Catalytic subunit; POLE, Polymerase (DNA Directed), Epsilon, Catalytic Subunit; HMP, hereditary mixed polyposis; GREM1, Gremlin 1; STK11, Serine/threonine kinase 11; PJS, Peutz-Jeghers syndrome; JPS, juvenile polyposis syndrome; SMAD4, SMAD family member 4; BMPR1A, Bone Morphogenetic Protein Receptor, Type IA.
1. Lynch Syndrome/HNPCC Syndrome
Lynch syndrome (also known HNPCC) is the most common inherited colon cancer syndrome, accounting for approximately 3% of all CRCs.14,37 It is inherited in an autosomal dominant manner and characterized by an increased risk for CRC and endometrial cancer as well as a lower risk for some other cancers (ovary, gastric, small intestine, hepatobiliary tract, upper urinary tract, brain and skin).6,38,39 Lynch syndrome is formally defined as the presence of a germline mutation in one of the 4 MMR genes: MSH2, MLH1, MSH6, or PMS2, with 90% of the mutations involving either MLH1 (50%) or MSH2 (40%),18,40,41 compared to MSH2 mutataions, MLH1 mutations have been associated with an earlier age of presentation with CRC.42 PMS2 mutations are rarely detected. PMS2 carriers have been observed to present with CRC at an older age and to have a lower overall risk for CRC.35,36 Recently, germline deletions in the epithelial cell adhesion molecule gene (EpCAM), also known as the TACSTD1 gene, upstream of MSH2, were detected in a subset of families with Lynch syndrome.43 In these Lynch syndrome families, findings included hypermethylation of the MSH2 promoter without MMR gene mutations and germline deletions in the 3' region of EpCAM, resulting in EpCAM-hMSH2 fusion transcripts.36,43 One estimate suggests that EpCAM deletion is present in 6.3% of genetically proven Lynch syndrome cases.44
