

Abstract
To develop potent and selective nNOS inhibitors, a new series of double-headed molecules with chiral linkers that derive from natural amino acid derivatives have been designed and synthesized. The new structures integrate a thiophenecarboximidamide head with two types of chiral linkers, presenting easy synthesis and good inhibitory properties. Inhibitor (S)-9b exhibits a potency of 14.7 nM against nNOS and is 1134 and 322-fold more selective for nNOS over eNOS and iNOS, respectively. Crystal structures show that the additional binding between the aminomethyl moiety of 9b and propionate A on the heme and tetrahydrobiopterin (H4B) in nNOS, but not eNOS, contributes to its high selectivity. This work demonstrates the advantage of integrating known structures into structure optimization, and it should be possible to more readily develop compounds that incorporate bioavailability with these advanced features. Moreover, this integrative strategy is a general approach in new drug discovery.
Keywords: nitric oxide synthase, selective inhibition, thiophenecarboximidamide, heme, tetrahydrobiopterin, protein crystallography
Nitric oxide (NO), an inorganic free radical, has diverse roles in both normal and pathological processes, including the regulation of blood pressure, neurotransmission, and macrophage defense systems.1 NO is synthesized from the enzymatic catalysis of L-arginine to L-citrulline by three isoforms of nitric oxide synthase (NOS): neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS). Although NO plays numerous physiological roles beneficial to the host organism, the overproduction of NO has been implicated in numerous disease states.2 In particular, excess NO produced by nNOS has been shown to play an important role in the development of several disorders including stroke,3 pain (migraine, chronic tension-type headache, visceral, and neuropathic),4 and neurodegenerative disorders5 (Parkinson's,6 Alzheimer's,7 Huntington's diseases8). However, any inhibitors to treat these conditions must be selective for nNOS since inhibition of eNOS and iNOS could result in unwanted effects, such as hypertension, atherogenesis, and interference with normal immune system functions.9 Therefore, the development of selective nNOS inhibitors is of critical interest and derives both from a therapeutic perspective and from their use as specific pharmacological tools.10
Early NOS inhibitors were structural analogs of the natural substrate L-arginine, which lack isoform selectivity.11, 12 Some later L-arginine mimetic NOS inhibitors are potent and selective.13,14 Although many recent reports disclose the diversity of NOS inhibitors,15 very few provided integrative profiles because of either low selectivity or poor bioavailability. Therefore, investigations into the synthesis and chemistry of novel isoform-selective NOS inhibitors and improvement of structure optimization and bioavailability still remain as ongoing challenges.
Our research group has developed two lead compounds, 216 and 317 (Figure 1), which feature easy synthesis and potent and selective nNOS inhibitory properties. To develop an integrative strategy and diversify the scope of NOS inhibitors, an early nNOS-selective inhibitor with good bioavailability properties (118) was combined with our two natural chiral linkers in 2 and 3 to explore new opportunities for structure optimization. These new compounds are expected to incorporate the features of easy accessibility, excellent inhibitory properties, and improved bioavailability.
Figure 1.
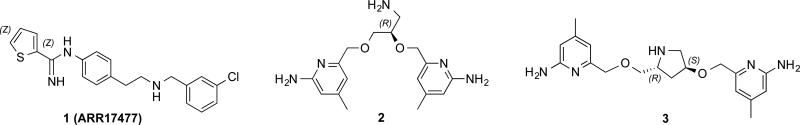
Structures of lead compounds 1, 2 and 3
The synthesis of 9 began with a commercially available amino compound, which was protected as the dimethylpyrrole (4) followed by a one-step ether synthesis, connecting two 3-nitrobenzyl heads simultaneously (5, Scheme 1). The nitro groups in 5 were then reduced to amines with hydrazine in the presence of Raney-Ni to afford 6. Coupling with 7 in ethanol19 led to double substituted 8. After removal of the protecting group under microwave conditions, 9 was successfully obtained.
