

Abstract
Background:
Familial Mediterranean Fever (FMF), also inherited with autosomal recessive trait, is characterized by recurrent episodes of fever, arthritis, and serositis. Congenital Byler Syndrome (Progressive Familial Intrahepatic Cholestasis) inherited with autosomal recessive trait and characterized by defective secretion of bile acids. FMF associated Amyloid A deposition occurs in many tissues and organs, but amyloid goiter is a rare entity that leads to enlargement and dysfunction of the thyroid.
Case Report:
We present a rare case of 24 year old male patient who had liver and kidney transplantation due to Byler Syndrome and secondary amyloidosis related to FMF, diagnosed as rapidly growing large amyloid goiter. Deposits of extracellular amyloid and dense adipose metaplasia diagnostic for amyloid goiter are determined upon histopathological examination of thyroidectomy material.
Conclusion:
When goiter was detected in cases with history of systemic amyloidosis and rapidly growing goitre, amyloid goiter should be remembered at first. This case is unique since two autosomal genetic disorders are together in the same patient and important as it emphasizes the consequences of consanguineous marriage, early diagnosis and treatment compliance of FMF and the awareness of amyloid goiter in patients followed by primary care physicians and healthcare professionals.
Keywords: Amyloid goiter, Byler disease, colchicine therapy, Familial Mediterranean Fever
Familial Mediterranean Fever (FMF) is an autosomal recessive inherited disease characterized by recurrent episodes of fever, peritonitis, sinovitis, pleuritis, and rarely pericarditis and meningitis (1). It is common in ethnic groups originating from around the Mediterranean Sea. The incidence of FMF and the frequency of FMF-associated amyloidosis are quite high in Turkey (1, 2). The diagnosis of FMF is based on typical clinical findings supported by family history, laboratory findings, and identification of genetic mutations. Regular colchicine treatment reduces the frequency and/or severity of the inflammatory episodes and prevents the development of amyloidosis (1).
Congenital Byler Syndrome (progressive familial intrahepatic cholestasis) is another autosomal recessive inherited disease presenting during infancy or childhood, characterized by the defective secretion of bile acids leading to cholestatic progressive liver failure. Ursodeoxycholic acid (UDCA) and partial external biliary diversion may be palliative treatment modalities, whereas liver transplantation is curative (3).
Clinically apparent amyloid goiter rarely develops due to secondary amyloidosis associated with inflammatory diseases. Amyloid deposits in the thyroid gland were first reported by von Rokitansky in 1855, and enlargement of thyroid caused by amyloid accumulation was first described by Beckman in 1858 and termed “amyloid goiter” by Eiselberg in 1904 (4).
We aim to present a rare case of a patient with two inherited diseases, Byler Syndrome and FMF, who had received liver and kidney transplantations, and then developed a rapidly growing large amyloid goiter related to secondary amyloidosis of FMF.
CASE PRESENTATION
In May 2013, a 24-year-old man presented to our outpatient clinic with the complaints of an enlargement in his throat and feeling compression in the supine position for approximately two years. He had no additional complaints except for occasional episodes of diarrhea, secondary to the use of colchicine since childhood.
His family history revealed that his parents had consanguineous marriage and his sibling with cholestasis with a partial clinical improvement with UDCA therapy had died in early infancy. His medical history revealed that he had been followed at Pediatric Gastroenterology and General Surgery clinics with a diagnosis of congenital Byler Syndrome. At the age of 3, he underwent liver transplantation from a live donor (his mother).
A diagnosis of Familial Mediterranean Fever was made in 2000 based on the presence of typical clinical symptoms, including abdominal pain, intermittent episodes of fever, and arthritis that responded to colchicine treatment. Before the diagnosis, he had undergone unnecessary appendectomy. Genetic testing for the FMF gene was not performed. Because of persistent proteinuria, a renal biopsy was performed in 2005 and revealed secondary amyloidosis. A second renal biopsy, performed in 2007 to rule out other causes of his massive proteinuria, confirmed amyloidosis as the only cause. Although colchicine therapy was prescribed at a dose of 1 gram three times a day, he was not compliant with treatment, and his renal disease progressed to severe nephrotic syndrome. In March 2011, medical nephrectomy was performed with high dose indomethacin and he then underwent renal replacement therapy with hemodialysis. Renal transplantation from the same live donor was performed in July 2011.
Upon his admission to our clinic in May 2013, his physical examination revealed that he had a bilateral palpable, painless, and multinodular thyroid gland. According to the classification of World Health Organization, his thyroid gland was classified as Grade 3. Thyroid functions were normal. Table 1 shows his initial laboratory findings.
TABLE 1.
