

Abstract
Human pluripotent stem cells (hPSCs) have great potential for studying human embryonic development, for modeling human diseases in the dish and as a source of transplantable cells for regenerative applications after disease or accidents. Neural crest (NC) cells are the precursors for a large variety of adult somatic cells, such as cells from the peripheral nervous system and glia, melanocytes and mesenchymal cells. They are a valuable source of cells to study aspects of human embryonic development, including cell fate specification and migration. Further differentiation of NC progenitor cells into terminally differentiated cell types offers the possibility to model human diseases in vitro, investigate disease mechanisms and generate cells for regenerative medicine. This article presents the adaptation of a currently available in vitro differentiation protocol for the derivation of NC cells from hPSCs. This new protocol requires 18 days of differentiation, is feeder-free, easily scalable and highly reproducible among human embryonic stem cell (hESC) lines as well as human induced pluripotent stem cell (hiPSC) lines. Both old and new protocols yield NC cells of equal identity.
Keywords: Neuroscience, Issue 87, Embryonic Stem Cells (ESCs), Pluripotent Stem Cells, Induced Pluripotent Stem Cells (iPSCs), Neural Crest, Peripheral Nervous System (PNS), pluripotent stem cells, neural crest cells, in vitro differentiation, disease modeling, differentiation protocol, human embryonic stem cells, human pluripotent stem cells
Introduction
Human embryonic stem cells (hESC) and human induced pluripotent stem cells (hiPSC) have shown immense potential, in particular for the investigation and future treatment of human diseases for which neither good animal models nor primary tissues are available. Application examples for the hESC/hiPSC technology are the following: Cells of particular interest can be generated from hESC/hiPSCs for regenerative medicine at unlimited quantity1. Cells can be produced from patients carrying a specific disease and used to establish in vitro disease models2,3. Such disease models can then be employed for large-scale drug screening in the quest for new drug compounds4 as well as testing of existing drugs for efficacy and toxicity5. In vitro disease models can lead to the identification of novel disease mechanisms. For all applications of the hESC/iPSC technology it is important to work with specific, well-defined cell types affected in the disease of interest. Thus, the availability of solid and reproducible in vitro differentiation protocols is crucial for all applications of the hESC/hiPSC technology. Protocols are desirable that show minimal variability, time expense, effort, difficulty and cost as well as maximal reproducibility among hESC/hiPSC lines and different researchers.
Neural crest (NC) cells emerge during vertebrate neurulation between the epidermis and the neural epithelium. They proliferate and migrate extensively throughout the developing embryo and give rise to an impressive diversity of progeny cell types, including bone/cartilage, the craniofacial skeleton, sensory nerves, Schwann cells, melanocytes, smooth muscle cells, enteric neurons, autonomic neurons, chromaffin cells, cardiac septum cells, teeth and adrenal/thyroid glandular cells6. Thus, NC cells are an attractive cell type for the stem cell field and important for the modeling of a variety of diseases, such as Hirschsprung's disease7, Familial Dysautonomia8 as well as cancers such as neuroblastoma9. Furthermore, they offer the possibility to study aspects of human embryonic development in vitro.
The currently available and widely applied in vitro differentiation protocol for the derivation of NC cells from hESCs10,11 requires up to 35 days of differentiation and it involves neural induction on stromal feeder cells such as MS5 cells and is thus performed under poorly defined conditions. While it can be up-scaled to generate large quantities of NC cells, for example required for high-throughput drug screening4, this is labor and cost intensive. Furthermore, it involves manual passaging of neural rosettes, which can be difficult to reproduce and thus is subject to overall variability, in particular when it is applied to a large variety of hESC or hiPSC lines. Here, the stepwise derivation of NC cells in an 18-day protocol that is free of feeder cells is shown. This method is shorter and more defined than the currently used protocol. Furthermore, it is very robust in generating NC cells among different hiPSC lines. Importantly, it is shown that the NC cells yielded by both protocols emerge at the border of neural rosettes (hereafter termed rosette-NC or R-NC). The cells derived using either of the two protocols look morphologically identical, they express the same NC markers and cluster together in microarray analysis. NC cells derived using the new protocol (R-NC) are functional, similar to NC cells derived using the old protocol (MS5-R-NC) such that they can migrate and further differentiate into neurons. Therefore, the cells can be used concurrently with the MS5-R-NC cells. The R-NC cell protocol for the derivation of NC cells from hESC/iPSC will be useful for all applications of the hESC/iPSC technology involving the NC lineage.
Protocol
1. Preparation of Culture Media, Coated Dishes and Maintenance of hPSCs
1.1 Media preparation
Note: Filter all media for sterilization and store at 4 °C in the dark for up to 2 weeks. Reagent names, company and catalog numbers are listed in the Materials Table.
DMEM/10%FBS: Combine 885 ml DMEM, 100 ml FBS, 10 ml Pen/Strep and 5 ml L-Glutamine.