Scheme 1.

Synthesis of 9a,b. Reagents and conditions: (a) p-TsOH, toluene, 2,5-hexanedione, reflux, 77-79%; (b) 3-nitrobenzyl bromide, NaH, NaI, DMF, 0 °C, 75-78%; (c) Raney-Ni, hydrazine hydrate, CH3OH, 98%; (d) 7, EtOH, 75-83%; (e) HCl in EtOH, microwave, 120 °C, 61-63%.
Four isomers of 14 followed a similar synthetic route as 9 except for starting compounds 10a-d, which were previously reported.17 Compounds 14 were obtained in good yields after removal of the Boc protecting group in 13 under acidic conditions (Scheme 2).
Scheme 2.

Synthesis of 14a-d. Reagents and conditions: (a) 3-nitrobenzyl bromide, NaH, NaI, DMF, 0 °C, 57-85%; (b) Raney-Ni, hydrazine hydrate, CH3OH, 97-99%; (c) 7, EtOH, 66-87%; (d) 1.25 M HCl in CH3OH, 89-96%.
The synthesis of 21 is described in Scheme 3. Starting from 15, a Mitsunobu reaction was used to give 16, followed by reduction of the methyl carboxylic ester to alcohol 17 with LiBH4. Coupling of the fluorobenzyl group to the prolinyl linker moiety afforded 18. After reduction of the nitro group to the amine, coupling with 7, and removal of the Boc protecting group, 21 was obtained in good yield.
Scheme 3.

Synthesis of 21. Reagents and conditions: (a) PPh3, DIAD, 3-nitrophenol, THF, 83%; (b) LiBH4, THF, 98%; (c) 3-fluorobenzylbromide, NaH, NaI, DMF, 0 °C, 91%; (d) Raney-Ni, hydrazine hydrate, CH3OH, 97%; (e) 7, EtOH, 77%; (f) 1.25 M HCl in CH3OH, 85%.
The synthetic route to 25a,b uses intermediate 17 from the synthesis of 21 (Scheme 4). A second Mitsunobu reaction connected the second nitrophenyl head (either 3- or 4-substituted) to the prolinyl linker moiety, providing 22. Upon reduction of the nitro groups, the resulting 23 was allowed to react with amidothiol ether 7 to generate the double-headed thiophene scaffold (24). Compound 25 was obtained after removal of the Boc protecting group.
Scheme 4.

Synthesis of 25. Reagents and conditions: (a) PPh3, DIAD, 3-nitrophenol, THF, 72-82%; (b) Raney-Ni, hydrazine hydrate, CH3OH, 98%; (c) 7, EtOH, 74-77%; (d) 1.25 M HCl in CH3OH, 86-94%.
Compound 32 has a similar structure as 21 (Scheme 5), but with transposition of the two head groups. Starting from 26, coupling of the fluorobenzyl group to the prolinyl linker via an ether synthesis was achieved prior to the Mitsunobu reaction to install the nitrophenyl group. The remaining synthetic steps are the same as those employed for 21 with similar yields.
Scheme 5.

Synthesis of 32. Reagents and conditions: (a) 3-fluorobenzylbromide, NaH, NaI, DMF, 0 °C, 68%; (b) LiBH4, THF, 98%; (c) PPh3, DIAD, 3-nitrophenol, THF, 76%; (d) Raney-Ni, hydrazine hydrate, CH3OH, 98%; (e) 7, EtOH, 71%; (f) 1.25 M HCl in CH3OH, 93%.
