

Abstract
Therapies based on RNA interference, using agents such as siRNA, are limited by the absence of safe, efficient vehicles for targeted delivery in vivo. The barriers to siRNA delivery are well known and can be individually overcome by addition of functional modules, such as conjugation of moieties for cell penetration or targeting. But, so far, it has been impossible to engineer multiple modules into a single unit. Here, we describe the synthesis of degradable nanoparticles that carry eight synergistic functions: 1) polymer matrix for stabilization/controlled release; 2) siRNA for gene knockdown; 3) agent to enhance endosomal escape; 4) agent to enhance siRNA potency; 5) surface-bound PEG for enhancing circulatory time; and surface-bound peptides for 6) cell penetration; 7) endosomal escape; and 8) tumor targeting. Further, we demonstrate that this approach can provide prolonged knockdown of PLK1 and control of tumor growth in vivo. Importantly, all elements in these octa-functional nanoparticles are known to be safe for human use and each function can be individually controlled, giving this approach to synthetic RNA-loaded nanoparticles potential in a variety of clinical applications.
Keywords: PLGA, Nanoparticle, Gene delivery, RNA interference
1. Introduction
The discovery of RNA interference (RNAi) with small-interfering RNA (siRNA) has led to more than fourteen clinical trials in the past decade [1]. Still clinical success of RNAi therapeutics is uncertain; four clinical trials were dropped recently because of toxicity or lack of efficacy [2-5]. Delivery remains the most significant challenge for RNAi therapeutics. Among the current clinical trials, six employ nanocarriers as delivery vehicles: five are lipid-based and one is polymer-based. In general, lipid-based nanocarriers deliver siRNA with high efficiency, but their clinical use is limited by toxicity, particularly at high therapeutic doses during systemic administration (which was the reason for the recent failure of one clinical trial [5]). Polyplexes of a cyclodextrin-containing polymer are the only polymer-based systems in current clinical trials, as well as the only targeted carrier [6]. No obvious immune response was observed during systemic administration using this carrier at low doses, although elevated levels of blood urea nitrogen and creatinine were observed, suggesting kidney toxicity at high doses [7,8]. However, the stability of polyplexes during long-term storage on shelf or long-term circulation post-administration is unclear. In addition, polyplexes are not able to produce controlled release mechanism, which is essential for a persistent therapeutic effect. To advance beyond the current point, better delivery systems for siRNA are needed.
An ideal delivery vehicle for siRNA needs to have several features, including: 1) the ability to persistently activate RNAi with high efficiency, 2) the ability to target delivery to specific organs or tissues, 3) the ability to maintain stable structure, and 4) an excellent safety profile. Recently, progress has been made in engineering multifunctional nanocarriers such as lipid nanoparticles [9,10] to meet some of these criteria. However, compared to most other types of delivery systems, solid polymeric nanoparticles such as those synthesized from poly(lactic-co-glycolic acid) (PLGA) have important advantages. PLGA was approved by the Food and Drug Administration (FDA) in 1969 and, since that time, has been in continuous, safe clinical use. Drug-releasing particles of PLGA were first approved in 1989 (Lupron Depot®), and are widely used, in large measure because of their reliability in controlling the release of agents over periods ranging from days to weeks, which is important for producing a persistent therapeutic effect. Furthermore, because nanoparticles of PLGA are solid (unlike lipid and polyplexes), they are stable and able to protect nucleic acids from degradation during circulation in blood stream, making them good carriers for oligonucleotides such as siRNA [11-14]. The solid phase also allows long-term storage and convenient use in clinic. Additionally, PLGA nanoparticles can be modified for targeted delivery [15,16]. The major drawback of PLGA nanoparticles is low siRNA delivery efficiency, although new strategies for improvement by enhancing cell uptake [17-21], increasing endosomal escape [21,22], increasing nucleus translocation [23,24] and enhancing RNAi potency [25] have been reported. In this study, we combine these new approaches into a multifunctional system, with eight separately controllable functions, which we evaluated for effectiveness in cultured cells and in animals.
2. Materials and methods
2.1. Materials
All chemicals were purchased from Sigma–Aldrich unless otherwise noted. Plasmid DNA expressing luciferase, pGL4.13, was purchased from Promega. All peptides were synthesized at the W.M. Keck Facility at Yale University. siRNAs were synthesized through Dharmacon. siRNA target sequences used were: PLK1: 5′-AGATCACCCTCCTTAAATATT-3′ as previously reported [26]; Luciferase (pGL4.13): 5′-GCTATGAAGCGCTATGGGC-3′; Scramble: 5′-GTCAAGTCTCACTTGCGTC-3′.
2.2. Cell culture
Human embryonic kidney 293 (HEK293T) and lung cancer A549 cell lines were obtained from ATCC (American Type Culture Collection). Cells were grown in DMEM medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen), in a 37 °C incubator containing 5% CO2.
2.3. Synthesis of PLGA-PLL
Synthesis of PLGA-PLL was accomplished via coupling using dicyclohexyl carbodiimide (DCC) as previously reported [14,27]. In a typical reaction, PLGA (3 g, 50:50 PLGA Acid End Group; i.v. ~0.67 dL/g; Absorbable Polymers: Pelham, AL) and poly(ε-carbobenzoxyl-l-lysine) in 5 M excess (200 mg; 1000–4000 MW, Sigma–Aldrich, St Louis, MO) were dissolved in dimethlyformamide (6 mL, DMF; Sigma–Aldrich) in a dry round-bottom flask under argon. DCC (58 mg; Sigma–Aldrich) and dimethylaminopyridine (0.31 mg, DMAP; Sigma–Aldrich) were dissolved in 2 mL DMF under argon, then added to the polymer solution and allowed to stir for ~48 h. The reacted solution was diluted by the addition of chloroform and precipitated in methanol. The dried polymer was then re-dissolved in chloroform, precipitated in ether, and dried under vacuum for 24 h. Unconjugated PLL was removed during the precipitation and subsequent washes. Dried protected product was placed in a round bottom flask, purged with argon, and dissolved in 10 mL hydrogen bromide, 30% wt in acetic acid (Sigma–Aldrich) and allowed to stir for 90 min for deprotection. The polymerwas precipitated in ether and washed until the product changed from a yellow to off-white appearance. The product was then dissolved in chloroform and precipitated in ether. The polymer was vacuum dried for 24 h to remove all traces of ether. Samples before and after deprotection were collected to confirm modification of the polymer and subsequent removal of protecting carbobenzoxyl (CBZ) groups. The samples were dissolved in trifluoroethanol (TFE; Sigma–Aldrich) and evaluated from 200 to 350 nm using spectroscopy (Cary 50 Bio UV–Vis Spectrophotometer, Varian, Palo Alto, CA).