HES-medium: Combine 800 ml DMEM/F12, 200 ml KSR, 5 ml L-Glutamine, 5 ml Pen/Strep, 10 ml MEM minimum essential amino acids solution, 1 ml β-Mercaptoethanol. Add 10 ng/ml FGF-2 after filtering the medium. CAUTION: β-Mercaptoethanol is toxic, avoid inhalation, ingestion and skin contact.
KSR-differentiation medium: Combine 820 ml Knockout DMEM, 150 ml KSR, 10 ml L-Glutamine, 10 ml Pen/Strep, 10 ml MEM minimum essential amino acids solution and 1 ml β-Mercaptoethanol.
N2-differentiation medium: Dissolve 12 g DMEM/F12 powder in 980 ml dH2O, add 1.55 g Glucose, 2 g Sodium Bicarbonate and 100 mg APO human transferrin. Mix 2 ml dH2O with 25 mg human insulin and 40 μl 1 N NaOH, add the dissolved solution to the medium. Add 100 μl putrescine dihydrochloride, 60 μl selenite, 100 μl progesterone and bring the volume up to 1 L with dH2O.
1.2 Coating of culture dishes
Matrigel coating: Thaw 1 ml frozen matrigel aliquot by pipetting 19 ml DMEM/F12 over the aliquot until it has dissolved. Remove clumps by passing it through a 40 μm cell strainer and plate 8 ml/10 cm dish. Incubate the dishes for 1 hr at RT. Aspirate the matrigel immediately before plating the cells. Note: Work quickly with matrigel because it can clump at temperatures above 4 °C.
PO/Lam/FN coating: Add 10 ml of 1x PBS containing 15 μg/ml Poly-L Ornithin hydrobromide to a 10 cm dish. Incubate the dish over night at 37 °C. Wash plates with 1x PBS once and add 10 ml/10 cm dish of 1x PBS containing 2 μg/ml mouse Laminin-I and 2 μg/ml fibronectin. Incubate the dishes over night at 37 °C. Before plating the cells, completely remove the solution and let the plates dry thoroughly at RT for 15-20 min by standing them up onto the tissue culture hood wall without the lid on. Note: The dishes are completely dry and ready for cell plating when one can see crystal structures on the surface (visible by eye). The plates can be kept at RT for a few hours in this dried state. PO/Lam/FN dishes have to be prepared 2 days ahead of time. In emergency cases Lam/FN can be incubated for 2-4 hr only. However, this may risk suboptimal differentiation/survival results.
1.3 Maintenance of hPSCs
Note: hPSCs are maintained on 0.1% gelatin and mitotically inactivated mouse embryonic fibroblasts (MEFs) in HES-medium supplemented with 10 ng/ml FGF-2 as described previously 10,12. The cells should be split every 6-8 days.
Coat a 10 cm dish with 8 ml 0.1% gelatin (in 1x PBS without Magnesium or Calcium) at RT for 5 min.
Thaw frozen MEFs quickly in a 37 °C water bath. Add 1 million MEFs to 10 ml DMEM/10% FBS.
Aspirate the gelatin and plate the cells. Incubate at 37 °C for at least 6 hr.
Aspirate the DMEM/10% FBS from the prepared MEF plate, wash plate with 1x PBS once and add 10 ml HES-medium supplemented with 10 μM Y-27632 dihydrochloride. Allow the medium to warm up at 37 °C for 20 min. Note: For manual splitting of hPSCs a laminar flow hood with an embedded microscope is used. However, the cells can be passaged using appropriate alternative methods.
Under a laminar flow hood with an embedded microscope, detach separate colonies using a cell lifter and let them float.
Use a 1 ml syringe to aspirate the floating colonies and dispatch them onto the fresh, warm MEF plate. Transfer approximately one fourth of the cells to the new dish. Incubate at 37 °C.
Feed cells daily with fresh HES medium.
2. Plating of hPSCs for Differentiation
Note: hPSCs should be split or plated for differentiation when the colonies are large, but still have sharp edges with as little as possible differentiating cells at their borders (see Figure 1B). When the cells are maintained using manual passaging the colonies should be large enough to easily be seen by eye. To get the right feel for this time point one can maintain a separate hPSC dish for two weeks without passaging and watch the cells reach and pass the ideal time point for passaging/differentiating them.
Prepare 10 cm dishes with matrigel 1 hr before starting the differentiation. Successful matrigel coating can be verified at 4x magnification.
When the hPSCs are ready to be split, aspirate the medium and add 4 ml of 0.05% trypsin-EDTA to the cells.
Shake the dish horizontally for 2 min vigorously until one can see the MEFs as single cells lifting off the plate under the microscope. The hPSC colonies remain attached as colonies.
Immediately and thoroughly, aspirate the trypsin and let the plate stand at RT for 2-4 min.
Add 2 ml of HES-medium to the plate and using a P1000 pipette, detach the cells. Note: If the cells cannot be lifted off the surface easily, the plate can be incubated empty at RT for another 2-3 min.