The Amsterdam criteria and revised Bethesda guidelines are used in clinical practice to identify individuals at risk for Lynch syndrome (Table 2).36 Studies evaluating the performance of clinical criteria in populations at high risk for Lynch Syndrome, have demonstrated that the Bethesda guidelines have a higher sensitivity compared to the Amsterdam and Amsterdam II criteria.45 Patients that meet only the Bethesda guidelines should first have their tumors assessed for MSI and/or stained for MMR protein by immunohistochemistry, followed by gene sequencing if positive. Tumors that display MSI and loss of MLH1 protein expression by immunohistochemistry should then be subjected to reflex testing for BRAF V600E mutation status and MLH1 promoter hypermethylation to help distinguish sporadic MSI tumors from Lynch syndrome (Table 3).46
Table 2.
Clinical Guidelines for the Diagnosis of Lynch Syndrome35
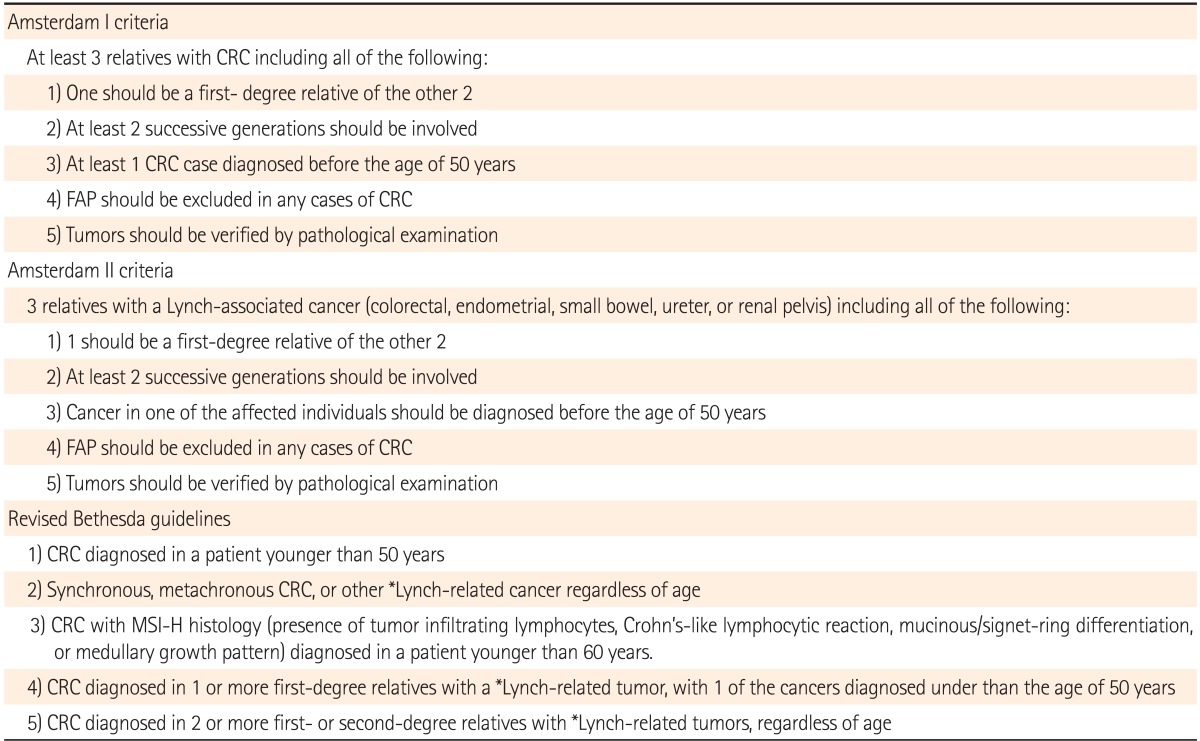
*Includes endometrial, ovarian, gastric, small bowel, urinary tract, biliary tract, pancreas, brain, and sebaceous gland.
CRC, colorectal cancer; FAP, familial adenomatous polyposis; MSI-H, microsatellite instability high.
Table 3.
Biomarkers Used in the Diagnosis of Lynch Syndrome46
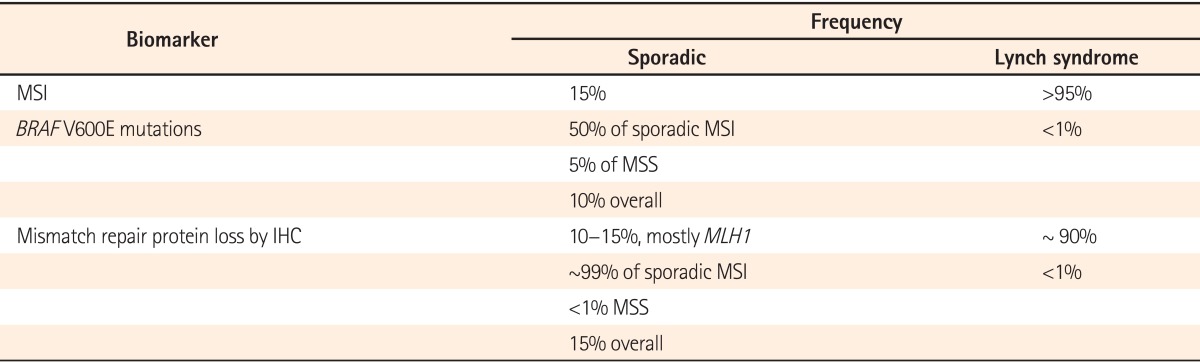
MSI, microsatellite instability; BRAF, B-type Raf; MSS, microsatellite stable; IHC, immunohistochemistry; MLH1, MutL homolog 1.
