

Abstract
Birdshot chorioretinopathy (BSCR) is a rare form of autoimmune uveitis that can lead to severe visual impairment. Intriguingly, >95% of cases carry the HLA-A29 allele, which defines the strongest documented HLA association for a human disease. We have conducted a genome-wide association study in 96 Dutch and 27 Spanish cases, and 398 unrelated Dutch and 380 Spanish controls. Fine-mapping the primary MHC association through high-resolution imputation at classical HLA loci, identified HLA-A*29:02 as the principal MHC association (odds ratio (OR) = 157.5, 95% CI 91.6–272.6, P = 6.6 × 10−74). We also identified two novel susceptibility loci at 5q15 near ERAP2 (rs7705093; OR = 2.3, 95% CI 1.7–3.1, for the T allele, P = 8.6 × 10−8) and at 14q32.31 in the TECPR2 gene (rs150571175; OR = 6.1, 95% CI 3.2–11.7, for the A allele, P = 3.2 × 10−8). The association near ERAP2 was confirmed in an independent British case–control samples (combined meta-analysis P = 1.7 × 10−9). Functional analyses revealed that the risk allele of the polymorphism near ERAP2 is strongly associated with high mRNA and protein expression of ERAP2 in B cells. This study further defined an extremely strong MHC risk component in BSCR, and detected evidence for a novel disease mechanism that affects peptide processing in the endoplasmic reticulum.
INTRODUCTION
Birdshot chorioretinopathy (BSCR; MIM605808) is a rare ocular disorder that has a very strong HLA association, with >95% of cases carrying the HLA-A29 allele (1–3). BSCR manifests as a severe progressive intraocular inflammation of the posterior eye segment, typically leading to extensive retinal atrophy and blindness (4,5). The disease is most often observed in middle-aged and elderly individuals of European descent and has a slight female predominance (3,6). Current treatment strategies include various immunosuppressive medications but fail to prevent or cease the retinal atrophy. HLA-A29 is relatively common in European populations, but only a tiny subset of HLA-A29-positive individuals develops BSCR (6). It is therefore assumed that other genes than HLA-A and additional exogenous factors are involved in the development of the disease.
RESULTS
To identify BSCR susceptibility genes and to fine-map the association within the MHC region, we conducted a genome-wide association study in 96 Dutch and 27 Spanish BSCR cases and 398 unaffected Dutch and 380 Spanish controls from European ancestry. After quality control and genome-wide SNP imputation, we analyzed a total of 9,932,851 SNPs in 117 cases and 693 controls (Materials and Methods; Supplementary Material, Fig. S1). We did not see evidence for population stratification (λGC = 0.96; Supplementary Material, Fig. S2).
The strongest association signal was located within the HLA class I region for rs142115394 (P = 6.3 × 10−17). We next imputed and tested classical alleles and amino acid polymorphisms in HLA-A, HLA-B, HLA-C, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1 and HLA-DRB1 following a recently described procedure (7). The strongest association of all variants tested mapped to the classical HLA-A*29:02 allele (P = 6.6 × 10−74) (Fig. 1 and Table 1; Supplementary Material, Table S1). We found HLA-A*29:01 to be nominally associated (P = 0.02) but not significant once the A*29:02 effect was accounted for (P = 0.093), indicating that the classical A29 effect can be primarily attributed to the A*29:02 allele. No polymorphic amino acid position could explain the data better than A*29:02 (Supplementary Material, Table S2). Controlling for the HLA-A*29:02 effect, no other classical HLA-A allele was significantly associated (best adjusted P = 3.0 × 10−4 for HLA-A*30:01).
Figure 1.
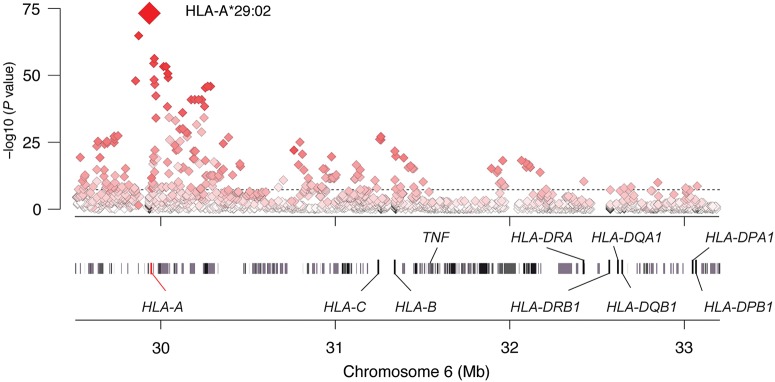
Association tests within the MHC region to birdshot chorioretinopathy. The strongest MHC signal mapped to HLA-A*29:02 allele. The shading depicts the strength of the correlation (r2) between HLA-A*29:02 (red diamond) and the SNPs tested in the region. Gene positions are obtained from the human genome build 37 (GRCh37/hg19).