2.4. Activation of PEG
PEG (molecular weight ~8000 Da, Acros Organics) was activated with CDI. Briefly, PEG was dissolved in dioxane at 37 °C. An 8:1 (CDI:PEG) molar excess of CDI was added and the resulting mixture was stirred under argon at 37 °C for 2 h. Unreacted CDI was removed by dialysis in deionized water for 12 h. The dialysate was changed every hour. The resulting solution was frozen in liquid nitrogen and freeze-dried for 3 days [15].
2.5. Synthesis of PLGA-PLL-PEG
A mixture of excess activated PEG and PLGA-PLL (5:1 molar ratio PEG:PLGA-PLL) was dissolved in anhydrous DMF and allowed to stir under argon. After 48 h, the polymer solution was diluted with chloroform and precipitated in methanol. Unconjugated PEG is soluble in methanol and was therefore removed. Polymer dissolution and precipitation was repeated two times to ensure the removal of unconjugated PEG. Successful synthesis was verified using methods that we have previously reported [15].
2.6. Synthesis of PLGA-PLL-PEG-iRGD
In a typical reaction, 100 mg of PLGA-PLL-PEG was mixed with 10.1 mg of iRGD peptide (3 times molar excess) in 3 ml of anhydrous dimethylformamide (DMF) with trifluoroethanol (TFE) and stirred. After 24 h, the polymer solution was diluted and dialyzed against deionized water for 12 h to remove unconjugated peptide. The dialysate was changed every hour. The PLGA-PLL-PEG-iRGD was then lyophilized and stored for use. Successful synthesis was verified using methods that we have previously reported [15].
2.7. Synthesis of avidin–palmitic acid
Synthesis of avidin–palmitic acid was performed as described previously. Briefly, avidin at 10 mg/mL was reacted with 10-fold excess of NHS–palmitic acid in PBS containing 2% deoxycholate buffer. The mixture was sonicated briefly and gently mixed at 37 °C overnight. Reactants were dialyzed against PBS containing 0.15% deoxycholate to remove excess fatty acid and hydrolyzed ester [16].
2.8. Synthesis of nanoparticles
Polymers were synthesized and verified as previous reported with slightly modifications [14,15,27]. Nanoparticles loaded with DNA or siRNA were synthesized by a double-emulsion solvent evaporation technique. In a typical synthesis, 100 mg of PLGA (50:50, Polysciences and Birmingham) was dissolved in 2 ml dichloromethane (DCM) to form the oil phase (O). 200 ml of deionized water containing 500 μg DNA or 100 nmol of siRNA as the inner aqueous phase (W1) was added dropwise to the polymer solution and sonicated to form the primary emulsion (W1/O). The W1/O emulsion was then added dropwise to 4 ml of 2.5% polyvinyl alcohol (PVA) as the outer aqueous phase (W2) and sonicated to form the second emulsion. The final emulsion was poured into a beaker containing aqueous 0.3% (ν/ν) PVA and stirred for 3 h to allow the DCM to evaporate and the particles to harden. Particles were collected by centrifugation, washed and applied for peptide conjugation.
Small molecules, including TCHD, CQ and QC, are water soluble and were included in the inner aqueous phase (W1) for incorporation into nanoparticles. ENX has limited solubility in water and was first dissolved in a mixed solvent of DCM/methanol (ν/ν, 90:10). The ENX solution was then further used to dissolve the polymer and finally used as the oil phase (O). To display avidin on the surface of the nanoparticles, avidin–palmitic acid was added to the outer aqueous phase (W2) containing 2.5% polyvinyl alcohol (PVA). During all syntheses, the total volume of the inner aqueous phase (W1), the outer aqueous phase (W2) and the oil phase (O) remained unchanged.
2.9. Conjugation of peptides to the surface of nanoparticles
Conjugation of peptides was conducted after evaporation of the organic solvent, DCM. For conjugation of peptide X, avidin–palmitic acid was used during the synthesis of nanoparticles. Avidin–palmitic acid was synthesized as reported before [16]. Typically, 3 h after the emulsion steps, the nanoparticles were collected by centrifugation and washed twice. Nanoparticles were then resuspended in 5 ml of PBS. Next, a 10× molar excess of the biotinylated peptide X was added to the nanoparticle suspension and rotated for 1 h at room temperature for conjugation.
For conjugation of peptide Y, PLGA-PLL was used for the synthesis of nanoparticles. Three hours after the emulsion steps, the nanoparticles were collected by centrifugation and resuspended in 5 ml phosphate buffer solution, pH 7.3. For a typical conjugation, 20 mg of NHS-PEG-Mal (JenKem Technology) was added to the nanoparticle suspension and rotated for 1 h at room temperature. Subsequently, nanoparticles were washed to remove unconjugated NHS-PEG (5000 Da)-Mal and resuspended in 5 ml phosphate buffer solution. A 3× molar excess of cysteine-terminated peptide Y was then added to the nanoparticle suspension and rotated for another hour at room temperature, for the conjugation of peptide Y.
For the synthesis of nanoparticles with both peptides X and Y, avidin–palmitic acid and PLGA-PLL were used for particle synthesis, respectively. The conjugation began with cysteine-terminated peptide Y, after which peptide X was conjugated. Conjugations were done using the same procedures as described above.
For the displaying of peptides X, Y, and Z (iRGD), the same procedures were used as for the conjugation of peptides X and Y, except that PLGA-PLL was replaced with a mixture of PLGA-PLL and PLGA-PLL-PEG-iRGD.
2.10. Quantification of surface ligand density
The density of avidin (for peptide X conjugation) on the nanoparticle surface was determined using the micro bicinchoninic acid assay (Pierce, Thermo Scientific) as previously reported [16,28]. The density of NH2 groups available for peptide Y conjugation and activated −OH groups available for peptide Z (iRGD) conjugation were quantified using the fluorescence probes NHS-PEG-FITC and NH2-PEG-FITC, respectively. Conjugation of PEG-FITC to nanoparticles was performed as described above. Conjugation of NHS-PEG-FITC to the nanoparticles was performed as previously described [15].
2.11. Characterization of nanoparticle loading
To determine loading and encapsulation efficiency of siRNA and small molecules, 3–5 mg of nanoparticles were completely dissolved in 1 ml of DMSO at room temperature. For quantification of the amount of siRNA encapsulated, the polymer solution was diluted (100×) in TE buffer and subjected to siRNA quantification using the Quant-iT™ RiboGreen® RNA Kit according to the manufacturer’s instructions (Invitrogen). Blank nanoparticles were used as the background control. A serial dilution of free siRNA was used to make a standard curve.