All of the inhibitors were assayed against the three different isoforms of NOS, including rat nNOS, bovine eNOS, and murine macrophage iNOS using L-arginine as a substrate (Table 1). The (S)- enantiomer of 9 (9b) was found to be four times more potent (14.7 nM) than the (R)-isomer with 5-fold enhancement of eNOS selectivity (1134-fold) and 2.5-fold enhancement of iNOS selectivity (322-fold). Of the four isomers of 14 with a pyrrolidine linker, compound 14a is the most potent (13.2 nM; although the other three range from 21.4-28.1 nM), but 14d is the most selective (658-fold for n/e and 98-fold for n/i). Again, the (S)-stereochemistry at the amino group is favored. Two analogs of 25, with two different bis-substituted phenyl linkages, show similar binding behaviors. Replacing one of the phenyl thiophenecarboximidamide moieties in 25a with a fluorobenzene does not lead to any improvement, as shown by 21 and 32.
Table 1.
Kia values of inhibitors for rat nNOS, bovine eNOS and murine iNOS

| |||||
|---|---|---|---|---|---|
| Compounds |
Ki [μM] |
Selectivityb |
|||
| nNOS | eNOS | iNOS | e/n | i/n | |
| 9a | 0.0590 | 12.44 | 8.11 | 210 | 137 |
| 9b | 0.0147 | 16.68 | 4.73 | 1134 | 322 |
| 14a | 0.0132 | 2.47 | 1.11 | 190 | 85 |
| 14b | 0.0281 | 3.63 | 1.99 | 130 | 71 |
| 14c | 0.0214 | 3.17 | 1.23 | 151 | 58 |
| 14d | 0.0221 | 1.45 | 2.11 | 658 | 96 |
| 21 | 0.0684 | 5.89 | 6.17 | 86 | 90 |
| 25a | 0.0282 | 3.34 | 2.42 | 119 | 86 |
| 25b | 0.0270 | 1.15 | 4.40 | 142 | 163 |
| 32 | 0.101 | 1.86 | 9.28 | 18 | 92 |
The IC50 values were measured for three different isoforms of NOS including rat nNOS, bovine eNOS, and murine macrophage iNOS using L-arginine as a substrate with a standard deviation of ±10%. The corresponding Ki values were calculated from the IC50 values using the equation Ki = IC50 / (1 + [S] / Km) with known Km values (nNOS, 1.3 μM; iNOS, 8.3 μM; eNOS, 1.7 μM).
The Ki values are used to calculate the selectivity.
The only change between the two double headed inhibitors, 9a and 9b, is the chirality at the position of the aminomethyl moiety, but their potencies and selectivities show significant differences. To reveal the structural basis for these observations we have determined the crystal structures of both nNOS and eNOS in complex with 9a and 9b. As shown in Figure 2 for the nNOS structures, both 9a and 9b use the carboximidamide moiety to H-bond with the Glu592 side chain, and the rest of the heme distal side is occupied by the thiophene and phenyl rings. However, the orientations of 9a and 9b are flipped 180° to one another. For 9a, it is the phenyl thiophenecarboximidamide moiety that is two atoms away from the chiral center, whereas for 9b it is the one that is three atoms away from the chiral center that binds to the NOS active site near Glu592. The phenyl thiophenecarboximidamide moiety is important in NOS active site recognition and thus is the fundamental source of the inhibitory potency. Because of different binding orientations, the aminomethyl group at the chiral center exhibits two distinct interactions with the protein: the one in 9b is sandwiched in between the THB and heme propionate A, making H-bonds with both and replacing a water molecule normally bound at this position (Figure 2B); however, the one in 9a points away from the water site and thus the water remains (Figure 2A). These extra H-bonds with 9b are likely the main source of the 4-fold better potency of 9b compared to 9a. The second phenyl thiophenecarboximidamide moiety makes non-bonded contacts with Leu337 and Trp306 of chain B, a little tighter in the case of 9b than in 9a, but this portion of the inhibitors is partially disordered in both structures, and hence it is difficult to make a comparison with high certainty.
Figure 2.