Table 1.
HLA-A29:02 and newly associated loci identified in this study
| Region | Dutch cohort |
Spanish cohort |
Mega-analysis | British cohort (replication) | Meta-analysis | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele | Case MAF | Control MAF | OR (95% CI) | P | Case MAF | Control MAF | OR (95% CI) | P | OR (95% CI) | P | Case MAF | Control MAF | OR (95% CI) | P | OR (95% CI) | P | |
| HLA | HLA-A29:02 | 0.422 | 0.026 | 303.0 (124.4, 845.6) | 2.0 × 10−63 | 0.548 | 0.084 | 25.4 (8.9, 94.9) | 9.3 × 10−12 | 157.5 (91.6, 272.6) | 6.6 × 10−74 | ||||||
| ERAP2-LNPEP | rs7705093-T | 0.618 | 0.405 | 2.6 (1.8, 3.7) | 2.7 × 10−7 | 0.554 | 0.399 | 2.0 (1.0, 3.7) | 3.8 × 10−2 | 2.3 (1.7, 3.1) | 8.6 × 10−8 | 0.621 | 0.442 | 2.0 (1.1, 3.6) | 0.011 | 2.2 (1.7–2.9) | 1.7 × 10−9 |
| TECPR2 | rs150571175-A | 0.075 | 0.017 | 5.9 (2.5, 14.2) | 6.4 × 10−5 | 0.183 | 0.041 | 6.0 (2.2, 16.5) | 6.0 × 10−4 | 6.1 (3.2, 11.7) | 3.2 × 10−8 | 0.033* | 0.036* | 1.0 (0.2–1.4)* | 1* | 4.6 (2.6–8.3)* | 1.6 × 10−7* |
*This SNP failed genotyping by Sanger sequencing, so replication genotyping for this locus was at rs7159505, which had r2 = 0.83 as calculated from Go NL.
Outside the MHC region, a novel association was found at 5q15 for rs7705093 (P = 8.6 × 10−8) within the LNPEP gene encoding leucyl/cystinyl aminopeptidase (Table 1). This association was further strengthened in 30 British cases and 2793 control samples (combined meta-analysis P = 1.7 × 10−9). Due to linkage disequilibrium (LD), several other SNPs across the locus also showed strong association (Supplementary Material, Fig. S3). This locus includes the three members of the M1 family of aminopeptidases; ERAP1, ERAP2 and the LNPEP gene (8). ERAP1 and ERAP2 encode endoplasmic reticulum aminopeptidase 1 and 2, respectively, and are both involved in antigen processing and presentation by HLA class I molecules to T cells (8–10). The LNPEP gene encodes leucyl/cystinyl aminopeptidase that specifically functions in peptide trimming for cross-presentation in dendritic cells (11).
To explore the biological relevance of the associated variation in the 5q15 locus, we first investigated whether the associated SNPs also influence expression levels of these three genes using the expression quantitative trait loci (eQTL) database GeneVar (12). The strongest QTL for ERAP2 expression in LCL cells (P = 2.0 × 10–114 from Genevar) is observed for rs10044354 (P = 1.2 × 10−7 for association with BSCR), which is almost in perfect LD with the lead SNP rs7705093 (r2 = 0.98 in the controls; Supplementary Material, Fig. S4). These SNPs are statistically indistinguishable with respect to BSCR risk (P > 0.2). The rs10044354 (and SNPs in LD with rs10044354) appeared to have no significant impact on LNPEP or ERAP1 expression (P > 0.2 from GeneVar).
Subsequently, we tested if this variant is also correlated to protein levels of ERAP2 in B-cell lines from individuals from the Centre d'Etude du Polymorphisme Humain (CEPH) panel (13) and five BSCR patients. Indeed, homozygous carriers for the rs10044354 C allele showed little or no ERAP2 protein expression in a quantitative western blot analysis, whereas hetero- or homozygous carriers of the rs10044354T risk allele showed higher protein levels of ERAP2 (P = 0.009) in cases and controls (Fig. 2). In contrast, we did not observe an effect of this SNP on LNPEP expression, consistent with the mRNA findings from GeneVar (Fig. 2, bottom).
Figure 2.

Expression of ERAP2 and LNPEP in CEPH control and BSCR according to rs10044354 genotype. B-cell lines from individuals from the CEPH panel (13) and five BSCR patients were lysed and ERAP2 and LNPEP expression was assessed after SDS–PAGE and western blotting with antibodies to ERAP2 and LNPEP. The endogenous levels of both α- and β-tubulin total protein were analyzed as a loading control. The horizontal lines indicate the means. Kruskal–Wallis test with Dunn's multiple-comparison post hoc test was used to assess differences in the levels of ERAP2 between B-cell line groups according to rs10044354 genotype.