2. Familial Adenomatous Polyposis
Familial adenomatous polyposis (FAP) is the most common polyposis syndrome, with a prevalence of approximately 0.5% of all CRCs.14,35 FAP is characterized by the presence of hundreds or thousands of adenomas, and carries a 100% lifetime risk of CRC. In FAP patients, CRC develops around the age of 40 years, or 10 to 15 years after the initial development of polyposis, compared to a median age of diagnosis of 70 years for sporadic cases.35 It is inherited in an autosomal dominant fashion. Attenuated FAP is a less-severe form of the disease, characterized by an average 69% lifetime risk of CRC, an oligopolyposis of less than 100 adenomas with right-sided predominance and a flat morphology, and polyp and CRC development at a later age.14,36,47 Patients with FAP may manifest several important extracolonic malignancies or signs such as upper gastrointestinal tract polyps and carcinomas, congenital hypertrophy of the retinal pigment epithelium, desmoid tumors, thyroid cancer, and hepatoblastomas.14,48,49
FAP and attenuated FAP are caused by germline mutations in the adenomatous polyposis coli (APC) gene on chromosome 5q21, which encodes a tumor suppressor. The APC gene normally blocks the transition from the G1 to the S phase of the cell cycle. Unmutated APC induces the degradation of β-catenin and therefore functions as a negative regulator of the Wnt signaling pathway. Sustained levels of intracellular β-catenin result in the prolonged activation of the Wnt pathway in APC mutated CRC cells.17 Distinctive phenotypic correlations exist for specific mutations in the APC gene.35 Classic FAP is associated with mutations between codons 169 and 1393, with a particularly severe phenotype observed with mutations between codons 1250 and 1464.14,35,50 By contrast, the attenuated FAP phenotype results from mutations either in the 5'part of the APC gene (5'to codon 158), exon 9, or in the 3'part of the gene beyond codon 1595.51
A diagnosis of FAP is made when at least 100 colonic adenomas are identified. The identification of APC mutations in a proband confirms the diagnosis. If a mutation is found in the proband, other at-risk family members (particularly first-degree relatives) should be tested for this specific mutation.35,36 Though most patients have a family history of the disease, approximately 25% emerge as having 'de novo' APC gene mutations.6,52
3. MUTYH-Associated Polyposis
MUTYH-associated polyposis (MAP) is a more recently described hereditary cancer syndrome, and is transmitted in an autosomal recessive fashion. While the true incidence remains unknown, MAP may account for 0.5%-1% of all CRCs.14,52 The colonic phenotype of MAP mimics that of attenuated FAP (AFAP), including a propensity for proximal colonic neoplasms. Extracolonic manifestations similar to those observed in FAP/AFAP have been reported.6,35
This condition is caused by a biallelic germline mutation in the base-excision repair gene MUTYH, which is involved in defending against oxidative DNA damage.6,36 Individuals with more than 10 adenomatous polyps (particularly those with a family history of colon cancer consistent with recessive inheritance) and significant polyposis similar to that observed in AFAP/FAP who test negative for APC mutations should be tested for MAP. If a mutation is identified in the proband, siblings should be offered testing as well.35,36
4. Hamartomatous Polyposis
The hamartomatous polyposis syndromes are characterized by an overgrowth of cells native to the area in which they normally occur.53 Peutz-Jeghers syndrome (PJS) and juvenile polyposis syndrome (JPS) are 2 major hamartomatous polyposis conditions, and are both inherited in an autosomal dominant fashion. It is now known that many of these syndromes carry a substantial risk for developing CRC as well as other gastrointestinal and pancreatic cancers. The lifetime risk of CRC is 39% in patients with PJS (70%-90% lifetime risk of cancer) and 10%-38% in patients with JPS.35,54,55
PJS has been associated with germline mutations or deletions in LKB1 (STK11), a serine-threonine kinase that regulates p53-mediated apoptosis and the mammalian target of rapamycin pathway, whereas JPS is caused by mutations in SMADH4, BMPR1A, and ENG related to the transforming growth factor-beta (TGF-β)/SMAD pathway.35,56,57
The diagnostic criteria of PJS and JPS are summarized in Table 4.36,58 The correct diagnosis of JPS is complicated by its morphological similarities to hyperplastic polyps, lymphocytic infiltrates, and dysplastic components. Positive genetic testing in an affected individual helps guide testing in at-risk relatives. If no mutation is identified, first-degree relatives should undergo thorough, regular physical examinations from birth to vigilantly monitor signs and symptoms.35
Table 4.