Patient initial laboratory findings
| Initial values | Normal range | |
|---|---|---|
| TSH (µIU/mL) | 0.423 | 0.27–4.2 |
| Free T4 (ng/dL) | 1.32 | 0.7–1.48 |
| Free T3 (pg/mL) | 2.55 | 1.71–3.71 |
| Anti-microsomal antibody (anti-TPO) (IU/mL) | 0.2 | <5.61 |
| Anti-thyroglobulin (IU/mL) | 1.71 | 0–4.11 |
| PTH (pg/mL) | 68.99 | 15–68 |
| Creatinine | 0.93 | 0.5–1.3 |
| CRP (sensitive) (mg/L) | 7.46 | 0–10 |
| Hemoglobin (g/dL) | 13.1 | 12–16 |
| ALT (U/L) | 27 | 0–41 |
TSH: thyroid-stimulating hormone; Free T4: free thyroxine; Free T3: free triiodothyronine; PTH: parathyroid hormone; CRP: C-reactive protein; ALT: alanine aminotransferase
On thyroid ultrasonography, the right lobe of the thyroid gland was 41×33×72 mm, the left lobe was 25×25×62 mm and the isthmus was 7.8 mm. There were hypo-echoic solid nodules with smooth contours at the posterior lower zone (11×8 mm) and middle-lower zone (26×13×24 mm and 16×11 mm) of the right lobe. A 10×7 mm, hypo-echoic solid nodule with smooth contours was detected in the posterior middle zone of the left lobe. A 5×3 mm anechoic cystic nodule with smooth contours and a thin wall was seen on the right lateral isthmus (Figure 1). Fine needle aspiration biopsy of the 26×13×24 mm hypo-echoic nodule at the middle lower zone of the right lobe revealed amyloid-negative staining.
FIG. 1.

Ultrasonography of the heterogeneous right lobe of the enlarged thyroid gland.
In June 2013, the patient underwent total thyroidectomy due to bilateral enlargement of the thyroid gland and compression symptoms. There were no post-operative complications and levothyroxin was initiated for the treatment of post-operative hypothyroidism. Histopathological examination of the thyroidectomy specimen revealed deposits of extracellular amyloid and dense adipose metaplasia, which are diagnostic for amyloid goiter (Figure 2). Informed consent was obtained from the patient for this case report. This case report was approved by the Baskent University Institutional Review Board (Project No: KA14/117).
FIG. 2.
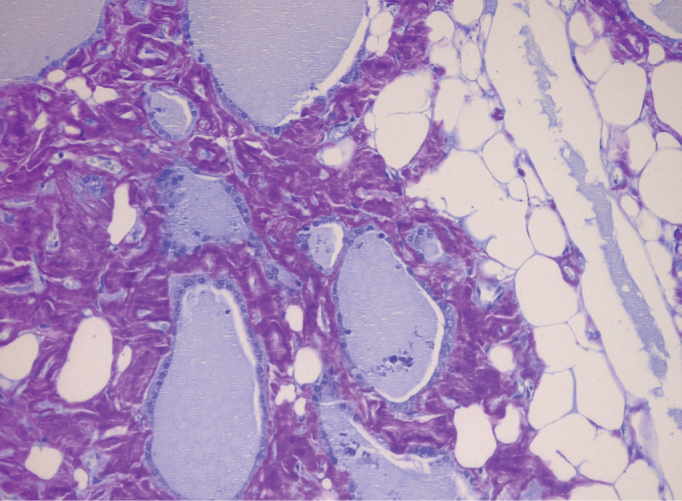
Amyloid accumulation at interfollicular sites and focal lipid infiltration in the interstitium (crystal violet, original magnification ×200)
DISCUSSION
Familial Mediterranean Fever has an autosomal recessive inheritance with gene polymorphism and can result in amyloidosis. Accumulation of amyloid A protein (AA) occurs in secondary amyloidosis associated with FMF. Colchicine treatment prevents and retards episodes as well as renal failure when it is initiated early after FMF diagnosis and used regularly (5). In a multicenter study by the Turkish FMF Working Group conducted on 2838 cases, it was reported that the mean age at onset of FMF was 9 years and the mean age at diagnosis was 16 years (6). The most frequent gene mutation (52%) in FMF patients is M694V, and 12.9% of patients are reported to have amyloidosis that might cause chronic renal failure. In this study, it was emphasized that renal failure due to systemic amyloidosis associated with FMF could be prevented by early diagnosis (6).
In communities with high rates of consanguineous marriage and FMF carriage, children are likely to be carriers or born with the illness. As the rate of FMF carriage is high in the Turkish population and consanguineous marriages are common, the risk of multiple inherited diseases is high (2, 6). In our case, the coexistence of Byler Syndrome and FMF disease led to double organ failure, and liver and kidney transplantations were required. Although our case was diagnosed with FMF when he was 11, unfortunately, amyloidosis could not be prevented due to the irregular use of colchicine. The diagnosis of Byler Syndrome had been based on clinical findings and histopathological features on liver biopsy, but was not confirmed by genetic analysis in our patient. To the best of our knowledge, there is no previously published case of concurrent FMF and Byler Syndrome in the literature, which makes our patient a unique case.
Amyloid goiter can occur in association with primary or secondary amyloidosis. Amyloid accumulation was demonstrated in the thyroid gland of 80% of patients with secondary amyloidosis in autopsy examination (4). Systemic amyloidosis was detected in 84 (7.6%) of 1100 FMF patients followed in a university clinic in Turkey; amyloid goiter was detected in only three (0.27%) of them (7).