Another novel association was detected at 14q32.31 in the TECPR2 gene (rs150571175, P = 3.2 × 10−8) (Table 1; Supplementary Material, Fig. S3). However, this association could not be confirmed in the British cohort (P = 1.0; combined meta-analysis P = 1.6 × 10−7). The protein encoded by TECPR2 (tectonin beta-propeller repeat-containing 2) interacts with Atg8 orthologs and functions in autophagosome accumulation for autophagy (14).
Because BSCR is considered an autoimmune disease and shares signatures of T helper 17-cell responses with other autoimmune disorders (15–19), we hypothesized that known risk loci for other autoimmune diseases may also be associated with BSCR. Therefore, we tested 289 bona fide non-HLA SNP associations in autoimmune disorders (Supplementary Material, Table S3). For all BSCR Dutch and Spanish cases and controls, we computed a weighted genetic risk score based on the observed number of risk alleles and the known effect sizes for each autoimmune disease separately. We performed a similar test for SNPs associated to either height or LDL levels as two control traits. After correction for multiple testing, we obtained no significant association between increased risk scores and BSCR risk for any of the autoimmune diseases tested, except for wGRS Psoriasis (higher for BSCR) and LDL (lower in BSCR) (Supplementary Material, Table S4). However, these associations were only nominally significant in the Dutch cohort but not in the Spanish cohort. Although this would suggest that BSCR shares no additional susceptibility loci with other autoimmune diseases, it is possible that the sample size and power was simply too low to detect such an overlap.
DISCUSSION
To date, the association of HLA-A29 with BSCR is the strongest association described between an HLA class I allele and human disease (2,6). Although this association has been known for over three decades (1), lack of understanding of the contribution of HLA-A29 to the pathogenesis of BSCR, impeded it to become an absolute criterion for diagnosis (22). HLA-A29 can be subdivided in at least 17 subtypes (23), but most common in Caucasians are the subtypes HLA-A*29:01 and HLA-A*29:02 that have both been most associated with BSCR (24). However, the imputation analyses of the HLA region revealed that the nominal association of HLA-A*29:01 disappeared once the A*29:02 effect was accounted for, indicating that the classical A29 effect in BSCR can be primarily attributed to the A*29:02 allele and comes with unusually large effect size in terms of explained variance (Nagelkerke's index = r2[controlled for top 4 PCs and cohorts] = 0.595). However, since HLA-A29 is relatively common in European populations (7%) and very few HLA-A29-positive individuals develop BSCR (6), other additional genetic or exogenous factors are required for the development of the disease. Here, we provide evidence for an association with ERAP2 and speculate about a possible association of TECPR2.
The ERAP2 gene is located on chromosome 5 and is positioned between the closely related ERAP1 and LNPEP genes (8,9). The ERAP2 gene encodes for an aminopeptidase located in the endoplasmic reticulum (ER) that is involved in the final processing and presentation of antigenic precursors, together with the closely related and better characterized ERAP1 (10). In fact, ERAP2 and ERAP1 were shown to form a heterodimer to combine their restricted sequence specificities for peptide trimming (10). Notably, some antigenic peptides, including the immunodominant epitope derived from HIV gag protein, have been shown to be solely dependent upon ERAP2 trimming (25).
Recent genome-wide association studies have implicated a role for ERAP1 and ERAP2 in several HLA-associated autoimmune diseases such as psoriasis (26), ankylosing spondylitis (20,21) and Crohn's disease (27), or juvenile idiopathic arthritis (28), suggesting an important functional role for these genes in the immunopathology of autoimmune diseases. Of note, all these ERAP1- or ERAP2-associated diseases share a major HLA class I association that contrasts the major HLA class II association found in many other autoimmune diseases not associated to ERAP1 or ERAP2. In this respect, our study substantiates an emerging concept as we also found a strong link between ERAP2 and BSCR, an autoimmune disease affecting the eye that manifests almost exclusively in HLA-A29-positive Caucasians.
Similar to HLA genes, ERAP2 has been shown to undergo balancing selection, which maintains two main haplotypes A and B, with highly differential protein expression due to alternative mRNA splicing (29). We discovered that the SNPs associated with BSCR at 5q15 also serve as tags for these haplotypes, with high expression levels of ERAP2 in BSCR patients and no or low expression in homozygous negative healthy individuals. Based on our results, and given the fact that ERAP2 has nonoverlapping trimming capacities (10) with ERAP1, it is likely that ERAP2 has a specific contribution to the immunopathology of BSCR, by differential generation or destruction of immunodominant epitopes capable of binding to HLA-A29. Modulation of the antigen processing and presentation pathway by targeting ERAP2 in BSCR is particularly of interest since, in contrast to ERAP1, a rather high frequency of ERAP2-deficient individuals is maintained in the population without any clear adverse effects on their immunity and healthy (29). However, the efficacy of selective inhibition of ERAP2 in BSCR, while remaining ERAP1 function, remains to be clarified and involves the use of novel inhibitors that targets this family of pleiotropic aminopeptidases (30).