Diagnostic Criteria of Hereditary Gastrointestinal Polyposis Syndromes58

FAP, familial adenomatous polyposis; CHRPE, congenital hypertrophy of the retinal pigment epithelium; AFAP, attenuated familial adenomatous polyposis; MAP, MUTYH-associated polyposis; PJS, Peutz-Jeghers syndrome; PJP, Peutz-Jeghers polyp; JPS, juvenile polyposis syndrome; JP, juvenile polyp.
5. Polymerase Proofreading Associated Polyposis
Polymerase proofreading associated polyposis (PPAP) is another polyposis syndrome characterized by multiple colorectal adenomas and/or early onset carcinoma, and shows autosomal dominant inheritance.59
It is associated with germline mutations in the proofreading domains of 2 DNA polymerases, POLE and POLD1.60 POLE encodes the catalytic and proofreading activities of the leading-strand DNA polymerase ε (Pol ε). POLD1 is the catalytic and proofreading subunit of the lagging-strand polymerase δ (Pol δ).61 Their proofreading (exonuclease) function detects and removes misincorporated bases in the daughter strand through failed complementary pairing with the parental strand.59 The phenotype of PPAP is similar to that of MAP or Lynch syndrome, except that tumors in PPAP are microsatellite stable.61 In particular, female POLD1 carriers have a greatly increased risk of endometrial cancer.59,62
6. Hereditary Mixed Polyposis Syndrome
Hereditary mixed polyposis syndrome (HMPS) is an autosomal dominantly inherited polyposis syndrome presenting with polyps of multiple and mixed morphologies including serrated lesions, Peutz-Jeghers polyps, juvenile polyps, conventional adenomas and CRC without any identifiable extra-colonic features.63
HMPS is associated with a 40kb duplication spanning the 3' end of the SCG5 gene and a region upstream of the GREM1 locus, subsequently increasing allele-specific GREM1 expression.63 In the colon, GREM1 is one of several bone morphogenetic protein antagonists produced by sub-epithelial myofibroblasts (ISEMFs).64 Polyposis is associated with stem cell expansion and crypt fission, reflecting a crucial homeostatic role for bone morphogenetic proteins in limiting intestinal stem cell self-renewal.65
CLINICAL APPLICATION OF GENETICS TO CRC TREATMENT AND THE PREDICTION OF PROGNOSIS
Currently, the tumor biomarkers show the greatest promise for guiding adjuvant chemotherapy with conventional drugs in patients with CRC. It is now recognized that MSI-positive CRC is a distinct subgroup of CRC with a favorable stage-adjusted prognosis compared to microsatellite stable CRC or CIN CRC.6,66 MSI CRC shows resistance to fluorouracil-based treatment, which is even potentially harmful in such cases.67 Recently, a large randomized trial of stage III CRC demonstrated improved outcomes in MSI patients treated with an irinotecan-containing regimen that included 5-fluorouracil compared with 5-fluorouracil/leucovorin alone.68 MSI CRC appear to be more responsive to irinotecan-based adjuvant chemotherapy.46,69
In the last few years, the endothelial growth factor receptor (EGFR)-targeted monoclonal antibody (mAb) cetuximab and the fully humanized mAb panitumumab, have proven to be effective in patients with metastatic CRC both as single agents and in combination with traditional fluorouracil treatment.3,46 EGFR is a transmembrane tyrosine kinase that transduces signals through two parallel intracellular pathways to activate cellular proliferation and survival. Following the dimerization of EGFR by endothelial growth factor (EGF) binding, the intracellular domain of EGFR is autophosphorylated and activates multiple downstream proteins of the RAS/RAF/MARK and PI3K/AKT pathway.17 This signaling cascade induce cell proliferation, angiogenesis, cell motility, and metastasis. EGFR-targeted mAbs block this signal pathway. Therefore, KRAS mutations are predictive of resistance to anti-EGFR mAbs, and the response is also negatively affected by NRAS, BRAF and PI3KCA mutations. Recently, randomized trials reported that the benefit of cetuximab was limited to patients with KRAS wild-type tumors, and established the use of KRAS mutational analysis as a predictive marker for anti-EGFR mAb resistance in patients with metastatic CRC.70,71 In addition, a recent meta-analysis showed that the BRAF mutation is associated with a poor response to anti-EGFR mAbs and that it is an adverse prognostic biomarker of survival in patients with metastatic CRC.72