Amyloid goiter is usually asymptomatic. Although thyroid functions were normal in our case, the patient complained of his enlarged thyroid, which caused compression on his throat. The histopathological findings after thyroidectomy were consistent with amyloid goiter and adipose metaplasia associated with systemic amyloidosis. (8–10).
In conclusion, when goiter is detected in patients with a history of systemic amyloidosis and especially in cases with a rapidly growing goiter without any functional abnormalities, amyloid goiter should be considered first. Fine needle aspiration biopsy may be inadequate, as was the case in our patient. Histopathological examination of thyroidectomy material is required for a definitive diagnosis (8–10).
We think that this case is unique and has important implications for primary care physicians and healthcare professionals since: 1) it draws attention to the risk of concurrent hereditary disorders as a consequence of consanguineous marriage, 2) it emphasizes the importance of early diagnosis and continuous treatment of FMF, and 3) it increases awareness regarding the diagnosis of amyloid goiter in patients with FMF.
Footnotes
Ethics Committee Approval: Ethics committee approval was received for this report from Başkent University Institutional Review Board.
Informed Consent: Written informed consent was obtained from the patients for the publication of this case report and any accompanying images.
Peer-review: Externally peer-reviewed.
Author contributions: Concept - Ö.C., A.D.Ü., H.P., E.D., O.T.; Design - Ö.C., A.D.Ü., H.P., E.D., O.T.; Supervision - Ö.C., A.D.Ü., H.P., E.D., O.T.; Resource - Ö.C., A.D.Ü., H.P., E.D., O.T.; Materials - Ö.C., A.D.Ü., E.D., O.T.; Data Collection&/or Processing - Ö.C., A.D.Ü., H.P., E.D., O.T.; Analysis&/or Interpretation - Ö.C., A.D.Ü., H.P., E.D., O.T.; Literature Search - Ö.C., A.D.Ü.; Writing - Ö.C., A.D.Ü., H.P.; Critical Reviews - Ö.C., A.D.Ü., H.P., E.D., O.T.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this report has received no financial support.
REFERENCES
- 1.Koné-Paut I, Hentgen V, Touitou I. Current data on familial Mediterranean fever. Joint Bone Spine. 2011;78:111–4. doi: 10.1016/j.jbspin.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Chetrit E, Touitou I. Familial Mediterranean Fever in the world. Arthritis Rheum. 2009;63:1447–53. doi: 10.1002/art.24458. [DOI] [PubMed] [Google Scholar]
- 3.Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2012;36(Suppl 1):S26–35. doi: 10.1016/S2210-7401(12)70018-9. [DOI] [PubMed] [Google Scholar]
- 4.Villa F, Dionigi G, Tanda ML, Rovera F, Boni L. Amyloid goiter. Int J Surg. 2008;6(Suppl 1):S16–18. doi: 10.1016/j.ijsu.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 5.Sevoyan MK, Sarkisian TF, Beglaryan AA, Shahsuvaryan GR, Armenian HK. Prevention of amyloidosis in familial Mediterranean fever with colchicine: A case-control study in Armenia. Med Princ Pract. 2009;18:441–6. doi: 10.1159/000235892. [DOI] [PubMed] [Google Scholar]
- 6.Tunca M, Akar S, Onen F, Ozdogan H, Kasapcopur O, Yalcinkaya F, et al. Familial Mediterranean fever (FMF) in Turkey: results of a nationwide multicenter study. Medicine (Baltimore) 2005;84:1–11. doi: 10.1097/01.md.0000152370.84628.0c. [DOI] [PubMed] [Google Scholar]
- 7.Altiparmak MR, Pamuk ON, Pamuk GE, Apaydin S, Ataman R, Serdengeçti K. Amyloid goitre in familial Mediterranean fever: Report on three patients and review of the literature. Clin Rheumatol. 2002;21:497–500. doi: 10.1007/s100670200122. [DOI] [PubMed] [Google Scholar]
- 8.Turhan İyidir Ö, Altay M, Konca Degertekin C, Altınova A, Karakoç A, Ayvaz G, et al. Diffuse amyloid deposition in thyroid gland: A cause for concern in familial Mediterranean fever. Amyloid. 2012;19:161–2. doi: 10.3109/13506129.2012.687701. [DOI] [PubMed] [Google Scholar]
- 9.Ori Y, Halpern M, Sadov R, Feinmesser R, Ramadan R, Korzets A. Familial Mediterranean fever, an amyloid thyroid goiter and an enlarged parathyroid gland. Isr Med Assoc J. 2012;14:232–3. [PubMed] [Google Scholar]
- 10.Eren R, Sayar H, Çiralik H, Ezberci F. [Diffuse lipid infiltration and squamous metaplasia accompanying amyloid goiter: Case report] Turk Patoloji Derg. 2013;29:146–9. doi: 10.5146/tjpath.2013.01167. [DOI] [PubMed] [Google Scholar]