QC is a compound which has excellent fluorescent properties and can be excited at 436 nm with an emission peak at 525 nm. Based on this, QC in polymer solution was quantified, with a serial dilution of QC serving as the standard curve. Blank nanoparticles were used as the background control.
Quantification of ENX was based on the ability of ENX to sensitively enhance the fluorescence intensity of Tb3+ - sodium dodecylbenzene sulfonate (SDBS) as previously reported [29]. To quantify the content of ENX encapsulated in nanoparticles, 25 ul of solution was added to a mixed solution of 25 μl of glycine (5%) - NaOH buffer solution (pH 8.0), 25 ul of 0.3 mM Tb3+ solution, 25 μl of 5 mM SDBS solution and 100 μl of deionized water and allowed to stand for 20–30 min. The fluorescence intensity was measured with an excitation wavelength of 290 nm and an emission wavelength of 545 nm using a spectrophotometer (Spectromax M5, Molecular Devices). Because QC also emits fluorescence when combined with Tb3+ - SDBS, the signal from ENX was obtained by subtracting the fluorescence intensity of QC from the total fluorescence intensity. Serial dilutions of QC and ENX were used to generate respective standard curves.
2.12. In vitro controlled release
Nanoparticles (3–5 mg) were suspended in 1.0 ml of phosphate buffer (pH 7.4), and incubated at 37° with gentle shaking (70 rpm). Release of siRNA or small molecules was monitored at several time intervals over 7 days. At each sampling time, the nanoparticle suspension was centrifuged for 10 min at 12,000 rpm. The supernatant was removed for quantification of siRNA or small molecules and an equivolume of PBS was replaced for continued monitoring of release. Detection of siRNA or small molecules was conducted using the same methods as described above.
2.13. Scanning electron microscopy (SEM)
Particle size was characterized by scanning electron microscopy (SEM). Briefly, samples were mounted on carbon tape and sputter-coated with gold, under vacuum, in an argon atmosphere, using a sputter current of 40 mA (Dynavac Mini Coater, Dynavac, USA). SEM analysis was carried out with a Philips XL30 SEM using a LaB electron gun with an accelerating voltage of 3 kV. The mean particle diameter and size distribution of the nanoparticles was determined by image analysis of >1000 particles using image analysis software (ImageJ, National Institute of Health). These micrographs were also used to assess particle morphology.
2.14. In vitro transfection and evaluation
In a typical transfection experiment for both lipofectamine and nanoparticles, cells were plated in 48 well plates, in 0.25 ml medium without penicillin or streptomycin, at a density of 8–10 × 104 cells/ml 24 h before transfection. Transfection using either Lipofectamine 2000 or Lipofectamine RNAiMAX (Invitrogen) followed the standard protocols described in the manufacturer’s manual. For DNA transfection, 1 μl of Lipofectamine 2000 and 0.4 μg DNAwas used. For siRNA delivery, 1 μl of Lipofectamine RNAiMAX and 6 pmol of siRNA was used. For transfection using nanoparticles, nanoparticles were resuspended in cell culture medium at a concentration of 2 mg/ml. After brief sonication, 0.25 ml of nanoparticle suspension was added to the cells. After 24 h of incubation, the solution containing nanoparticles was removed and replaced with fresh cell culture medium.
The effects of transfection on cell proliferation was quantified by the standard MTT assay. Expression of luciferase was quantified using Luciferase Assay Reagent (Promega), in accordance with the standard protocol described in the manufacturer’s manual.
2.15. Quantitative RT-PCR
Twenty-four hours after transfection, cells were washed and collected in cold PBS. Total RNA (1 μg) was isolated using an RNeasy® Mini Kit (Qiagen) according to the manufacturer’s protocol. Purified total mRNA was reverse transcribed using the iScriptTM cDNA Synthesis Kit (Biorad) to generate cDNA. Real-time PCR was performed on 2 μl of cDNA combined with the iQ SyBrTM Green (Biorad) reagents for fluorescent detection of PCR products. All reactions were performed in a 50 μl volume, in duplicate. Primers used for RT-PCR [30] are: GAPDH forward: 5′-TCGCTCTCTGCTCCTCCTGTTC-3′; GAPDH reverse: 5′-CGCCCAATACGACCAAATCC-3′; PLK1 forward: 5′-GACAAGTACGGCCTTGGGTA-3′; PLK1 reverse: 5′-GTGCCGTCACGCTCTATGTA-3′. PCR parameters consisted of a 5 min activation of the DNA polymerase at 95 °C, followed by 40 cycles of 95 °C × 20 s, 60 °C × 30 s, and 72 °C × 20 s. Standard curves were generated and the relative amount of PLK1 mRNA was normalized to GAPDH mRNA. Specificity was verified by melt curve analysis and agarose gel electrophoresis.
For analysis of PLK1 expression in residual tumors after treatment, tumors was collected and stored on dry ice immediately. Tumor tissues were then broken down using a homogenizer (IKAWorks, Inc). RNA extraction and RT-PCR were performed as described above.
2.16. In vivo tumor homing of nanoparticles
Female athymic (NCr-nu/nu) nude mice were used for this study and maintained in a sterile environment. This project was approved by the Yale University Institutional Animal Care and Utilization Committee (IACUC). To establish tumors, mice received subcutaneous flank injections of 1 × 106 A549 tumor cells. When tumor volumes reached ~100 mm3, the mice were randomly divided into three groups, with five mice per treatment group, as follows: group 1: nanoparticles without PEG (X conjugation only); group 2: nanoparticles with PEG (X and Y conjugations); and group 3: nanoparticles with PEG and iRGD (X, Y and Z conjugations). All nanoparticles also encapsulated coumarin-6 (C6), with a loading of approximately 3.0–3.4 mg per 100 mg polymer. Mice received a single intravenous injection of nanoparticles, at a dose of 2 mg per animal, through the tail vein. Two hours after injection, mice were sacrificed and tumors were excised, weight, and homogenized in DMSO. The fluorescence of the extracted C6 (in DMSO) was measured with an excitation wavelength of 444 nm and an emission wavelength of 538 nm, using a spectrophotometer (Spectromax M5, Molecular Devices).