We found a significant association for TECPR2 in the Dutch and Spanish cohorts, but these findings could not be replicated in British patients. TECPR2 encodes for TECPR2 (tectonin beta-propeller repeat-containing 2) that interacts with Atg8 orthologs and is a positive regulator of autophagy and functions in autophagosome accumulation (14). A mutation in the TECPR2 gene was recently implicated in hereditary spastic paraparesis (31). The eye maintains an unconventional form of autophagy that is crucial for the function and health of the photoreceptors in the retina (32,33). Interestingly, interfering with autophagy by rapamycin, a potent inducer of autophagy, can paradoxically reduce of exacerbate uveitis (34,35). Moreover, autophagy-related genes have also been associated with other autoimmune diseases, including SLE and Crohn's disease (36,37). Further investigation of the association of the TECPR2 gene in other BSCR populations is necessary to clarify its possible contribution to the pathogenesis of BSCR.
In conclusion, we identified HLA-A*29:02 as the principal MHC association and report variants near the ERAP2 gene at 5q15 that predispose to BSCR. Expression studies indicate that the disease-associated variants at 5q15 correlate to mRNA and protein expression levels of ERAP2, suggesting that differential peptide processing and antigen presentation to T cells is an essential disease mechanism for BSCR.
MATERIALS AND METHODS
Subject collection
A total of 96 unrelated Dutch BSCR cases, 27 Spanish BSCR cases and 398 unrelated Dutch healthy controls and 380 Spanish controls, all from European ancestry, are enrolled in this study. BSCR diagnosis was based on criteria established by international consensus (22). Dutch cases were recruited at the Department of Ophthalmology at the University Medical Center Utrecht, the Eye Hospital Rotterdam and Radboud University Nijmegen Medical Center, the Netherlands. These three institutes represent the largest registry of BSCR patients in the Netherlands; we estimate that >90% of all BSCR cases in the Netherlands are included in this cohort. Spanish cases were recruited at the department of ophthalmology at the Hospital Clinico San Carlos, Madrid, Hospital Clinico San Cecilio, Granada, Hospital de Cruces, Bilbao, Hospital Clinic Barcelona, Hospital Carlos Haya, Malaga and Hospital Universitario, Leon. Dutch controls were collected at UMC Utrecht and Spanish controls were recruited from the National bloodbank. For replication, we included an additional 30 UK BSCR cases recruited at the department of ophthalmology at the Moorfields Eye Hospital NHS Foundation Trust, London. As controls, we used genotype data from the UKBS and 58BC controls, which are part of the WTCCC study (38). The UKBS cohort contained 1397 control individuals selected from a UK sample of blood donors, and the 58BC contained 1396 control individuals from the 1958 British Birth Cohort (39). This study was performed in compliance with the guidelines of the Declaration of Helsinki and has the approval of the local Institutional Review Boards. Study details were explained to all cases and controls, and the collection of blood was carried out after obtaining written informed consent from each participant.
Genotyping, quality control and association analysis
All Dutch and Spanish samples were genotyped using the Ilumina Human OmniExpress BeadChip, which contains 730 525 SNPs. Data for the British 1958 Birth Cohort and UKBS were obtained from the European Genome-Phenome Archive at https://www.ebi.ac.uk/ega/. These individuals were genotyped using the Affymetrix GeneChip 500K platform and the Illumina Immunochip platform. We used PLINK (40) to clean the genotype data using standard quality control parameters: we removed SNPs with low frequency (<0.02), out of Hardy–Weinberg equilibrium (P < 10−6), or with excess missingness (>5%). We also checked for relatedness and inconsistent gender. Principal components analysis was performed to detect population stratification. Overall, we excluded two Dutch and four Spanish cases, and four Dutch and 85 Spanish controls, and kept a total of 393 831 SNPs for subsequent analyses. Imputation of untyped SNPs was performed using Minimac (41). As reference panel, we used the 998 phased haplotypes from the Genome of the Netherlands Project release 4 encompassing 19 763 454 SNPs (42). We included the top four principal components as covariates in logistic regression models using PLINK (version 1.08) and R (version 2.11.1). SNPs with MAF <1% or with imputation quality (info score) <0.01 were excluded from analyses. Genome-wide distribution of the test statistic indicated no evidence for population stratification (QQ plots Supplementary Material, Fig. S2). As previously described (7), we imputed classical alleles and amino acid polymorphisms in HLA-A, HLA-B, HLA-C, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1 and HLA-DRB1 at a four-digit resolution, as well as an additional 3,117 SNPs across the MHC, using a large reference panel of 2,767 individuals of European descent. We imputed cases and controls together. For the association analyses in the MHC, we assessed the significance of the improvement in fit by calculating the deviance (defined as −2 × the log likelihood), which follows a χ2 distribution.