Transfer the cells to 8 ml HES-medium supplemented with 10 μM Y-27632 dihydrochloride.
Aspirate the matrigel from a previously prepared 10 cm dish. Plate the cells at a 1:1 or 1:2 ratio (for example: The cells from one 10 cm dish are split into two 10 cm dishes) onto the matrigel plate. Incubate at 37 °C over night. Note: This plating should result in approximately 100,000 cells/cm2.
3. Induction of Neural Differentiation
Note: The differentiation can be initiated (day 0) when the cells are 90-100% confluent (see Figure 1C), usually the following day. If the accurate confluency is not reached yet, the cells can be fed daily with HES-medium until they are ready for differentiation. Alternatively, the initial number of cells plated can be increased.
On day 0 to 3, feed the cells daily with 10 ml/10 cm dish KSR-differentiation medium containing 0.1 μM LDN193189 and 10 μM SB431542.
On day 4 and 5 feed the cells with 75% KSR-differentiation medium and 25% N2-differentiation medium both containing LDN193189 and SB431542.
On day 6 and 7 feed the cells with 50% KSR-differentiation medium and 50% N2-differentiation medium both containing LDN193189 and SB431542.
On day 8 and 9 feed the cells with 25% KSR-differentiation medium and 75% N2-differentiation medium both containing LDN193189 and SB431542.
On day 9 and 10 prepare PO/Lam/FN dishes as indicated in 1.2.2 for replating of the cells on day 11.
On day 10 feed cells with N2-differentiation medium containing LDN193189 and SB431542.
4. Replating in Droplets for NC Specification
On day 11 aspirate Lam/FN from the previously prepared plates and let them dry completely at RT for 20-30 min, as explained in 1.2.2.
Remove medium from the differentiating cells, wash the cells once with 1x PBS and add 8 ml accutase/10 cm dish. Incubate for 20 min at 37 °C.
Using a cell lifter, detach the cells and resuspend them in accutase with a 5 ml pipette. Transfer the suspension to a 15 ml tube.
Add 5 ml of 1x PBS and spin the cells for 5 min at 114 x g.
Resuspend the cells in 10 ml 1x PBS. To ensure a single cell suspension, filter them through a 40 μm cell strainer and count the cells using a hemocytometer or equivalent technique.
Spin the cells down and resuspend them in N2-differentiation medium containing 200 μM Ascorbic Acid (AA), 20 ng/ml BDNF, 100 ng/ml FGF8, 20 ng/ml SHH, 10 μM Y-27632 dihydrochloride at the concentration of 100,000-150,000 cells/10 μl.
Using a repeat-pipettor, plate 10 μl droplets close to each other without them touching onto the dried PO/Lam/FN 15 cm dishes. Note: One 10 cm dish of differentiated cells normally results in approximately 2-3 times 15 cm dishes or approximately 100 x 10 μl droplets/15 cm dish.
Let the droplets stand at RT for 20-30 min, then very carefully (not to disturb the droplets) add 30 ml of N2/AA/BDNF/FGF8/SHH/Y and incubate at 37 °C.
On day 12, carefully feed the cells with 20 ml/15 cm dish N2/AA/BDNF/FGF8/SHH.
From day 14-17 feed the cells every second or third day with N2/AA/BDNF/FGF8.
On day 16 and 17 prepare PO/Lam/FN plates to replate NC cells after FACS sorting.
5. Fluorescence Activated Cell Sorting (FACS) of NC cells
Note: The preparation of the cells for FACS requires approximately 2 hr.
On day 18 remove medium, wash the cells once with 1x PBS in the dish and add 12 ml accutase. Incubate the cells for 20 min at 37 °C.
Using a cell lifter, detach the cells from the surface and let them float. Pipette the cells into suspension using a 5 ml pipette, transfer them to a 50 ml tube and add 30 ml of 1x PBS. Spin the cells for 5 min at 114 x g.
Using a P1000 pipette, resuspend the cells in 1 ml of 2% FBS/HBSS (the HBSS contains 15 mM HEPES). Add 19 ml of 2% FBS/HBSS and filter the cells through a 40 μm cell strainer to ensure a single cell suspension. Incubate the cells on ice for 15 min for antibody blocking and then count them using a hemocytometer.
Spin the cells down and resuspend them at the concentration of 10 million cells/ml in 2% FBS/HBSS.
Set aside 1 million cells each for the unstained, single-stained (HNK-1 only and p75 only) and secondary antibody stained only (APC only and 488 only) FACS controls.
Add 5 μl of HNK-1 and 5 μl of p75 primary antibody per 10 million cells in 1 ml suspension to the proper samples and incubate for 20 min on ice.
Wash cells in 10 ml 1x PBS (centrifuge 5 min at 114 x g) and resuspend the cells in 1 ml 2% FBS/HBSS. Add 2 μl of APC and 1 μl of 488 secondary antibody per 10 million cells in 1 ml suspension to the proper samples and incubate for 20 min on ice in the dark. Note: APC and 488 are the secondary antibodies used here for the HNK-1 and p75 staining, respectively.