With regard to aspirin use for CRC risk reduction,73 regular aspirin use was associated with a lower risk of BRAF wild - type colorectal cancer but not BRAF mutated cancer in a recently published cohort study. These findings suggest that BRAF mutant colon tumor cells may be less sensitive to the effect of aspirin.74 In another study, regular aspirin use after diagnosis was associated with longer survival among patients with mutated-PIK3CA CRC, but not among patients with wild-type PIK3CA cancer. The findings from this molecular pathological epidemiology study suggest that the PIK3CA mutation in CRC may serve as a predictive molecular biomarker for adjuvant aspirin therapy.75
CONCLUSIONS
Although we do not yet possess an in depth and comprehensive understanding, interesting biological insights and promising translational tools have enhanced our knowledge of genetic and epigenetic mechanisms involved in tumor progression.
Regarding inherited disease, specific genetic testing can help identify at-risk patients and at-risk relatives. Using a simple algorithmic approach based on the genetic and epigenetic mechanisms of CRC, such as MSI testing with a sequential BRAF mutation or MLH1 promoter methylation test, inheritable diseases can be distinguished from sporadic CRC. Moreover, rapid advances in our understanding of the molecular mechanisms of CRC are useful in predicting the response to chemotherapy and prognosis. The use of assays for mutant KRAS and BRAF reduced medical costs and improved patient outcomes by enabling the application of targeted therapies such as anti-EGFR mAbs, the use of which has recently increased, to selected CRC patients. However, a fair number of additional oncogenes and tumor suppressor genes with roles in the pathogenesis of CRC remains, and the identification of these genes and characterization of their contribution to cancer will be an important yet challenging task.
Footnotes
Financial support: This study was supported by Samsung Biomedical Research Institute grant (GL1-B2-091-1).
Conflict of interest: None.
References
- 1.Jung KW, Won YJ, Kong HJ, Oh CM, Lee DH, Lee JS. Cancer statistics in Korea: incidence, mortality, survival, and prevalence in 2011. Cancer Res Treat. 2014;46:109–123. doi: 10.4143/crt.2014.46.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coppede F. Epigenetic biomarkers of colorectal cancer: Focus on DNA methylation. Cancer Lett. 2014;342:238–247. doi: 10.1016/j.canlet.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 3.Coppede F, Lopomo A, Spisni R, Migliore L. Genetic and epigenetic biomarkers for diagnosis, prognosis and treatment of colorectal cancer. World J Gastroenterol. 2014;20:943–956. doi: 10.3748/wjg.v20.i4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marisa L, de Reynies A, Duval A, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsang AH, Cheng KH, Wong AS, et al. Current and future molecular diagnostics in colorectal cancer and colorectal adenoma. World J Gastroenterol. 2014;20:3847–3857. doi: 10.3748/wjg.v20.i14.3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bogaert J, Prenen H. Molecular genetics of colorectal cancer. Ann Gastroenterol. 2014;27:9–14. [PMC free article] [PubMed] [Google Scholar]