Protein expression assays
We assessed ERAP2 and LNPEP protein levels in EBV transformed B cells (EBV-LCL), from 19 individuals from the CEPH panel and EBV-LCLs generated from peripheral blood mononuclear cells of five BSCR patients, by western blotting (13). From the CEPH panel, we selected six homozygotes for the C allele, six heterozygotes and seven homozygotes for the T allele (NA07022, NA07029, NA07056, NA07345, NA07357, NA10838, NA10846, NA10854, NA10863, NA11839, NA12003, NA12056, NA12144, NA12145, NA12239, NA12249, NA12812, NA12815 and NA12872). Protein extracts (20 μg/lane) from EBV-LCL cells were separated on a 4–12% NuPageBis–Tris gel (Invitrogen) and transferred to a PVDF membrane. Proteins were detected using a 1:2500 dilution of primary antibody [goat anti-ERAP2 polyclonal antibody (AF3830, R&D Systems), mouse anti-ERAP2 polyclonal (ab69037, Abcam), sheep anti-LNPEP polyclonal antibody (AF6386, R&D Systems) and anti-α/β-tubulin monoclonal prepared in Rabbit (#2148, Cell Signaling)]. Anti- and anti-rabbit secondary antibodies conjugated to Alexa Fluor 680 (Molecular Probes; 1:2500) and anti-goat secondary antibody conjugated to IRDye800 (Li-Cor; 1:2500) were used to probe primary antibodies. Protein bands were detected and quantified by western blotting with the Odyssey system (Li-Cor). The ratio of the intensity of the ERAP2-α/β-tubulin band of CC-homozygotes to CT-heterozygotes and TT-homozygotes was calculated using ImageJ.
Transcriptional analysis
eQTL analysis was performed based on data available from the Sanger Institute GENEVAR project for lymphoblastoid-cell lines from 726 HapMap 3.0 CEU individuals and three tissue types (LCL cell type was used in the analysis) collected from 856 healthy female twins of the MuTHER resource (12,43). Differences in the levels of ERAP2 in B-cell line between groups according to rs10044354 genotype were analyzed using non-parametric one-way ANOVA; Kruskal–Wallis test with Dunn's multiple-comparison post hoc test in GraphPad Prism 5 software. Statistical significance was accepted at a significance level of P < 0.05.
Genetic risk score
To test the cumulative effect of validated SNPs associated with other autoimmune diseases (ankylosing spondylitis, rheumatoid arthritis, Crohn's disease, ulcerative colitis, psoriasis, vitiligo, type 1 diabetes and multiple sclerosis) (20,26,44–49), we computed a weighted genetic risk score in all cases and controls of our study. We summed the estimated dosages of the known risk alleles (for a given autoimmune disease) weighted by the natural log of the published odds ratios (Supplementary Material, Table S3). We tested the genetic risk scores for each disease as a continuous variable for association to BSCR status using logistic regression, including the top four principal components as covariates. We corrected for multiple testing of 10 traits in total (including height (50–52) and LDL (53,54) as a negative control).
SUPPLEMENTARY MATERIAL
FUNDING
Genotyping of Dutch control sample was supported by NIH/NIMH MH090553 to R.A.O. P.I.W.d.B. is a recipient of a VIDI Award from the Netherlands Organization for Scientific Research (N.W.O. project number 016.126.354).