Wash the cells in 10 ml 1x PBS twice, resuspend 10 million cells in 500 μl of 2% FBS/HBSS containing DAPI (at 0.5 ng/μl) or alternative appropriate live cell stain to exclude dead cells from the FACS analysis.
Transfer the samples to appropriate FACS tubes.
Prepare FACS tubes with 0.5 ml 2% FBS/HBSS for cell collection. Note: The cells and collection tubes are kept on ice in the dark during the sorting time.
Using a cell sorting machine with lasers that can detect DAPI, APC and 488 sort DAPI-/HNK-1+/P75+ double positive cells. Note: Preparation time and handling of the cells leads to a small percentage of cell death, DAPI stains dead cells only and thus allows for these cells to be excluded from the sorted population. Any alternative live/dead stain works equally well for this purpose. DAPI itself does not cause cytotoxicity in the live cell population.
6. Replating of Sorted Cells, NC Maintenance and Expansion
Note: FACS sorted cells should be handled with special care to ensure optimal survival. Keep them on ice until replating them. Do not vortex or pipette them harshly. The cells can be resuspended by flicking the tube.
After FACS sorting, count the NC cells, then spin them down and resuspend in N2-differentiation medium supplemented with 10 ng/ml FGF2, 20 ng/ml EGF and 10 μM Y-27632 dihydrochloride in the volume appropriate to replate them at 30,000 cells/10 μl droplets.
Thoroughly dry previously prepared PO/Lam/FN plates and plate approximately 50-70 droplets per 10 cm dish. Let the dishes stand at RT for 20-30 min.
Very carefully add 20 ml/10 cm dish of N2/FGF2/EGF/Y-27632 without disturbing the droplets. Incubate the cells at 37 °C.
Feed the cells every 2-3 days with N2/FGF2/EGF. Note: NC cells are expanded or passaged approximately every 4-5 days or when they start piling up within the droplets.
To passage the NC cells, remove the medium, wash the cells once with 1x PBS and add 8 ml accutase/10 cm dish. Incubate for 20 min at 37 °C.
Pipette the cells off the plate using a 5 ml pipette and transfer them to a 15 ml tube. Add 6 ml 1x PBS.
Spin the cells for 5 min at 114 x g.
Resuspend the cells in N2-differentiation medium supplemented with 10 ng/ml FGF2, 20 ng/ml EGF and 10 μM Y-27632 dihydrochloride in order to have 20,000 cells/10 μl droplet.
Replate the cells in 10 μl droplets onto dried PO/Lam/FN plates. Let the dishes stand at RT for 20-30 min.
Carefully add 20 ml/10 cm dish of N2/FGF2/EGF/Y-27632. Incubate at 37 °C. Note: NC cells can be maintained or expanded for up to 2 weeks as a relatively homogeneous progenitor population. Longer culture may lead them to differentiate into various progeny cell types.
Representative Results
The two most important improvements of the R-NC protocol over the MS5-R-NC protocol 11 are the feeder-free, defined differentiation conditions and the overall shortening of the time requirement. MS5 feeder cells 13 are murine bone-marrow derived stromal cells that have been shown to support neural differentiation from hESCs 14. HESCs cultured on MS5 feeder cells at low density form epithelial structures and neural rosettes 15, at the periphery of which NC cells emerge 10, thus mimicking early human neural development. However, it is not clear what signaling molecules and growth factors are released from MS5 feeder cells. Therefore, the differentiation conditions are poorly defined, making it difficult to reproduce them in recurrent experiments and across hPSC lines. For adequate neural induction, hESCs have to be seeded at low density in small colonies onto MS5 feeder cells, complicating up-scaling and reaching high yields of differentiated cells. In 2009, a method was developed that aimed at neuralizing hPSCs very efficiently 12. In this method hPSCs are seeded at high density, in monolayers in the absence of MS5 feeder cells, achieving high yields of neural induction in 10 days. We followed the scheme of this method in adapting the MS5-R-NC differentiation protocol (Figure 1A). HPSCs cultured in colonies on MEFs (Figure 1B) are dispersed and seeded onto matrigel as single cells in a monolayer culture at high density (Figure 1C). At day 11 of differentiation, the majority of cells have successfully neuralized (pax6-positive, not shown 12). Replating the cells at high density in droplets allows the formation of condensed neural rosettes within the droplet (Figure 1D and Figure 2). NC cells emerge at the borders of neural rosettes (Figure 1D) and migrate out of the droplet (Figure 1E). After 7 days of further culture the NC cells can be isolated by FACS sorting. HNK-1/p75 double positive NC cells can be isolated at the efficiency of 20-40% (Figure 1F). This protocol requires 18 days, while the MS5-R-NC protocol requires up to 35 days, making the R-NC cell protocol more useful for a variety of downstream applications.