- 7.Pancione M, Remo A, Colantuoni V. Genetic and epigenetic events generate multiple pathways in colorectal cancer progression. Patholog Res Int. 2012;2012:509348. doi: 10.1155/2012/509348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michor F, Iwasa Y, Lengauer C, Nowak MA. Dynamics of colorectal cancer. Semin Cancer Biol. 2005;15:484–493. doi: 10.1016/j.semcancer.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 10.Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138:2059–2072. doi: 10.1053/j.gastro.2009.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003;3:695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- 12.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 13.Al-Sohaily S, Biankin A, Leong R, Kohonen-Corish M, Warusavitarne J. Molecular pathways in colorectal cancer. J Gastroenterol Hepatol. 2012;27:1423–1431. doi: 10.1111/j.1440-1746.2012.07200.x. [DOI] [PubMed] [Google Scholar]
- 14.Legolvan MP, Taliano RJ, Resnick MB. Application of molecular techniques in the diagnosis, prognosis and management of patients with colorectal cancer: a practical approach. Hum Pathol. 2012;43:1157–1168. doi: 10.1016/j.humpath.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Walther A, Houlston R, Tomlinson I. Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut. 2008;57:941–950. doi: 10.1136/gut.2007.135004. [DOI] [PubMed] [Google Scholar]
- 16.Bardhan K, Liu K. Epigenetics and colorectal cancer pathogenesis. Cancers (Basel) 2013;5:676–713. doi: 10.3390/cancers5020676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Armaghany T, Wilson JD, Chu Q, Mills G. Genetic alterations in colorectal cancer. Gastrointest Cancer Res. 2012;5:19–27. [PMC free article] [PubMed] [Google Scholar]
- 18.Umar A, Risinger JI, Hawk ET, Barrett JC. Testing guidelines for hereditary non-polyposis colorectal cancer. Nat Rev Cancer. 2004;4:153–158. doi: 10.1038/nrc1278. [DOI] [PubMed] [Google Scholar]
- 19.Narayan S, Roy D. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Mol Cancer. 2003;2:41. doi: 10.1186/1476-4598-2-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kloor M, Staffa L, Ahadova A, von Knebel Doeberitz M. Clinical significance of microsatellite instability in colorectal cancer. Langenbecks Arch Surg. 2014;399:23–31. doi: 10.1007/s00423-013-1112-3. [DOI] [PubMed] [Google Scholar]
- 21.Suk KT, Kim HS, Lee JH, et al. Clinicopathological characteristics of colorectal cancer according to microsatellite instability. Intest Res. 2009;7:14–21. [Google Scholar]
- 22.Schnekenburger M, Diederich M. Epigenetics offer new horizons for colorectal cancer prevention. Curr Colorectal Cancer Rep. 2012;8:66–81. doi: 10.1007/s11888-011-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu WK, Sung JJ. MicroRNA dysregulations in gastrointestinal cancers: pathophysiological and clinical perspectives. Intest Res. 2012;10:324–331. [Google Scholar]
- 24.Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol. 2005;45:629–656. doi: 10.1146/annurev.pharmtox.45.120403.095832. [DOI] [PubMed] [Google Scholar]
- 25.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 26.Daniel FI, Cherubini K, Yurgel LS, de Figueiredo MA, Salum FG. The role of epigenetic transcription repression and DNA methyltransferases in cancer. Cancer. 2011;117:677–687. doi: 10.1002/cncr.25482. [DOI] [PubMed] [Google Scholar]
- 27.Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. 2001;20:3139–3155. doi: 10.1038/sj.onc.1204341. [DOI] [PubMed] [Google Scholar]
- 28.Sharp AJ, Stathaki E, Migliavacca E, et al. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 2011;21:1592–1600. doi: 10.1101/gr.112680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anier K, Malinovskaja K, Aonurm-Helm A, Zharkovsky A, Kalda A. DNA methylation regulates cocaine-induced behavioral sensitization in mice. Neuropsychopharmacology. 2010;35:2450–2461. doi: 10.1038/npp.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 31.Domingo E, Niessen RC, Oliveira C, et al. BRAF-V600E is not involved in the colorectal tumorigenesis of HNPCC in patients with functional MLH1 and MSH2 genes. Oncogene. 2005;24:3995–3998. doi: 10.1038/sj.onc.1208569. [DOI] [PubMed] [Google Scholar]