WEB RESOURCES
Summary statistics of all SNPs included in this study are available for download through: http://cmm.umcutrecht.nl/publications/koeleman/birdshot; Beagle Genetic Analysis Software, http://faculty.washington.edu/browning/beagle/beagle.html PLINK statistical software, http://pngu.mgh.harvard.edu/~purcell/plink/; R project, http://www.r-project.org/; ImageJ, http://rsbweb.nih.gov/ij/index.html; GeneVar, http://sanger.ac.uk/resources/software/genevar/; The 1000 Genomes Project, http://www.1000genomes.org/; LocusZoom, http://csg.sph.umich.edu/locuszoom/; SNP2HLA imputation pipeline, http://www.broadinstitute.org/mpg/snp2hla/; Ensemble Genome Browser, http://www.ensembl.org/.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all participants involved in this study, including the Birdshot Uveitis Society (http://birdshot.org.uk/), The Moorfield Biobank (UK) and Spanish Birdshot Group (GENUVE). This study was supported by the combined grants from the Dr F.P. Fischer Stichting, Amersfoort; the Algemene Nederlandse Vereniging Ter Voorkoming Van Blindheid, Doorn; the Landelijke Stichting Voor Blinden en Slechtzienden, Utrecht, the Stichting Nederlands Oogheelkundig Onderzoek (SNOO), Rotterdam and the Blindenpenning Stichting, Amsterdam, the Netherlands.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Nussenblatt R.B., Mittal K.K., Ryan S., Green W.R., Maumenee A.E. Birdshot retinochoroidopathy associated with HLA-A29 antigen and immune responsiveness to retinal S-antigen. Am. J. Ophthalmol. 1982;94:147–158. doi: 10.1016/0002-9394(82)90069-1. [DOI] [PubMed] [Google Scholar]
- 2.Priem H.A., Kijlstra A., Noens L., Baarsma G.S., De Laey J.J., Oosterhuis J.A. HLA typing in birdshot chorioretinopathy. Am. J. Ophthalmol. 1988;105:182–185. doi: 10.1016/0002-9394(88)90183-3. [DOI] [PubMed] [Google Scholar]
- 3.Shah K.H., Levinson R.D., Yu F., Goldhardt R., Gordon L.K., Gonzales C.R., Heckenlively J.R., Kappel P.J., Holland G.N. Birdshot chorioretinopathy. Surv. Ophthalmol. 2005;50:519–541. doi: 10.1016/j.survophthal.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Levinson R.D., Gonzales C.R. Birdshot retinochoroidopathy: immunopathogenesis, evaluation, and treatment. Ophthalmol. Clin. North Am. 2002;15:343–350. doi: 10.1016/s0896-1549(02)00031-7. [DOI] [PubMed] [Google Scholar]
- 5.Rothova A., Van Schooneveld M.J. The end stage of birdshot retinochoroidopathy. Br. J. Ophthalmol. 1995;79:1058–1059. doi: 10.1136/bjo.79.11.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wee R., Papaliodis G. Genetics of birdshot chorioretinopathy. Semin. Ophthalmol. 2008;23:53–57. doi: 10.1080/08820530701745231. [DOI] [PubMed] [Google Scholar]
- 7.Jia X., Han B., Onengut-Gumuscu S., Chen W.M., Concannon P.J., Rich S.S., Raychaudhuri S., de Bakker P.I. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS ONE. 2013;8:e64683. doi: 10.1371/journal.pone.0064683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evnouchidou I., Papakyriakou A., Stratikos E. A new role for Zn(II) aminopeptidases: antigenic peptide generation and destruction. Curr. Pharm. Des. 2009;15:3656–e63670. doi: 10.2174/138161209789271816. [DOI] [PubMed] [Google Scholar]
- 9.Haroon N., Inman R.D. Endoplasmic reticulum aminopeptidases: Biology and pathogenic potential. Nat. Rev. Rheumatol. 2010;6:461–467. doi: 10.1038/nrrheum.2010.85. [DOI] [PubMed] [Google Scholar]
- 10.Saveanu L., Carroll O., Lindo V., Del Val M., Lopez D., Lepelletier Y., Greer F., Schomburg L., Fruci D., Niedermann G., et al. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat. Immunol. 2005;6:689–697. doi: 10.1038/ni1208. [DOI] [PubMed] [Google Scholar]
- 11.Saveanu L., Carroll O., Weimershaus M., Guermonprez P., Firat E., Lindo V., Greer F., Davoust J., Kratzer R., Keller S.R., et al. IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science. 2009;325:213–217. doi: 10.1126/science.1172845. [DOI] [PubMed] [Google Scholar]
- 12.Stranger B.E., Montgomery S.B., Dimas A.S., Parts L., Stegle O., Ingle C.E., Sekowska M., Smith G.D., Evans D., Gutierrez-Arcelus M., et al. Patterns of cis regulatory variation in diverse human populations. PLoS. Genet. 2012;8:e1002639–0. doi: 10.1371/journal.pgen.1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dausset J., Cann H., Cohen D., Lathrop M., Lalouel J.M., White R. Centre d'etude du polymorphisme humain (CEPH): collaborative genetic mapping of the human genome. Genomics. 1990;6:575–577. doi: 10.1016/0888-7543(90)90491-c. [DOI] [PubMed] [Google Scholar]
- 14.Behrends C., Sowa M.E., Gygi S.P., Harper J.W. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown M.A. Genetics of ankylosing spondylitis. Curr. Opin. Rheumatol. 2010;22:126–132. doi: 10.1097/BOR.0b013e3283364483. [DOI] [PubMed] [Google Scholar]