The aim of this work is to generate NC cells in a shorter, more reproducible and cheaper protocol, yielding the same cells as the old NC protocol. We have used this new protocol to successfully differentiate at least 10 hESC and hiPSC lines derived from patients and healthy controls (data not shown), showing solid reproducibility of the protocol across different hPSC lines. The new protocol saves costs due to the use of LDN versus noggin, less use of SHH and the lack of MS5 production costs. The new protocol lasts 18 days compared to 35 days in the old protocol, which saves approximately 5 feedings and thus their associated media costs. To investigate whether the cells produced with the two protocols have similar identity, we show that NC cell development in both protocols follows the same differentiation pattern (Figure 2). The cells pass through a neural rosette stage and yield NC cells with identical morphology after FACS sorting. The smaller size of the neural rosettes in the new protocol compared to the old protocol is due to the high cell density after replating at day 11. In contrast, in the old protocol rosettes form within hPSC colonies, which are not as condensed. NC cells derived with the two protocols express the same biological NC markers, such as HNK-1, AP2 and nestin. We analyzed NC cells generated with the two protocols on a global gene expression level (Figure 3A) and found that NC cells generated with the two protocols cluster closely together. NC cells that were generated by activation of the wnt signaling pathway (wnt-NC) and were shown to have the potential of generating melanocytes 16, cluster separately when analyzed by global gene expression, indicating that this may be a different NC population. Indeed, we have been unable to generate melanocyte progenitor cells in vitro from R-NC or MS5-R-NC cells (data not shown). Similarly, neuroepithelial cells (LSB), differentiated for 10 days in LDN193189 and SB431542 only show a clearly distinct gene expression pattern. Neuroepithelial cells are early progenitors of the nervous system, and thus are less differentiated compared to NC cells and are included here as a negative control population. To assess if the R-NC cells generated here are similar to the cells made with the old protocol, we show their migration capacity in an in vitro scratch assay (Figure 3B). 48 hr after scratching a confluent well of sorted R-NC cells, the cells migrate into the scratch successfully. Lastly, we show that R-NC cells have the potential to differentiate into cells from the autonomic nervous system. The cells were spontaneously differentiated for 4 days post HNK1+/p75+ FACS and stained for Mash1 and Tuj1, genes that are expressed in autonomic neurons (Figure 3C).
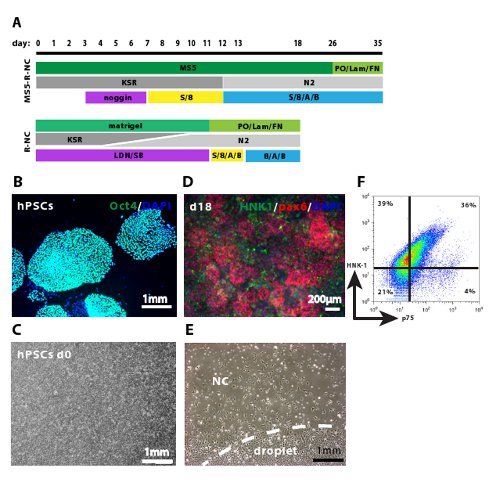 Figure 1. Critical steps in the R-NC differentiation protocol. A. MS5-R-NC10,11 and R-NC differentiation protocol scheme. The specific differentiation steps of the two protocols are shown. MS5: feeder stromal cells, KSR: KSR-differentiation medium, N2: N2-differentiation medium, LDN: LDN193189, SB: SB431542, S: sonic hedgehog, 8: FGF8, A: ascorbic acid, B: BDNF. B. Shows undifferentiated hPSC colonies stained with Oct-4 and DAPI before induction of differentiation. C. Shows the cell density (90-100% confluency) on day 0 of differentiation, typically 1 day after plating hPSCs. D. Shows neural rosettes stained with Pax6 and emerging NC cells stained with HNK-1 within the droplet. E. Shows day 13 cells, replated at day 11 in droplets and NC cells emerging from the edge of the droplet. F. Representative FACS sorting plot, showing HNK-1/p75 double positive NC cells at day 18 of differentiation. The gates were chosen based on the unstained and single stained controls (not shown). FACS sorting plots may differ between experiments, hPSC lines and the use of the old or new protocol in terms of the percentage of double positive and single positive cells. Thus, it is important to isolate the double positive population to ensure the proper NC cells are extracted. Images C to F were generated using the new R-NC protocol. Please click here to view a larger version of this figure.