View of the active site of nNOS complexed with inhibitor 9a (A)(PDB: 4UPM) and 9b (B)(PDB: 4UPN), showing also the omit Fo – Fc electron density for the bound inhibitor at the 2.5 δ contour level. Note that a different phenyl thiophenecarboximidamide moiety in each case is used to anchor the inhibitor to the active site. Key hydrogen bonds are depicted with dashed lines. Structural figures were prepared with PyMOL (www.pymol.org).
The structures of eNOS with 9a and 9b bound, shown in Figure 3, also provide an explanation for the isoform selectivity of 9b. Different from the case of the nNOS structures, the same phenyl thiophenecarboximidamide moiety, with 3-atoms from the chiral center, in both 9a and 9b anchors the inhibitor to the eNOS active site. The aminomethyl groups in both inhibitors, regardless the chirality, make a H-bond with the water molecule bound in between the THB and heme propionate A rather than replacing the water. Its capabilities of replacing the water and making direct H-bonds with both the THB and heme seems to be the underlying reasons for 9b, compared to 9a, being both a more potent nNOS inhibitor and more selective for nNOS over eNOS. Moreover, how the second phenyl thiophenecarboximidamide moiety that extends out of the active site in both of the inhibitor-bound structures depends on the protein environment in each isoform. In nNOS, the second thiophene ring points toward a pocket defined by Trp306 (chain B), Ser602, and Arg603 (Figure 2), while in eNOS, the second phenyl thiophenecarboximidamide is rather disordered but is pointing toward the opening of the active site access channel near Leu107, with some differences in exact positions between 9a and 9b (Figure 3). The aforementioned pocket in nNOS, where the second thiophene ring points, is surrounded in eNOS by Trp76 (chain B), His373, and Arg374 (Figure 3). The bulkier His373 (compared to Ser602 in nNOS) side chain makes this pocket too shallow to allow the thiophene to fit optimally. One additional, potentially important, difference between the two isoforms is another amino acid variant that is flanking this pocket, Asp309 in nNOS vs. Gly79 in eNOS. The exposed N atom of the carboximidamide group of 9b is less than 6.0 Å from Asp309 in the nNOS dimer (Figure 2B). This could provide more favorable electrostatic interactions in nNOS than in eNOS when the second thiophenecarboximidamide moiety fits in the pocket. Optimization of these interactions in nNOS requires 9b to adopt a compact conformation, which favors close interactions between the aminomethyl group and heme propionate. In sharp contrast, the lack of these additional interactions with the carboximidamide group in eNOS enables the tail end of 9b to adopt an extended conformation and clearly binds less tightly, given the poor quality of the electron density in this region. This extended tail conformation of 9b in eNOS could also be the reason why the aminomethyl group at the chiral center is pulled away from the heme propionate and THB, leaving a structural water untouched (Figure 3B).
Figure 3.

View of the active site of eNOS complexed with inhibitor 9a (A)(PDB: 4UPQ) and 9b (B)(PDB: 4UPR), showing also the omit Fo – Fc electron density for the bound inhibitor at the 2.5 δ contour level. Note that the same phenyl thiophenecarboximidamide moiety in both cases is used for active site recognition. Key hydrogen bonds are depicted with dashed lines.
Using chiral proline analogs (10a-d) as the linker, we synthesized inhibitors 14a-d. These four compounds show very similar binding behaviors, with 14a being the most potent (13.2 nM) but 14d the most selective (658-fold for nNOS over eNOS). We determined the structures of only 14d bound to both nNOS and eNOS (Figure 4). Again, 14d relies on the phenyl thiophenecarboximidamide moiety, the one with a 3-atom linker to the center pyrrolidine ring, to anchor itself to the NOS active site. The longer linker length pushes the pyrrolidine ring out so that in nNOS the ring nitrogen sits in between the two heme propionates at about 4.0 Å without making H-bonds (Figure 4A). The second phenyl thiophenecarboximidamide moiety of 14d in both nNOS and eNOS is poorly defined with weak density. The disordering is more severe in eNOS than in nNOS. However, the residual density is clear enough to indicate that the thiophene ring is heading out of the active site access channel and near Pro708 in nNOS (Pro479 in eNOS). That means that even in nNOS 14d is too lengthy, compared to 9a-b, to fit into the pocket next to Ser602 and Arg603.