- 16.Holtta V., Klemetti P., Sipponen T., Westerholm-Ormio M., Kociubinski G., Salo H., Rasanen L., Kolho K.L., Farkkila M., Savilahti E., et al. IL-23/IL-17 immunity as a hallmark of Crohn's disease. Inflamm. Bowel. Dis. 2008;14:1175–1184. doi: 10.1002/ibd.20475. [DOI] [PubMed] [Google Scholar]
- 17.Kuiper J.J.W., Mutis T., de J.W., de Groot-Mijnes J.D., Rothova A. Intraocular interleukin-17 and proinflammatory cytokines in HLA-A29-associated birdshot chorioretinopathy. Am. J. Ophthalmol. 2011;152:177–182. doi: 10.1016/j.ajo.2011.01.031. [DOI] [PubMed] [Google Scholar]
- 18.McInnes I.B., Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 19.Yang P., Foster C.S. Interleukin 21, interleukin 23, and transforming growth factor beta1 in HLA-A29-associated birdshot retinochoroidopathy. Am. J. Ophthalmol. 2013;156:400–406. doi: 10.1016/j.ajo.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Evans D.M., Spencer C.C., Pointon J.J., Su Z., Harvey D., Kochan G., Oppermann U., Dilthey A., Pirinen M., Stone M.A., et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat. Genet. 2011;43:761–767. doi: 10.1038/ng.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cortes A., Hadler J., Pointon J.P., Robinson P.C., Karaderi T., Leo P., Cremin K., Pryce K., Harris J., Lee S., et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet. 2013;45:730–738. doi: 10.1038/ng.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levinson R.D., Brezin A., Rothova A., Accorinti M., Holland G.N. Research criteria for the diagnosis of birdshot chorioretinopathy: results of an international consensus conference. Am. J. Ophthalmol. 2006;141:185–187. doi: 10.1016/j.ajo.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 23.Holdsworth R., Hurley C.K., Marsh S.G., Lau M., Noreen H.J., Kempenich J.H., Setterholm M., Maiers M. The HLA dictionary 2008: a summary of HLA-A, -B, -C, -DRB1/3/4/5, and -DQB1 alleles and their association with serologically defined HLA-A, -B, -C, -DR, and -DQ antigens. Tissue Antigens. 2009;73:95–170. doi: 10.1111/j.1399-0039.2008.01183.x. [DOI] [PubMed] [Google Scholar]
- 24.Levinson R.D., Rajalingam R., Park M.S., Reed E.F., Gjertson D.W., Kappel P.J., See R.F., Rao N.A., Holland G.N. Human leukocyte antigen A29 subtypes associated with birdshot retinochoroidopathy. Am. J. Ophthalmol. 2004;138:631–634. doi: 10.1016/j.ajo.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 25.Fruci D., Niedermann G., Butler R.H., van Endert P.M. Efficient MHC class I-independent amino-terminal trimming of epitope precursor peptides in the endoplasmic reticulum. Immunity. 2001;15:467–476. doi: 10.1016/s1074-7613(01)00203-5. [DOI] [PubMed] [Google Scholar]
- 26.Strange A., Capon F., Spencer C.C., Knight J., Weale M.E., Allen M.H., Barton A., Band G., Bellenguez C., Bergboer J.G., et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franke A., McGovern D.P., Barrett J.C., Wang K., Radford-Smith G.L., Ahmad T., Lees C.W., Balschun T., Lee J., Roberts R., et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hinks A., Cobb J., Marion M.C., Prahalad S., Sudman M., Bowes J., Martin P., Comeau M.E., Sajuthi S., Andrews R., et al. Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat. Genet. 2013;45:664–669. doi: 10.1038/ng.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andres A.M., Dennis M.Y., Kretzschmar W.W., Cannons J.L., Lee-Lin S.Q., Hurle B., Schwartzberg P.L., Williamson S.H., Bustamante C.D., Nielsen R., et al. Balancing selection maintains a form of ERAP2 that undergoes nonsense-mediated decay and affects antigen presentation. PLoS. Genet. 2010;6:e1001157–0. doi: 10.1371/journal.pgen.1001157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zervoudi E., Saridakis E., Birtley J.R., Seregin S.S., Reeves E., Kokkala P., Aldhamen Y.A., Amalfitano A., Mavridis I.M., James E., et al. Rationally designed inhibitor targeting antigen-trimming aminopeptidases enhances antigen presentation and cytotoxic T-cell responses. Proc. Natl. Acad. Sci. USA. 2013;49:19890–19895. doi: 10.1073/pnas.1309781110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oz-Levi D., Ben-Zeev B., Ruzzo E.K., Hitomi Y., Gelman A., Pelak K., Anikster Y., Reznik-Wolf H., Bar-Joseph I., Olender T., et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am. J. Hum. Genet. 2012;91:1065–1072. doi: 10.1016/j.ajhg.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J.Y., Zhao H., Martinez J., Doggett T.A., Kolesnikov A.V., Tang P.H., Ablonczy Z., Chan C.C., Zhou Z., Green D.R., et al. Noncanonical autophagy promotes the visual cycle. Cell. 2013;154:365–376. doi: 10.1016/j.cell.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodríguez-Muela N., Koga H., García-Ledo L., de la Villa P., de la Rosa E.J., Cuervo A.M., Boya P. Balance between autophagic pathways preserves retinal homeostasis. Aging Cell. 2013;12:478–488. doi: 10.1111/acel.