2.17. Antitumor activity in xenograft model
Female athymic (NCr-nu/nu) nude mice were purchased from the National Cancer Institute and maintained in a sterile environment. This project was approved by the Yale University Institutional Animal Care and Utilization Committee (IACUC). To establish tumors, mice received subcutaneous flank injections of 1 × 106 A549 tumor cells. When tumor volumes reached ~50 mm3, mice were randomly divided into three groups, with eight mice per treatment group, as follows: group 1: PBS control group; 2: nanoparticles loaded with scramble siRNA; group 3: nanoparticles loaded with siRNA against PLK1. Intravenous injections at 2 mg per mouse were performed through tail vein, three days a week (Monday, Wednesday and Friday), and stopped one week prior to the conclusion of the experiments. Tumor size was measured three times a week using traceable digital vernier calipers (Fisher). The tumor volume was determined by measuring the length (l) and width (w), and then calculating the volume (V) using the following formula: V = lw2/2. The growth curve was plotted using the mean of the tumor volumes for each treatment group, at each timepoint. The animals were scarified 7 days after the last treatment, at which point the tumors were excised and fixed in formalin for immunohistochemistry. Serial sections were obtained and stained with hematoxylin and eosin (H&E) and Terminal Deoxynucleotidyl Transferase (TUNEL) for analysis of therapeutic effect.
2.18. Statistical analysis
All data were taken in triplicate and reported as mean and standard deviation. Comparison of two conditions was evaluated by a paired Student’s t-test. One-way ANOVA analysis was performed to determine the statistical significance of treatment related changes in tumor volume, in athymic nude mice. A p ≤ 0.05 was considered to indicate a statistically significant difference.
3. Results and discussion
3.1. Synthesis of octa-functional nanoparticles
To produce PLGA nanoparticles in which we could separately control multiple functions, we synthesized particles with three levels of complexity (Fig. 1a–c). First, to add molecules to the nanoparticle surface, we used a versatile method for displaying biotinylated ligands on surface-bound palmitylated avidin (Fig. 1a). [16] Second, to provide distinct functional groups for linkage of other ligands, we prepared avidin-decorated particles from a copolymer of PLGA and poly(l-lysine) (PLL) [14] (Fig. 1b): the lysine groups allow surface functionalization and stabilize anions (such as nucleic acids) within the core. These two approaches allow post-fabrication modification of particles: we call ligands added via avidin “X peptides” and ligands added via conjugation to lysine “Y peptides.” To allow a third level of control, we conjugated some ligands—which we call “Z peptides”—directly to PLGA-PLL prior to particle fabrication (Fig. 1c). All three of these approaches yield spherical nanoparticles ~150 nm in diameter (Fig. 1a–c). Significantly, this approach allows for separate control of the binding of up to three different surface ligands. All approaches are compatible with our previously published methods for simultaneous encapsulation of nucleic acids and drugs (Fig. 1a–c): encapsulated siRNA and drugs such as quinacrine (QC) and enoxacin (ENX) are slowly released from the particles upon incubation in water (Fig. 1d,e). As a result of these methods, we were able to produce nanoparticles having up to eight separate functions, all of which were under individual control (Fig. 1f).
Fig. 1.
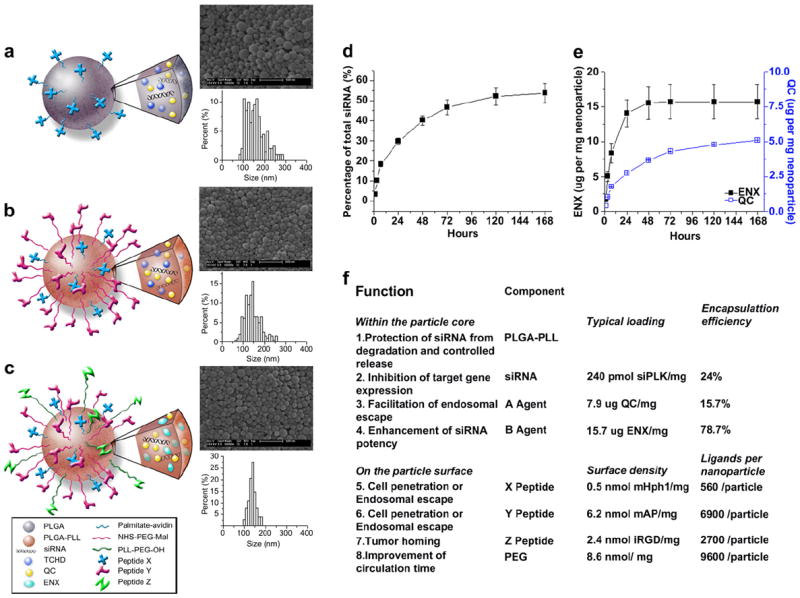
Characterization of nanoparticles. (a), (b), and (c): schematics, morphology – as captured by SEM, and size distribution of multifunctional nanoparticles conjugated with a single peptide X (a), two peptides X and Y (b) and three peptides X, Y, and Z (c). (d) Controlled release of siRNA from nanoparticles loaded with siRNA against PLK1. (e) Controlled release of QC and ENX from nanoparticles loaded with siRNA against PLK1. (f) Components of a functional nanoparticle loaded with siRNA against PLK1 and their individual functions.
3.2. Rational design of nanoparticles for intracellular delivery
We first used these particles to test the hypothesis that certain peptides—which are known to facilitate cellular delivery in other settings—would enhance intracellular delivery of degradable nanoparticles. All nanoparticles were loaded with a plasmid encoding luciferase to facilitate detection of successful intracellular delivery. The TAT peptide, originally derived from a human immunodeficiency virus (HIV)-1 protein, is a well-studied cell penetration peptide (CPP) that is widely used to enhance cellular uptake. Recently, the TAT peptide was modified by adding histidine (H) residues that can be protonated under acidic conditions, such as those found in endosomes, to form helical structures that destabilize the endosomal membrane and promote escape. [21] One of these peptides, mTAT (Fig. 2a), significantly improved gene delivery efficiency, by up to 7000 fold [21]. We conjugated mTAT to PLGA nanoparticles as X peptides and tested their gene delivery effectiveness when exposed to HEK293T cells at 1 mg/ml. Addition of mTAT significantly improved gene delivery (Fig. 2b). Gene delivery efficiency peaked with addition of 5 mg avidin/mTAT per 100 mg PLGA, where the luciferase expression was 270 fold greater than that obtained using nanoparticles without mTAT. Further increases in surface treatment did not increase gene delivery, possibly due to interference in the release of DNA due to avidin, as we observed previously [31].
Fig. 2.