Figure 1. Critical steps in the R-NC differentiation protocol. A. MS5-R-NC10,11 and R-NC differentiation protocol scheme. The specific differentiation steps of the two protocols are shown. MS5: feeder stromal cells, KSR: KSR-differentiation medium, N2: N2-differentiation medium, LDN: LDN193189, SB: SB431542, S: sonic hedgehog, 8: FGF8, A: ascorbic acid, B: BDNF. B. Shows undifferentiated hPSC colonies stained with Oct-4 and DAPI before induction of differentiation. C. Shows the cell density (90-100% confluency) on day 0 of differentiation, typically 1 day after plating hPSCs. D. Shows neural rosettes stained with Pax6 and emerging NC cells stained with HNK-1 within the droplet. E. Shows day 13 cells, replated at day 11 in droplets and NC cells emerging from the edge of the droplet. F. Representative FACS sorting plot, showing HNK-1/p75 double positive NC cells at day 18 of differentiation. The gates were chosen based on the unstained and single stained controls (not shown). FACS sorting plots may differ between experiments, hPSC lines and the use of the old or new protocol in terms of the percentage of double positive and single positive cells. Thus, it is important to isolate the double positive population to ensure the proper NC cells are extracted. Images C to F were generated using the new R-NC protocol. Please click here to view a larger version of this figure.
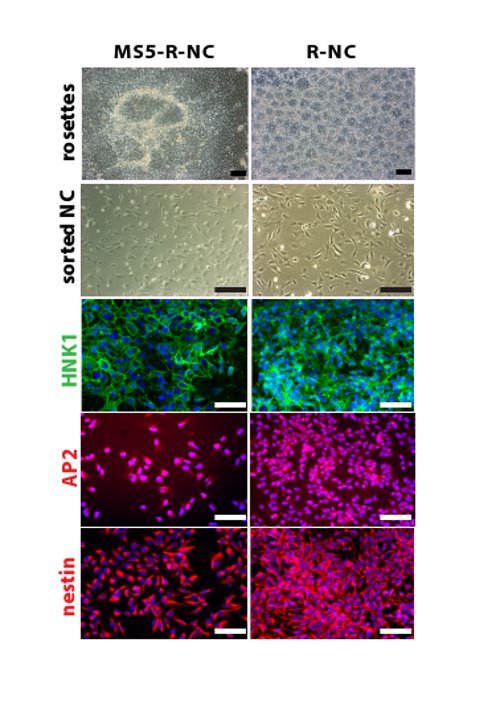 Figure 2. Comparable neural crest cell identity of the cells derived with the MS5-R-NC or the R-NC protocol. In both protocols the cells pass through a neural rosette stage. Sorted NC cells look identical by morphology and by expression of NC markers such as HNK-1 and AP2. NC cells are nestin-positive, showing that they are progenitor cells. Scale bar: 200 µm. All fluorescence pictures are counterstained for DAPI. Please click here to view a larger version of this figure.
Figure 2. Comparable neural crest cell identity of the cells derived with the MS5-R-NC or the R-NC protocol. In both protocols the cells pass through a neural rosette stage. Sorted NC cells look identical by morphology and by expression of NC markers such as HNK-1 and AP2. NC cells are nestin-positive, showing that they are progenitor cells. Scale bar: 200 µm. All fluorescence pictures are counterstained for DAPI. Please click here to view a larger version of this figure.
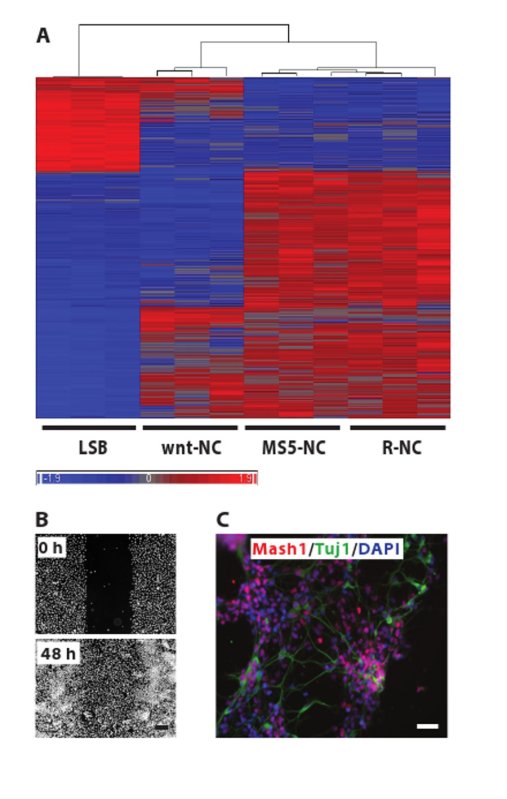 Figure 3. NC cells generated with the old and the new protocol have similar identity.A. Unsupervised clustering of Illumina microarray gene expression data comparing NC cells derived with the MS5-R-NC and the R-NC protocol. NC cells derived with the MS5-R-NC or the R-NC protocol at day 35 or day 18 respectively were analyzed in triplicates by hybridization to a Illumina human 12 Oligonucleotide array. Data analysis was conducted using the Partek Genomic Suite software. Significant differences were defined as those with a fold change greater than 2 and FDR less than 0.05. 1,421 genes were analyzed. Default settings, such as Euclidean sample dissimilarity, average linkage cluster method and 25 for dendrogram length were used. NC cells induced by activating wnt signaling (wnt-NC) were included (the cells were differentiated in LDN193189 day 0-3, SB431542 day 0-4, CHIR99021 day 0-11 and FACS sorted at day 11 for sox10) 16. These cells cluster separately from R-NC cells, indicating that wnt-NC cells and R-NC cells are distinct populations. Cells differentiated for 11 days in LDN193189 and SB431542 were included as a neuroepithelial control (LSB). R-NC and MS5-R-NC cells cluster closely together compared to the control cells. Raw gene expression data are available on GEO (www.ncbi.nlm.nih.gov/geo/) accession #: GSE50643. B. Migration assay. HNK1+/p75+ FACS sorted R-NC cells were plated on PO/Lam/FN in 96-wells and scratched manually 24 hr later. The 0 hr picture was taken immediately after scratching and post staining with Hoechst. The remaining wells were allowed to migrate for 48 hr before the second picture was taken. Scale bar: 200 µm C. Sorted R-NC cells were allowed to spontaneously differentiate for 4 days and were stained for Mash1, Tuj1 and DAPI. Scale bar: 500 µm. Please click here to view a larger version of this figure.