Figure 4.

View of the active site of nNOS (A)(PDB: 4UPO) and eNOS (B)(PDB: 4UPS) complexed with inhibitor 14d, showing also the omit Fo – Fc electron density for the bound inhibitor at the 2.5 δ contour level. Key hydrogen bonds are depicted with dashed lines.
To gain information on how 14a binds to nNOS, we simply placed a model of 14a in the structure of nNOS-14d according to the binding mode observed for 14d. Considering the structural similarity between these two analogs, it is not surprising to see that the first phenyl thiophenecarboximidamide moiety including the pyrrolidine ring nitrogen of 14a can mimic the exact position seen for 14d (Figure S1). Differences only start from the different chirality at the pyrrolidine ring. Even so, the second thiophene ring can still end up with the same position seen for 14d. Apparently, in the cases of 14a-d, the different stereochemistry does not impact the inhibitor binding mode large enough to significantly change the potency or isoform selectivity.
The linker length in 14a-d is too long to allow pyrrolidine interactions with the heme propionates, and is flexible enough to compensate for any differences generated at the chiral centers. Therefore, we further developed inhibitors 25a,b to include the same chiral pyrrolidine at the center as in 14a but with one less methylene in the linker than their parent compound, in the hope that a shorter structure might fit the substrate pocket of NOS better. We also tried both meta- (25a) and para-substitution (25b) at the phenyl ring to adjust the inhibitor binding conformation. The structures of 25b bound to nNOS and eNOS (Figure 5) show that the para-substituted phenyl thiophenecarboximidamide moiety with a 2-atom linker to the pyrrolidine is not only able to anchor the inhibitor to the NOS active site in both cases but also allows the pyrrolidine nitrogen to make direct H-bonds (~ 3.0 Å) with both heme propionates in nNOS (Figure 5A). To our surprise, the additional H-bonds in 25b do not translate to better binding affinity of 25b to nNOS compared to 14a-d (Table 1). The same H-bonds in eNOS cannot be established (Figure 5B), most likely because the second thiophene head is pointing in a different direction in eNOS from that seen in nNOS. Similar to what was seen in nNOS-9a the second thiophene of 25b fits into the pocket defined by Trp306 (chain B), Ser602, and Arg603 (Figure 5A). In contrast, the second thiophene of 25b in eNOS is more disordered than that in the nNOS-25b structure, and the residual density at the lower contour level guides the tail toward the opening of the active site access channel (Figure 5B). This tail orientation pulls the pyrrolidine ring away from the heme in eNOS.
Figure 5.

View of the active site of nNOS (A)(PDB: 4UPP) and eNOS (B)(PDB: 4UPT) coordinated with inhibitor 25b, showing also the omit Fo – Fc electron density for the bound inhibitor at the 2.5 δ contour level. Key hydrogen bonds are depicted with dashed lines.
By superimposing the meta-substituted ring of 25a and 25b (Figure S2), we found that 25a can still orient the pyrrolidine nitrogen in the same position seen for that of 25b, thus making H-bonds with both heme propionates. The second thiophene head can also fall into the same pocket, although the exact position of the second meta-substituted phenyl ring is different in the two cases (Figure S2). Overall, the model provides a good explanation as to why the two compounds show similar binding affinity to nNOS (Table 1), even though the conformation around the phenyl ring is different.