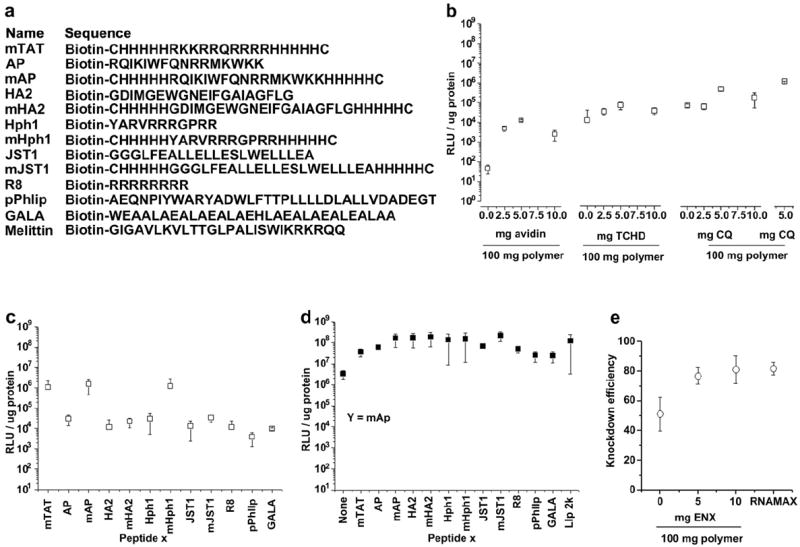
Effect of peptides and small molecule enhancers for DNA and siRNA delivery. (a) Sequences for the biotinylated peptides used in screening for the optimal transfection enhancement effect. (b) Conjugation of mTAT and incorporation of TCHD and CQ or QC enhances the gene delivery efficiency of nanoparticles. (c) Transfection efficiency of nanoparticles conjugated with all peptides listed in (a). (d) Combination of two peptides for gene delivery. (e) Incorporation of ENX enhances knockdown efficiency of nanoparticles encapsulated with siRNA.
Next, we examined the simultaneous release of small molecules that facilitate either nuclear translocation or endosomal escape. The nuclear envelope consists of an impermeable double bilayer, perforated by many nuclear pore complexes (NPCs), each ~40 nm in diameter. NPCs strictly regulate nucleo-cytoplasmic transport. Molecules smaller than 30 kDa, or nucleic acids up to approximately 300 base pairs, passively transit through NPCs [23,24,32]. In contrast, active transport, mediated mainly by receptors of the importin family, is necessary for the translocation of large molecules [24,32]. Nuclear transport can be a substantial obstacle: less than 0.1% of the DNA microinjected into cytoplasm is able to enter the nucleus for transcription [32]. Peptides or proteins containing a nuclear localization sequence (NLS), which are recognized by importin active transporters, have been used to enhance transport, but with limited success [24,32]. As an alternative, we sought to enhance nuclear membrane permeability by release of trans-cyclohexane- 1,2-diol (TCHD), an amphipathic alcohol, which is able to reversibly collapse the permeability barrier of the NPCs [24]. When TCHD was added to the internal polymer phase of nanoparticles (Fig. 1a), gene delivery efficiency was enhanced by a factor of 6, with an optimal dose of 5 mg/100 mg polymer (Fig. 2b). Inadequate escape from endosomes is another significant barrier to delivery, which can be overcome by delivery of agents such as chloroquine (CQ), a weak base that buffers endosomal luminal pH [22]. Addition of CQ to nanoparticles significantly enhanced delivery efficiency by a factor of 7 at a dose of 5 mg/100 mg polymer (Fig. 2b). We screened a range of compounds with purported ability to promote endosomal escape: among them, the CQ analog quinacrine (QC) [22] produced even better enhancement (Fig. 2b).
Since TAT, the first cell penetrating peptide, was reported in 1988 [20], a variety of other fusigenic or endosomolytic peptides have been proposed and tested. One of the most significant advantages of the avidin–biotin system used for ligand conjugation is its versatility. We exploited the versatility and ease of coupling to the avidin–biotin system, and individually coupled a panel of peptides in the peptide X position (Fig. 2a). All experiments were performed with a common batch of nanoparticles, to which a different peptide was coupled. With the exception of Melittin, which was toxic in our experiments, addition of each peptide enhanced gene delivery, with AP, HA2, Hph1, and JST1 producing the best effects (Fig. 2c). However, none of these peptides performed better than mTAT, presumably due to their limited endosomolytic ability. To improve this, we modified these peptides by flanking them with five H residues, creating modified AP, HA2, Hph1, and JST1 (mAP, mHA2, mHph1, and mJST), which further enhanced their delivery effectiveness, particularly in mAP and mHph1, which yielded 50 and 40 fold increases in delivery, respectively, over AP and Hph1 (Fig. 2c). Amongst all peptides, mAP demonstrated superior delivery enhancement.
Despite the versatility of the avidin–biotin (peptide X) system, we have found it difficult to control the relative attachment of multiple peptides. Therefore, we developed the peptide Y modification, by making nanoparticles from PLGA-PLL. Amine groups of PLL are exposed on the surface of these nanoparticles, allowing for easy surface modification (Fig. 1b). Generally, the conjugation of peptides through this peptide Y approach yields higher densities than the peptide X approach (Fig. 1f), leading to higher efficiency of delivery when mAP is in the peptide Y position, compared to the peptide X position (compare mAPin Fig. 2d to c). Because they use different but compatible chemistries, the peptide X and Yapproaches can be used simultaneously, which we hypothesized would lead to synergistic effects. With mAP in the peptide Y position, we screened the same panel of peptides through the peptide X position. Nanoparticles with mAP in both peptide X and Y positions were able to transfect cells with an efficiency of 130 times greater than nanoparticles with mAP in the X position only, and 50 times higher than nanoparticles with mAP in the Y position only (Fig. 2d). Combinations of peptides also produced effective delivery, with mHA2, mHph1 and mJST1 providing the best synergy with mAP, resulting in gene delivery efficiencies comparable to lipofectamine 2000 (Fig. 2d).
Testing of these modifications was facilitated by delivery of plasmid DNA, which was evaluated using luciferase expression, but we sought to apply these approaches to delivery of siRNA. Since the regulation of RNA transcription by siRNA occurs in the cytoplasm, entry into the nucleus is not necessary for siRNA delivery, so TCHD was eliminated from these particles. HEK293T cells that stably expressed the luciferase gene were used for the evaluation of silencing effects due to siRNA directed against the luciferase gene. These nanoparticles (which contained mAP in the peptide X and Y positions) inhibited luciferase expression by 50% (Fig. 2e). To improve siRNA delivery, we added enoxacin (1-ethyl-6-fluoro-1,4-dihydro-4-oxo-7-(1-piperazinyl)-1,8-naphthyridine-3-carboxylic acid, ENX) to the polymer interior (Fig. 1c). ENX is a potent third-generation quinolone antibiotic, used to treat bacterial infections with minimal side effects, which is nontoxic in vitro at up to 150 μM [25,29]. Recently, ENX was found to significantly enhance siRNA activity both in vitro and in vivo, most likely by promoting the loading of siRNA duplexes onto RNAinduced silencing complex (RISC) by facilitating the interaction between trans-activation-responsive region RNA-binding protein (TRBP) and RNAs [25]. When 10 mg of ENX was added per 100 mg of polymer, knockdown was significantly enhanced: nanoparticles inhibited luciferase expression by 80%, comparable to RNAiMAX (Fig. 2e). The loading of ENX in these particles was 16 μg per mg nanoparticles, of which 14 μg was released during the first 24 h (Fig. 1e,f), which produces ~44.0 μM of ENX in cell culture. Interestingly, nanoparticles encapsulating siRNA maintained sustained gene knockdown compared to RNAiMAX: seven days after transfection, the knockdown of luciferase in cells transfected with RNAiMAX was about 24% of the control cells, in contrast to 36% in cells transfected with siRNA nanoparticles (p < 0.05).