- 32.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 33.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 34.Johns LE, Houlston RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol. 2001;96:2992–3003. doi: 10.1111/j.1572-0241.2001.04677.x. [DOI] [PubMed] [Google Scholar]
- 35.Gala M, Chung DC. Hereditary colon cancer syndromes. Semin Oncol. 2011;38:490–499. doi: 10.1053/j.seminoncol.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 36.Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010;138:2044–2058. doi: 10.1053/j.gastro.2010.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26:5783–5788. doi: 10.1200/JCO.2008.17.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boland CR, Troncale FJ. Familial colonic cancer without antecedent polyposis. Ann Intern Med. 1984;100:700–701. doi: 10.7326/0003-4819-100-5-700. [DOI] [PubMed] [Google Scholar]
- 39.Marra G, Boland CR. Hereditary nonpolyposis colorectal cancer: the syndrome, the genes, and historical perspectives. J Natl Cancer Inst. 1995;87:1114–1125. doi: 10.1093/jnci/87.15.1114. [DOI] [PubMed] [Google Scholar]
- 40.Bonis PA, Trikalinos TA, Chung M, et al. Hereditary nonpolyposis colorectal cancer: diagnostic strategies and their implications. Rockville, MD: Agency for Healthcare Research and Quality; 2007. Evidence Report/Technology Assessment No. 150. [PMC free article] [PubMed] [Google Scholar]
- 41.Peltomaki P. Lynch syndrome genes. Fam Cancer. 2005;4:227–232. doi: 10.1007/s10689-004-7993-0. [DOI] [PubMed] [Google Scholar]
- 42.Kastrinos F, Stoffel EM, Balmana J, Steyerberg EW, Mercado R, Syngal S. Phenotype comparison of MLH1 and MSH2 mutation carriers in a cohort of 1,914 individuals undergoing clinical genetic testing in the United States. Cancer Epidemiol Biomarkers Prev. 2008;17:2044–2051. doi: 10.1158/1055-9965.EPI-08-0301. [DOI] [PubMed] [Google Scholar]
- 43.Kovacs ME, Papp J, Szentirmay Z, Otto S, Olah E. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat. 2009;30:197–203. doi: 10.1002/humu.20942. [DOI] [PubMed] [Google Scholar]
- 44.Niessen RC, Hofstra RM, Westers H, et al. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chromosomes Cancer. 2009;48:737–744. doi: 10.1002/gcc.20678. [DOI] [PubMed] [Google Scholar]
- 45.Syngal S, Fox EA, Eng C, Kolodner RD, Garber JE. Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Genet. 2000;37:641–645. doi: 10.1136/jmg.37.9.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pritchard CC, Grady WM. Colorectal cancer molecular biology moves into clinical practice. Gut. 2011;60:116–129. doi: 10.1136/gut.2009.206250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burt RW, Leppert MF, Slattery ML, et al. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology. 2004;127:444–451. doi: 10.1053/j.gastro.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 48.Bjork J, Akerbrant H, Iselius L, et al. Periampullary adenomas and adenocarcinomas in familial adenomatous polyposis: cumulative risks and APC gene mutations. Gastroenterology. 2001;121:1127–1135. doi: 10.1053/gast.2001.28707. [DOI] [PubMed] [Google Scholar]
- 49.Bulow S, Bjork J, Christensen IJ, et al. Duodenal adenomatosis in familial adenomatous polyposis. Gut. 2004;53:381–386. doi: 10.1136/gut.2003.027771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dobbie Z, Spycher M, Mary JL, et al. Correlation between the development of extracolonic manifestations in FAP patients and mutations beyond codon 1403 in the APC gene. J Med Genet. 1996;33:274–280. doi: 10.1136/jmg.33.4.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23:6445–6470. doi: 10.1038/sj.onc.1207714. [DOI] [PubMed] [Google Scholar]