To further investigate the structure-activity relationship of the inhibitors, we designed and synthesized 21 and 32, which integrate features from the structure of 1, but with the linker from 25a. The transposed position of the thiophenecarboximidamide and fluorobenzyl heads was aimed to optimize the structure; however, neither provided improved results compared with 25a. From what we learned from the binding of 14d (Figure 4A) and 25b (Figure 5A) to nNOS, we expected that if 21 were bound to the nNOS active site via its phenyl thiophenecarboximidamide moiety, the short one-atom ether linker might not be able to bring the pyrrolidine nitrogen to an optimal position to make good H-bonds with heme as observed for 25b, which could lead to poorer binding. As for 32, with its phenyl thiophenecarboximidamide anchored to the nNOS active site, the 2-atom ether linker should be able to position the pyrrolidine to interact with the heme propionates. However, the fluorobenzyl group attached to a short linker might not make significant van der Waals contacts with the protein to establish a binding better than that with 25a.
In conclusion, we designed and synthesized a new series of double-headed inhibitors with two types of chiral linkers derived from amino acids. By combining a phenyl thiophenecarboximidamide head with our previously-developed skeleton, inhibitor 9b emerges as being both a potent nNOS inhibitor, Ki = 14.7 nM and exhibiting dual selectivities of 1134-fold (e/n) and 322-fold (i/n). Compared to 1, the redesigned structures contain two ether bonds, which allows for easy synthesis and structure-ready-to-optimize features. Crystallography shows that additional binding of the aminomethyl moiety of 9b to both the propionate A on heme and THB, replacing a structural water molecule there, explains the high selectivity. A similar feature, in which a secondary amine is bound to the same site leading to good potency and isoform selectivity, was also observed with another series of NOS inhibitors developed in our lab.20 Without synthesizing a large number of small molecules, we were able to discover new, potent and selective inhibitors, easily accessed, which confirm the efficiency of utilizing an integrative strategy.
Supplementary Material
Acknowledgments
The authors are grateful for financial support from the National Institutes of Health (GM049725 to R.B.S. and GM057353 to T.L.P.). We thank Dr. Bettie Sue Siler Masters (NIH grant GM52419, with whose laboratory P.M. and L.J.R. are affiliated). B.S.S.M. also acknowledges the Welch Foundation for a Robert A. Welch Distinguished Professorship in Chemistry (AQ0012). P.M. is supported by grants 0021620849 from MSMT of the Czech Republic. We also thank the beamline staff at SSRL and ALS for their assistance during the remote X-ray diffraction data collections.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data (detailed synthetic procedures and full characterization (1H-NMR, 13C NMR) of compounds 9, 14, 21, 25, and 32; crystal preparation, X-ray diffraction data collection and structure refinement procedures and statistics) associated with this article can be found, in the online version, at doi:
References
- 1.Mustafa AK, Gadalla MM, Synder SH. Sci. Signal. 2009;2:1. doi: 10.1126/scisignal.268re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vallance P, Leiper J. Nature Rev. Drug Discovery. 2002;1:939. doi: 10.1038/nrd960. [DOI] [PubMed] [Google Scholar]