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberge F.G., Xu D., Chan C.C., de Smet M.D., Nussenblatt R.B., Chen H. Treatment of autoimmune uveoretinitis in the rat with rapamycin, an inhibitor of lymphocyte growth factor signal transduction. Curr. Eye Res. 1993;12:197–203. doi: 10.3109/02713689308999487. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Z., Wu X., Duan J., Hinrichs D., Wegmann K., Zhang G.L., Hall M., Rosenbaum J.T. Low dose rapamycin exacerbates autoimmune experimental uveitis. PLoS ONE. 2012;7:e36589. doi: 10.1371/journal.pone.0036589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han J.W., Zheng H.F., Cui Y., Sun L.D., Ye D.Q., Hu Z., Xu J.H., Cai Z.M., Huang W., Zhao G.P., et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat. Genet. 2009;41:1234–e31237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 37.Parkes M., Barrett J.C., Prescott N.J., Tremelling M., Anderson C.A., Fisher S.A., Roberts R.G., Nimmo E.R., Cummings F.R., Soars D., et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat. Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., McCarthy M.I., Ouwehand W.H., Samani N.J., et al. Welcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Power C., Ellitott J. Cohort profile: 1958 British birth cohort (National Child Development Study) Int. J. Epidemiol. 2006;35:34–41. doi: 10.1093/ije/dyi183. [DOI] [PubMed] [Google Scholar]
- 40.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Howie B., Fuchsberger C., Stephens M., Marchini J., Abecasis G.R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boomsma D.I., Wijmenga C., Slagboom E.P., Swertz M.A., Karssen L.C., Abdellaoui A., Ye K., Guryev V., Vermaat M., van Dijk F., et al. The Genome of the Netherlands: design, and project goals. Eur. J. Hum. Genet. 2014;22:221–227. doi: 10.1038/ejhg.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grundberg E., Small K.S., Hedman Å.K., Nica A.C., Buil A., Keildson S., Bell J.T., Yang T.P., Meduri E., Barrett A., et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat. Genet. 2012;44:1084–1089. doi: 10.1038/ng.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson C.A., Boucher G., Lees C.W., Franke A., D'Amato M., Taylor K.D., Lee J.C., Goyette P., Imielinski M., Latiano A., et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011;43:246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barrett J.C., Clayton D.G., Concannon P., Akolkar B., Cooper J.D., Erlich H.A., Julier C., Morahan G., Nerup J., Nierras C., et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin Y., Birlea S.A., Fain P.R., Ferrara T.M., Ben S., Riccardi S.L., Cole J.B., Gowan K., Holland P.J., Bennett D.C., et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet. 2012;44:676–680. doi: 10.1038/ng.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reveille J.D., Sims A.M., Danoy P., Evans D.M., Leo P., Pointon J.J., Jin R., Zhou X., Bradbury L.A., Appleton L.H., et al. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat. Genet. 2010;42:123–127. doi: 10.1038/ng.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawcer S., Hellenthal G., Pirinen M., Spencer C.C., Patsopoulos N.A., Moutsianas L., Dilthey A., Su Z., et al. International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stahl E.A., Raychaudhuri S., Remmers E.F., Xie G., Eyre S., Thomson B.P., Li Y., Kurreeman F.A., Zhernakova A., Hinks A., et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lango Allen H., Estrada K., Lettre G., Berndt S.I., Weedon M.N., Rivadeneira F., Willer C.J., Jackson A.U., Vedantam S., Raychaudhuri S., et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–838. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu J.Z., Medland S.E., Wright M.J., Henders A.K., Heath A.C., Madden P.A., Duncan A., Montgomery G.W., Martin N.G., McRae A.F., et al. Genome-wide association study of height and body mass index in Australian twin families. Twin. Res. Hum. Genet. 2010;13:179–193. doi: 10.1375/twin.13.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Estrada K., Krawczak M., Schreiber S., van Duijn K., Stolk L., van Meurs J.B., Liu F., Penninx B.W., Smit J.H., Vogelzangs N., et al. A genome-wide association study of northwestern Europeans involves the C-type natriuretic peptide signaling pathway in the etiology of human height variation. Hum. Mol. Genet. 2009;18:3516–3524. doi: 10.1093/hmg/ddp296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teslovich T.M., Musunuru K., Smith A.V., Edmondson A.C., Stylianou I.M., Koseki M., Pirruccello J.P., Ripatti S., Chasman D.I., Willer C.J., et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waterworth D.M., Ricketts S.L., Song K., Chen L., Zhao J.H., Ripatti S., Aulchenko Y.S., Zhang W., Yuan X., Lim N., et al. Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2010;30:2264–2276. doi: 10.1161/ATVBAHA.109.201020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.