3.3. PLGA nanoparticles deliver siRNAs for cancer treatment
To test the activity of these octa-functional nanoparticles in vivo, we chose a cancer model using the human lung cancer cell line A549. To identify the best peptide combination for A549 cell transfection, we performed a two-round peptide screen. In the first round of screening, using the peptide X approach, mAP and mHph1 provided the highest effectiveness (Fig. 3a). In the second round, mAP was added via the peptide Y approach and the other peptides were screened as peptide X. Peptide mHph1 provided the best synergy with mAP, producing nanoparticles that transfected A549 cells at an efficiency comparable to, but slightly lower than, Lipofectamine 2000 (Fig. 3a).
Fig. 3.
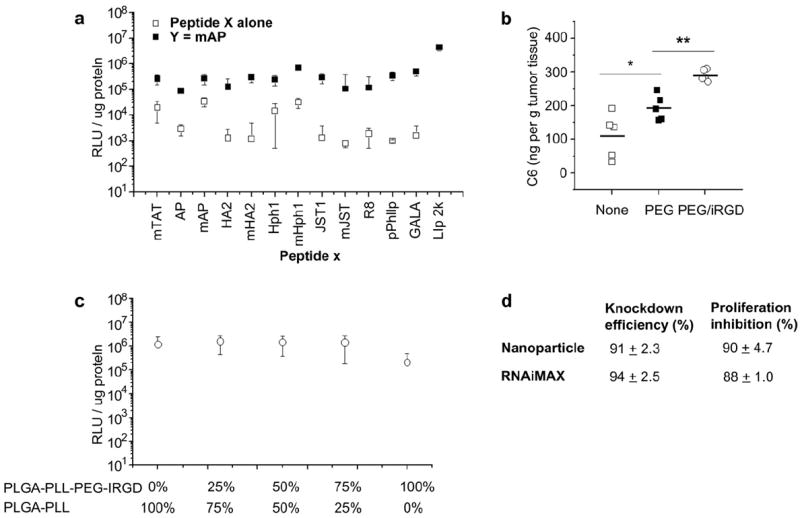
Nanoparticles for delivery of siRNA against PLK1 to A549 cells. (a) Conjugation of a single peptide or two peptides enhances gene delivery to A549 cells. (b) Distribution of nanoparticles loading with a fluorescence dye, Coumarin-6, with different surface modifications. (c) The effect of replacing mHph1 with iRGD on gene delivery efficiency. PLGA-PLL and PLGA-PLL-PEG-iRGD were mixed at the ratios indicated for display of the peptide iRGD. (d) Comparison of delivery efficiency and cytotoxicity of siRNA against PLK1, using nanoparticles and RNAiMAX, on A549 cells.
A major challenge for in vivo tumor delivery is the ability to reach tumor cells after intravenous injection. We employed a heterobifunctional PEG for conjugation of peptide Y, to take advantage of PEG’s potential to minimize binding to plasma proteins, thus improving circulation time and reducing the clearance of nanoparticles by the reticuloendothelial system (RES). Improved circulation of nanoparticles in the blood enhances the delivery to tumor tissues by the enhanced permeability and retention (EPR) effect [33]. For nanoparticles of the current formulation, ~9600 moieties of PEG were conjugated to the surface of each nanoparticle, which covers 8.6% of the nanoparticle surface (Fig. 1f). These nanoparticles, rendered fluorescent by addition of coumarin-6 to the polymer core, were injected via the tail vein in mice bearing A549 tumors in the flank: nanoparticles with PEG on the surface accumulated in tumors 1.8 times more efficiently than nanoparticles without modifications (Fig. 3b).
To further improve the delivery of nanoparticles to tumors in vivo, we introduced a third peptide, iRGD, to the surface of nanoparticles [15]. iRGD (CRGDKGPDC) is a modified form of RGD, which is reported to significantly enhance the penetration of nanoparticles into tumors. When nanoparticles conjugated with iRGD were administrated in vivo, iRGD homed them specifically to tumors through a three step process: binding to tumor endothelium through the interaction of RGD, avb3, and avb5, a proteolytic cleavage to expose a binding motif for neuropilin-1, which then facilitates penetration into tumor tissue [18]. Conjugation of iRGD to our nanoparticles was accomplished in the peptide Z position, using a variation on an established approach [15], in which iRGD (pepZ, peptide Z) was conjugated to PLGA-PLL-PEG prior to particle synthesis. When the PLGA-PLL-PEG-pepZ is added to the polymer phase of the emulsion during nanoparticle formation, peptide Z is displayed on the surface of the nanoparticles (Fig. 1c). To allow formation of particles with peptides in X, Y, and Z positions simultaneously, we developed procedures for mixing PLGA-PLL-PEG-pepZ with PLGA-PLL in the polymer blend. Interestingly, when mHph1 is in the X position and mAP is in the Y position, up to 75% of PLGA-PLL can be replaced with PLGA-PLL-PEG-iRGD without affecting gene delivery efficiency (Fig. 3c). It is known that A549 cells highly express both avb3 [34] and neuropilin-1 [35], molecules required by iRGD for penetration of cells and tissues, which provides the most likely explanation for why a large percentage of mHph1 can be replaced by iRGD without affecting gene delivery efficiency. We tested the ability of particles conjugated with iRGD to accumulate in tumors. When 25% of the PLGA-PLL was replaced with PLGA-PLL-PEG-iRGD, the amount of nanoparticles in the tumors increased by 1.5 fold, as compared to nanoparticles with only PEG and no iRGD on the surface (p < 0.01) (Fig. 3b). When these particles were loaded with siRNA against PLK1, they provided knockdown of PLK1 expression in A549 cells (91.0%), which was similar to the inhibition seen with Lipofectamine RNAiMAX (94.4%) (Fig. 3d). In addition, delivery siRNA against PLK1 significantly inhibited the in vitro proliferation of A549 cells (Fig. 3d).