- 3.Sims NR, Anderson MF. Neurochem. Int. 2002;40:511. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 4.Miclescu A, Gordh T. Acta Anaesthesiol. Scand. 2009;53:1107. doi: 10.1111/j.1399-6576.2009.02054.x. [DOI] [PubMed] [Google Scholar]
- 5.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield AD, Stella AMG. Nature Rev. Neurosci. 2007;8:766. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L, Dawson VL, Dawson TM. Pharmacol. Ther. 2006;109:33. doi: 10.1016/j.pharmthera.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Dorheim M-A, Tracey WR, Pollock JS, Grammas P. Biochem. Biophys. Res. Commun. 1994;205:659. doi: 10.1006/bbrc.1994.2716. [DOI] [PubMed] [Google Scholar]
- 8.Norris PJ, Waldvogel HJ, Faull RLM, Love DR, Emson PC. Neuronsicence. 1996;72:1037. doi: 10.1016/0306-4522(95)00596-x. [DOI] [PubMed] [Google Scholar]
- 9.Cayatte AJ, Palacino JJ, Horten K, Cohen RA. Arterioscler. Thromb. 1994;14:753. doi: 10.1161/01.atv.14.5.753. [DOI] [PubMed] [Google Scholar]
- 10.Maddaford S, Annedi SC, Ramnauth J, Rakhit S. Annu. Rep. Med. Chem. 2009;44:27. [Google Scholar]
- 11.Narayanan K, Spack L, McMillan K, Kilbourn RG, Hayward MA, Masters BSS, Griffith OW. J. Biol. Chem. 1995;270:11103. doi: 10.1074/jbc.270.19.11103. [DOI] [PubMed] [Google Scholar]
- 12.Ji H, Erdal EP, Litzinger EA, Seo J, Zhu Y, Xue F, Fang J, Huang J, Silverman RB. Reitz AB, Choudhary MI, Atta-ur-Rahman, editors. In Frontiers in Medicinal Chemistry. Bentham Science. 2009;54:842. [Google Scholar]
- 13.Ijuin R, Umezawa N, Nagai S, Higuchi T. Bioorg. Med. Chem. Lett. 2005;15:2881. doi: 10.1016/j.bmcl.2005.03.078. [DOI] [PubMed] [Google Scholar]
- 14.Ji H, Gomez-Vidal JA, Martásek P, Roman LJ, Silverman RB. J. Med. Chem. 2006;49:6254. doi: 10.1021/jm0604124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a Ramnauth J, Speed J, Maddaford SP, Dove P, Annedi SC, Renton P, Rakhit S, Andrews J, Silverman S, Mladenova G, Zinghini S, Nair S, Catalano C, Lee DKH, Felice MD, Porreca FJ. Med. Chem. 2011;54:5562. doi: 10.1021/jm200648s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Annedi SC, Ramnauth J, Maddaford SP, Renton P, Rakhit S, Mladenova G, Dove P, Silverman S, Andrews JS, Felice MD, Porreca F. J. Med. Chem. 2012;55:943. doi: 10.1021/jm201564u. [DOI] [PubMed] [Google Scholar]; c Liang G, Neuenschwander K, Chen X, Wei L, Munson R, Francisco G, Scotese A, Shutske G, Black M, Sarhan S, Jiang J, Morize I, Vaz RJ. Med. Chem. Commun. 2011;2:201. [Google Scholar]; d Silverman RB. Acc. Chem. Res. 2009;42:439. doi: 10.1021/ar800201v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jing Q, Li H, Chreifi G, Roman LJ, Martásek P, Poulos TL, Silverman RB. Bioorg. Med. Chem. Lett. 2013;23:5674. doi: 10.1016/j.bmcl.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jing Q, Li H, Roman LJ, Martásek P, Poulos TL, Silverman RB. ACS MedChemLett. 2014;5:56. doi: 10.1021/ml400381s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a Zhang ZG, Reif D, Macdonald J, Tang WX, Kamp DK, Gentile RJ, Shakespeare WC, Murray RJ, Chopp MJ. Cereb. Blood Flow Metab. 1996;16:599. doi: 10.1097/00004647-199607000-00009. [DOI] [PubMed] [Google Scholar]; b Salerno L, Sorrenti V, Di Giacomo C, Romeo G, Siracusa MA. Curr. Pharm. Des. 2002;8:177. doi: 10.2174/1381612023396375. [DOI] [PubMed] [Google Scholar]
- 19.The synthesis of 7 followed the reported method. Collins JL, Shearer BG, Oplinger JA, Lee S, Garvey EP, Salter M, Duffy C, Burnette TC, Furfine ES. J. Med. Chem. 1998;41:2858. doi: 10.1021/jm980072p.
- 20.Kang S, Tang W, Li H, Chreifi G, Martásek P, Roman LJ, Poulos TL, Silverman RB. J. Med. Chem. 2014;57:4382. doi: 10.1021/jm5004182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.