- 52.Bisgaard ML, Fenger K, Bulow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat. 1994;3:121–125. doi: 10.1002/humu.1380030206. [DOI] [PubMed] [Google Scholar]
- 53.Schreibman IR, Baker M, Amos C, McGarrity TJ. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol. 2005;100:476–490. doi: 10.1111/j.1572-0241.2005.40237.x. [DOI] [PubMed] [Google Scholar]
- 54.Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447–1453. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- 55.Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol. 1998;5:751–756. doi: 10.1007/BF02303487. [DOI] [PubMed] [Google Scholar]
- 56.Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 57.Howe JR, Roth S, Ringold JC, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- 58.Aretz S. The differential diagnosis and surveillance of hereditary gastrointestinal polyposis syndromes. Dtsch Arztebl Int. 2010;107:163–173. doi: 10.3238/arztebl.2010.0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Briggs S, Tomlinson I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013;230:148–153. doi: 10.1002/path.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Church JM. Polymerase proofreading-associated polyposis: a new, dominantly inherited syndrome of hereditary colorectal cancer predisposition. Dis Colon Rectum. 2014;57:396–397. doi: 10.1097/DCR.0000000000000084. [DOI] [PubMed] [Google Scholar]
- 61.Seshagiri S. The burden of faulty proofreading in colon cancer. Nat Genet. 2013;45:121–122. doi: 10.1038/ng.2540. [DOI] [PubMed] [Google Scholar]
- 62.Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jaeger E, Leedham S, Lewis A, et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat Genet. 2012;44:699–703. doi: 10.1038/ng.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tomlinson IP, Carvajal-Carmona LG, Dobbins SE, et al. Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part of the missing heritability of colorectal cancer. PLoS Genet. 2011;7:e1002105. doi: 10.1371/journal.pgen.1002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wakefield LM, Hill CS. Beyond TGFbeta: roles of other TGFbeta superfamily members in cancer. Nat Rev Cancer. 2013;13:328–341. doi: 10.1038/nrc3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 67.Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol. 2009;27:1814–1821. doi: 10.1200/JCO.2008.18.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fallik D, Borrini F, Boige V, et al. Microsatellite instability is a predictive factor of the tumor response to irinotecan in patients with advanced colorectal cancer. Cancer Res. 2003;63:5738–5744. [PubMed] [Google Scholar]
- 70.Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 71.Lyseng-Williamson KA. Cetuximab: a guide to its use in combination with FOLFIRI in the first-line treatment of metastatic colorectal cancer in the USA. Mol Diagn Ther. 2012;16:317–322. doi: 10.1007/s40291-012-0007-2. [DOI] [PubMed] [Google Scholar]
- 72.Xu Q, Xu AT, Zhu MM, Tong JL, Xu XT, Ran ZH. Predictive and prognostic roles of BRAF mutation in patients with metastatic colorectal cancer treated with anti-epidermal growth factor receptor monoclonal antibodies: a meta-analysis. J Dig Dis. 2013;14:409–416. doi: 10.1111/1751-2980.12063. [DOI] [PubMed] [Google Scholar]
- 73.Leshno A, Gat-Harlap A, Arber N. Can an aspirin a day keep the colorectal cancer away? Intest Res. 2012;10:229–234. [Google Scholar]
- 74.Nishihara R, Lochhead P, Kuchiba A, et al. Aspirin use and risk of colorectal cancer according to BRAF mutation status. JAMA. 2013;309:2563–2571. doi: 10.1001/jama.2013.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liao X, Lochhead P, Nishihara R, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012;367:1596–1606. doi: 10.1056/NEJMoa1207756. [DOI] [PMC free article] [PubMed] [Google Scholar]