Figure 3. NC cells generated with the old and the new protocol have similar identity.A. Unsupervised clustering of Illumina microarray gene expression data comparing NC cells derived with the MS5-R-NC and the R-NC protocol. NC cells derived with the MS5-R-NC or the R-NC protocol at day 35 or day 18 respectively were analyzed in triplicates by hybridization to a Illumina human 12 Oligonucleotide array. Data analysis was conducted using the Partek Genomic Suite software. Significant differences were defined as those with a fold change greater than 2 and FDR less than 0.05. 1,421 genes were analyzed. Default settings, such as Euclidean sample dissimilarity, average linkage cluster method and 25 for dendrogram length were used. NC cells induced by activating wnt signaling (wnt-NC) were included (the cells were differentiated in LDN193189 day 0-3, SB431542 day 0-4, CHIR99021 day 0-11 and FACS sorted at day 11 for sox10) 16. These cells cluster separately from R-NC cells, indicating that wnt-NC cells and R-NC cells are distinct populations. Cells differentiated for 11 days in LDN193189 and SB431542 were included as a neuroepithelial control (LSB). R-NC and MS5-R-NC cells cluster closely together compared to the control cells. Raw gene expression data are available on GEO (www.ncbi.nlm.nih.gov/geo/) accession #: GSE50643. B. Migration assay. HNK1+/p75+ FACS sorted R-NC cells were plated on PO/Lam/FN in 96-wells and scratched manually 24 hr later. The 0 hr picture was taken immediately after scratching and post staining with Hoechst. The remaining wells were allowed to migrate for 48 hr before the second picture was taken. Scale bar: 200 µm C. Sorted R-NC cells were allowed to spontaneously differentiate for 4 days and were stained for Mash1, Tuj1 and DAPI. Scale bar: 500 µm. Please click here to view a larger version of this figure.
Discussion
For the successful differentiation of R-NC cells from hESC/hiPSCs the following considerations should be made. It is critical to work under sterile culture conditions at all times. In particular, it is important to test hPSC cultures for mycoplasma contamination regularly, since this contamination will hinder successful differentiation, but cannot readily be detected visually in hPSC cultures. The R-NC differentiation should be initiated at 90-100% cell density; lower cell density affects cell survival and efficiency of R-NC differentiation. It is critical to empirically validate optimal concentrations and lot numbers of some of the reagents, in particular KSR for supporting efficient NC differentiation. When the cells are replated at day 11 in droplets, the cell density within the 10 µl droplet should be high and is best empirically determined. Proper rosette- and NC-formation depends on the appropriate cell density within the droplet. We empirically observed increased cell survival when the cells are washed at least twice after treatment with accutase. For the successful isolation of R-NC cells by FACS the proper experimental controls, such as unstained, single antibody stained and secondary antibody only stained cells are crucial. Dead cells can be excluded by DAPI, 7-AAD or Propidium Iodide staining. Furthermore, antibody dilutions should be empirically determined for each specific antibody lot. It is important to FACS purify R-NC cells by double staining with HNK-1 and p75, since single staining may lead to contamination of the NC population with unwanted cell types, such as p75-positive CNS cells, early mesoderm or placode. In our experience percentages of double versus single stained populations can vary between hPSC lines and differentiation experiments. R-NC cell survival post FACS can be increased by avoiding harsh pipetting of the cells, instead flicking of the tube to re-suspend the cells is advisable. Also, plating R-NC FACS isolated cells in conditioned medium (filtered medium the cells grew in before FACS) has been shown to improve survival 11. It is advisable to carry out a smaller scale differentiation in parallel that can be stained with the neural epithelial marker Pax6 at day 11 and day 14 to ensure efficient neutralization and rosette formation. NC markers, such as HNK-1 or AP2 at the rosette stage for proper NC differentiation should be assessed as well. Furthermore, isolated R-NC cells should be validated by morphology and staining for NC markers, such as AP2, HNK-1 and nestin (Figure 2).