We then tested the effectiveness of our nanoparticles for treatment of human cancer in vivo. PLK1 is an essential gene for mitosis and plays an important role in maintaining genomic stability. It is overexpressed in many types of cancer, where it is associated with tumorigenesis and correlated with poor patient prognosis. Therefore, PLK1 represents a promising therapeutic target for anticancer therapies [36]. siRNAs against PLK1 have been reported to significantly inhibit proliferation of a variety of tumor cells both in vitro and in vivo [26,30]. siRNA sequences against PLK1 have been screened previously: the best sequence significantly inhibits tumor cell proliferation in vitro and tumor growth in vivo by activation of RNAi machinery, as evidenced by the presence of RNAi-specific mRNA cleavage products [26]. We loaded this siRNA sequence into nanoparticles that were optimized for intravenous delivery. Intravenous injection of these nanoparticles significantly inhibited tumor growth (p < 0.05) (Fig. 4a) and the expression of the PLK1 gene in the treatment group was 47% of the expression in the control group, even 7 days after the last treatment (Fig. 4b). Histologically, tumors from control treatments revealed a highly cellular mass with prominent nucleoli; in contrast, tumors from animals treated with siPLK1 revealed a much less cellular mass with a lower nuclear-cytoplasmic ratio (Fig. 4c), and a marked increase in the number of apoptotic cells (Fig. 4d).
Fig. 4.
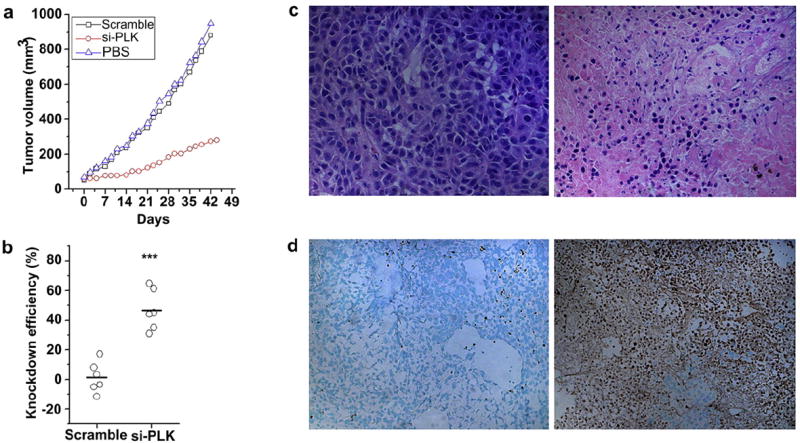
Systemic delivery of siRNA against PLK1, using nanoparticles, for inhibition of tumor growth in vivo. (a) Antitumor effects of nanoparticles loaded with siRNA against PLK1. (b) Expression of the PLK1 gene in residual tumors. (c) H&E staining of the control tumor (left) and the siPLK1 treated tumor (right); 40× magnification. (d) TUNEL staining of the control tumor (left) and the siPLK1 treated tumor (right); 20× magnification.
Since its discovery in the 1990s, there has been tremendous interest in using RNAi in the development of a new class of therapeutic agents. However, there are still no RNAi-based drugs approved for clinical use. The major obstacle to clinical utility is inadequate in vivo delivery. An ideal delivery vehicle needs to have several features, including: 1) the ability to deliver RNAi at high efficiency, 2) the ability to target delivery to specific organs, 3) the ability to maintain stable structure, and 4) an excellent safety profile. Unfortunately, the majority of delivery vehicles now proposed for clinical use, including many in clinical testing, fail to satisfy all of these requirements. The octa-functional PLGA nanoparticle system described in this study, which satisfies all four requirements, may be useful for future clinical applications.
4. Conclusions
We have developed PLGA-based nanoparticles that provide targeted and efficient delivery of siRNA. These nanoparticles are truly multifunctional and offer several advantages over other technologies for siRNA delivery. First, PLGA, the major biomaterial used in this nanoparticle synthesis, has been safely used in clinical applications for many decades. Secondly, PLGA is a biodegradable polymer. During the course of its degradation via hydrolysis, siRNA is slowly released providing sustained knockdown of the target gene. Finally, up to three different ligands can be precisely conjugated to the nanoparticle surface. This provides flexibility in modifying these nanoparticles for a variety of potential applications.
Acknowledgments
We thank Caroline Weller for assistance with animal experiments and Dr. Joseph M. Piepmeier for encouragement and helpful discussions. This work was supported by grants from the Chicago Institute of Neurosurgery and Neuroresearch (CINN) Foundation, the Voices Against Brain Cancer (VABC) Foundation, and a pilot grant from the Yale Institute for Nanoscience and Quantum Engineering (YINQE).
References
- 1.Vaishnaw AK, Gollob J, Gamba-Vitalo C, Hutabarat R, Sah D, Meyers R, et al. A status report on RNAi therapeutics. Silence. 2010;1:14. doi: 10.1186/1758-907X-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Opko Health, Inc. Safety and efficacy study evaluating the combination of Bevasiranib & Lucentis therapy in wet AMD (COBALT) 2011 Available from URL: http://clinicaltrials.gov/ct2/show/NCT00499590?term=bevasiranib&rank=1; Online.
- 3.Allergen, Inc. A study using intravitreal injections of a small interfering RNA in patients with age-related macular degeneration. 2011 Available from URL: http://clinicaltrials.gov/ct2/show/NCT00395057?term=macular+degeneration+siRNA&rank=1; Online.
- 4.Pfizer, Inc. Prospective, randomized, multi-center, comparator study evaluating efficacy and safety of PF-04523655 versus laser in subjects with diabetic macular edema (DEGAS) 2011 Available from URL: http://clinicaltrials.gov/ct2/show/NCT00701181?term=PF-04523655&rank=1; Online.
- 5.Tekmira Pharmaceuticals Corporation. Study to evaluate the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD), of Liposomal siRNA in subjects with high cholesterol. 2011 Available from URL: http://clinicaltrials.gov/ct2/show/NCT00927459?term=siRNA&rank=10; Online.
- 6.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–70. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu-Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ. Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing’s sarcoma. Cancer Res. 2005;65:8984–92. doi: 10.1158/0008-5472.CAN-05-0565. [DOI] [PubMed] [Google Scholar]
- 8.Heidel JD, Yu Z, Liu JY, Rele SM, Liang Y, Zeidan RK, et al. Administration in non-human primates of escalating intravenous doses of targeted nanoparticles containing ribonucleotide reductase subunit M2 siRNA. Proc Natl Acad Sci U S A. 2007;104:5715–21. doi: 10.1073/pnas.0701458104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Bathula SR, Yang Q, Huang L. Targeted nanoparticles deliver siRNA to melanoma. J Invest Dermatol. 2010;130:2790–8. doi: 10.1038/jid.2010.222. [DOI] [PubMed] [Google Scholar]
- 10.Pirollo KF, Rait A, Zhou Q, Hwang SH, Dagata JA, Zon G, et al. Materializing the potential of small interfering RNA via a tumor-targeting nanodelivery system. Cancer Res. 2007;67:2938–43. doi: 10.1158/0008-5472.CAN-06-4535. [DOI] [PubMed] [Google Scholar]
- 11.Shea LD, Smiley E, Bonadio J, Mooney DJ. DNA delivery from polymer matrices for tissue engineering. Nat Biotechnol. 1999;17:551–4. doi: 10.1038/9853. [DOI] [PubMed] [Google Scholar]
- 12.Jones DH, Corris S, McDonald S, Clegg JC, Farrar GH. Poly(DL-lactide-co-glycolide)- encapsulated plasmid DNA elicits systemic and mucosal antibody responses to encoded protein after oral administration. Vaccine. 1997;15:814–7. doi: 10.1016/s0264-410x(96)00266-6. [DOI] [PubMed] [Google Scholar]
- 13.Luo D, Woodrow-Mumford K, Belcheva N, Saltzman WM. Controlled DNA delivery systems. Pharm Res. 1999;16:1300–8. doi: 10.1023/a:1014870102295. [DOI] [PubMed] [Google Scholar]
- 14.Blum JS, Saltzman WM. High loading efficiency and tunable release of plasmid DNA encapsulated in submicron particles fabricated from PLGA conjugated with poly-L-lysine. J Control Release. 2008;129:66–72. doi: 10.1016/j.jconrel.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertram JP, Williams CA, Robinson R, Segal SS, Flynn NT, Lavik EB. Intravenous hemostat: nanotechnology to halt bleeding. Sci Transl Med. 2009;1:11ra22. doi: 10.1126/scitranslmed.3000397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fahmy TM, Samstein RM, Harness CC, Mark Saltzman W. Surface modification of biodegradable polyesters with fatty acid conjugates for improved drug targeting. Biomaterials. 2005;26:5727–36. doi: 10.1016/j.biomaterials.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 17.Cheng CJ, Saltzman WM. Enhanced siRNA delivery into cells by exploiting the synergy between targeting ligands and cell-penetrating peptides. Biomaterials. 2011;32:6194–203. doi: 10.1016/j.biomaterials.2011.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16:510–20. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328:1031–5. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–93. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 21.Lo SL, Wang S. An endosomolytic tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials. 2008;29:2408–14. doi: 10.1016/j.biomaterials.2008.01.031. [DOI] [PubMed] [Google Scholar]
- 22.Cheng J, Zeidan R, Mishra S, Liu A, Pun SH, Kulkarni RP, et al. Structure-function correlation of chloroquine and analogues as transgene expression enhancers in nonviral gene delivery. J Med Chem. 2006;49:6522–31. doi: 10.1021/jm060736s. [DOI] [PubMed] [Google Scholar]
- 23.Bastos R, Pante N, Burke B. Nuclear pore complex proteins. Int Rev Cytol. 1995;162B:257–302. doi: 10.1016/s0074-7696(08)62619-4. [DOI] [PubMed] [Google Scholar]
- 24.Vandenbroucke RE, Lucas B, Demeester J, De Smedt SC, Sanders NN. Nuclear accumulation of plasmid DNA can be enhanced by non-selective gating of the nuclear pore. Nucleic Acids Res. 2007;35:e86. doi: 10.1093/nar/gkm440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shan G, Li Y, Zhang J, Li W, Szulwach KE, Duan R, et al. A small molecule enhances RNA interference and promotes microRNA processing. Nat Biotechnol. 2008;26:933–40. doi: 10.1038/nbt.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Judge AD, Robbins M, Tavakoli I, Levi J, Hu L, Fronda A, et al. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J Clin Invest. 2009;119:661–73. doi: 10.1172/JCI37515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lavik EB, Hrkach JS, Lotan N, Nazarov R, Langer R. A simple synthetic route to the formation of a block copolymer of poly(lactic-co-glycolic acid) and polylysine for the fabrication of functionalized, degradable structures for biomedical applications. J Biomed Mater Res. 2001;58:291–4. doi: 10.1002/1097-4636(2001)58:3<291::aid-jbm1019>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 28.Steenblock ER, Fahmy TM. A comprehensive platform for ex vivo T-cell expansion based on biodegradable polymeric artificial antigen-presenting cells. Mol Ther. 2008;16:765–72. doi: 10.1038/mt.2008.11. [DOI] [PubMed] [Google Scholar]
- 29.Tong CL, Xiang GH. Sensitive determination of enoxacin by its enhancement effect on the fluorescence of terbium(III)-sodium dodecylbenzene sulfonate and its luminescence mechanism. J Luminescence. 2007;126:575–80. doi: 10.1007/s10895-006-0107-7. [DOI] [PubMed] [Google Scholar]
- 30.McNamara JO, 2, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, et al. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol. 2006;24:1005–15. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- 31.Cu Y, LeMoellic C, Caplan MJ, Saltzman WM. Ligand-modified gene carriers increased uptake in target cells but reduced DNA release and transfection efficiency. Nanomedicine. 2010;6:334–43. doi: 10.1016/j.nano.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Douglas KL. Toward development of artificial viruses for gene therapy: a comparative evaluation of viral and non-viral transfection. Biotechnol Prog. 2008;24:871–83. doi: 10.1021/bp070319o. [DOI] [PubMed] [Google Scholar]
- 33.Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Control Release. 2010;145:178–81. doi: 10.1016/j.jconrel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Achilefu S, Bloch S, Markiewicz MA, Zhong T, Ye Y, Dorshow RB, et al. Synergistic effects of light-emitting probes and peptides for targeting and monitoring integrin expression. Proc Natl Acad Sci U S A. 2005;102:7976–81. doi: 10.1073/pnas.0503500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frankel P, Pellet-Many C, Lehtolainen P, D’Abaco GM, Tickner ML, Cheng LL, et al. Chondroitin sulphate-modified neuropilin 1 is expressed in human tumour cells and modulates 3D invasion in the U87MG human glioblastoma cell line through a p130Cas-mediated pathway. Embo Rep. 2008;9:983–9. doi: 10.1038/embor.2008.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006;6:321–30. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]