The derivation of MS5-R-NC cells from hPSC has been described in 2007 by Lee et al. 10. It was shown that this precursor population can be propagated and further differentiated in vitro into derivatives of the NC lineage. MS5-R-NC cells can be spontaneously differentiated using the neural sphere method 10,17 or by directed in vitro differentiation. It was established that MS5-R-NC cells can give rise to cells of the peripheral nervous system (autonomic and sensory neurons), peripheral glia (Schwann cells), myofibroblasts, mesenchymal stem cells and their progeny and smooth muscle 10. Transplantation into chick and mice showed that MS5-R-NC cells migrate and differentiate in vivo 10. MS5-R-NC cells have further been implicated in the modeling of human diseases. The characteristic phenotype of the genetic disease Familial Disautonomia (FD) was modeled in vitro using patient specific hiPSC-derived MS5-R-NC cells 2. NC characteristic phenotypes, such as dysfunctional NC cell development and migration were shown in cells derived from FD patients but not in healthy control cells. MS5-R-NC cells have also been utilized to establish toxicological testing of compounds affecting neural crest migration during human embryonic development, with the potential to avoid the release of future drugs that could negatively affect neurodevelopment 5. An exciting application of the hPSC technology is high-throughput screening and testing of pharmaceutical treatment options for human diseases. MS5-R-NC cells were used to accomplish the first such screen in an iPSC-based disease model, i.e. Familial Dysautonomia 4. This screen led to the identification of new compounds that may be further evaluated in clinical trials.
For all applications of the hPSC field it is critical to have accurate, reproducible, defined and specific in vitro differentiation protocols available. In this report, we show the adaptation of an established in vitro differentiation protocol for the derivation of R-NC cells from hPSCs. The reported protocol yields the same cells as previously reported 10 in more defined culture conditions and a shorter time frame. This protocol can be employed to generate R-NC cells for human diseases affecting cells from the NC lineage, for compound screening, toxicology testing and the further development of directed differentiation protocols to cell types derived from the NC lineage.
Disclosures
The authors have no conflicting interests to disclose.
Acknowledgments
This work was supported by a fellowship for advanced researchers from the Swiss National Science Foundation and through grants from NYSTEM (C026446; C026447) and the Tri-institutional stem cell initiative (Starr Foundation).
References
- Lee G, Studer L. Induced pluripotent stem cell technology for the study of human disease. Nat Methods. 2010;7:25–27. doi: 10.1038/nmeth.f.283. [DOI] [PubMed] [Google Scholar]
- Lee G, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402–406. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert AD, et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, et al. Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol. 2012;30:1244–1248. doi: 10.1038/nbt.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer B, et al. Evaluation of developmental toxicants and signaling pathways in a functional test based on the migration of human neural crest cells. Environ Health Perspect. 2012;120:1116–1122. doi: 10.1289/ehp.1104489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupin E, Sommer L. Neural crest progenitors and stem cells: from early development to adulthood. Dev Biol. 2012;366:83–95. doi: 10.1016/j.ydbio.2012.02.035. [DOI] [PubMed] [Google Scholar]
- McKeown SJ, Stamp L, Hao MM, Young HM. Hirschsprung disease: a developmental disorder of the enteric nervous system. Wiley Interdiscip Rev Dev Biol. 2013;2:113–129. doi: 10.1002/wdev.57. [DOI] [PubMed] [Google Scholar]
- Slaugenhaupt SA, et al. Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am J Hum Genet. 2001;68:598–605. doi: 10.1086/318810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397–411. doi: 10.1038/nrc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, et al. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat Biotechnol. 2007;25:1468–1475. doi: 10.1038/nbt1365. [DOI] [PubMed] [Google Scholar]
- Lee G, Chambers SM, Tomishima MJ, Studer L. Derivation of neural crest cells from human pluripotent stem cells. Nat Protoc. 2010;5:688–701. doi: 10.1038/nprot.2010.35. [DOI] [PubMed] [Google Scholar]
- Chambers SM, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, et al. Reproducible establishment of hemopoietic supportive stromal cell lines from murine bone marrow. Exp Hematol. 1989;17:145–153. [PubMed] [Google Scholar]
- Barberi T, et al. Neural subtype specification of fertilization and nuclear transfer embryonic stem cells and application in parkinsonian mice. Nat Biotechnol. 2003;21:1200–1207. doi: 10.1038/nbt870. [DOI] [PubMed] [Google Scholar]
- Elkabetz Y, et al. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008;22:152–165. doi: 10.1101/gad.1616208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mica Y, Lee G, Chambers SM, Tomishima MJ, Studer L. Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell Rep. 2013;3:1140–1152. doi: 10.1016/j.celrep.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, et al. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962–967. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]