

Abstract
The core alpha1,6-fucosyltransferase (FUT8) catalyzes the transfer of a fucosyl moiety from GDP-fucose to the innermost asparagine-linked N-acetylglucosamine residue of glycoproteins. In mammals, this glycosylation has an important function in many fundamental biological processes and although no essential role has been demonstrated yet in all animals, FUT8 amino acid (aa) sequence and FUT8 activity are very well conserved throughout the animal kingdom. We have cloned the cDNA and the complete gene encoding the FUT8 in the Sf9 (Spodoptera frugiperda) lepidopteran cell line. As in most animal genomes, fut8 is a single-copy gene organized in different exons. The open reading frame contains 12 exons, a characteristic that seems to be shared by all lepidopteran fut8 genes. We chose to study the gene structure as a way to characterize the evolutionary relationships of the fut8 genes in metazoans. Analysis of the intron-exon organization in 56 fut8 orthologs allowed us to propose a model for fut8 evolution in metazoans. The presence of a highly variable number of exons in metazoan fut8 genes suggests a complex evolutionary history with many intron gain and loss events, particularly in arthropods, but not in chordata. Moreover, despite the high conservation of lepidoptera FUT8 sequences also in vertebrates and hymenoptera, the exon-intron organization of hymenoptera fut8 genes is order-specific with no shared exons. This feature suggests that the observed intron losses and gains may be linked to evolutionary innovations, such as the appearance of new orders.
Introduction
Glycosylation of proteins is a key process. Indeed, congenital disorders of glycosylation lead to severe dysfunction and disability. Maturation of glycoproteins in the Golgi apparatus requires hundreds of enzymes (i.e., glycosyltransferases, glycosidases), known as Carbohydrate-Active Enzymes (CAZymes) [1], and also chaperones that act through complex protein-protein interactions.
Fucosylation is one of the most common post-translational modifications. Fucosylated glycans are involved in various biological processes, such as cell adhesion, growth factor receptor modulation, microbial and viral infections, cancer and atherosclerosis [for review 2, 3]. Several fucosyltransferases (FucTs) have been identified and classified in the CAZy glycosyltransferase (GT) families GT-11 (α1,2-FucTs), GT-10 (α1,3/4-FucTs), GT-23 (core α1,6-FucTs, known as fut8), GT-37 (α1,2-FucTs), GT-65 (O-FucTs pofut1 and fut12) and GT-68 (O-FucTs pofut2 and fut13). A previous phylogenetic analysis of vertebrates, invertebrates and bacterial FucTs genes highlighted their ancient and divergent evolution from one or two ancestral genes [4].
While α1,2- and α1,3/4-FucTs are implicated in terminal fucosylation (e.g., histo-blood group antigens) [5], core α1,6-FucT (FUT8) adds fucose to the innermost asparagine-linked N-acetylglucosamine (GlcNAc) of the chitobiose disaccharide-core unit of glycoproteins. This core fucosylation has an essential role in regulating the function of many glycoproteins, such as activation of growth factor receptors [6] and modulation of antibody-dependent cell-mediated cytotoxicity (ADCC) in vitro and in vivo [7]. In mammals, FUT8 physiological relevance has been demonstrated by genetic ablation of the fut8 gene: 80% of these mice die three days after birth [8] and the survivors present severe growth retardation.
The crystal structure of human FUT8 has been resolved [9]. It is a type-II glycoprotein with a short N-terminal cytoplasmic tail, a transmembrane domain, a stem region and a C-terminal catalytic domain. The latter encompasses three peptidic consensus sequences (motif I, motif II and motif III) [4], [9] located in the Rossmann fold [10], a protein motif that binds to nucleotides. These motifs are conserved among α1,2-, α1,6- and O-FucTs, strongly suggesting their implication in the fucose transfer reaction [4], [11]–[14]. Site-directed mutagenesis [9], [12] confirmed this hypothesis demonstrating that most of the highly conserved aa in this region are essential for enzymatic activity. Four disulfide bonds may be important for protein folding and stability [9]. A SH3 domain is also present at the C-terminus in all known FUT8 sequences [15], [16], but its role has not been elucidated yet.
Lepidopteran cells can perform most of the post-translational modifications, including N- and O-glycosylation. N-glycan processing [for review, 17] is comparable to what observed in mammalian cells during the early stages of the glycosylation pathway with the synthesis of GlcNAcβ1,2Manα1,3[Manα1,6]Manβ1,4GlcNAcβ1,4[Fucα1,6]GlcNAc [18]. The glycan structures most frequently identified on glycoproteins synthesized in insect cells are fucosylated paucimannose structures (Man3(Fuc)GlcNAc2) and, to a lesser extent, oligomannose-type glycans [19]. The absence of complex oligosaccharides correlates with the absence of β1,4-galactosyltransferase I (β1,4GalTI) [20], [21] and sialyltransferases [18]. Despite the in silico identification of α2,6-sialyltransferase (st6gal) gene sequences in some lepidopterans, such as Bombyx mori [22], no sialyltransferase activity has been detected in insect cell lines, such as Sf9 (S. frugiperda) cells [18], [23]. In addition, the very low expression level of N- acetylglucosaminyltransferase I (GNT-I) [18], [24], [25] and GNT-II [26] and the presence of a Golgi-associated N-acetylglucosaminidase (FDL) [27], [28] explain the formation of paucimannose structures. In contrast to mammals, two core-fucosylation events with α1,6- and α1,3-linkage have been reported in insects and in plants [29]. Analysis of fucosyltransferase activities in several lepidopteran cell lines showed distinct activities in each cell line. While core α1,3- and core α1,6-fucose are found on glycoproteins expressed in Masmestra brassicae [20], [30], [31] and Trichoplusia ni (High five) cell lines [32], only core α1,6-fucose was found in B mori and Sf9 cells [20], [30], [33].
Although its essential role has not yet been demonstrated in all animals, FUT8 aa sequence and FUT8 enzymatic activity are well conserved throughout the animal kingdom as testified by several molecular cloning and functional studies in vertebrates and invertebrates [34], [35], [36], [37], [38]. In the present study, we report the molecular cloning and functional characterization of a cDNA encoding fut8 from the Sf9 lepidopteran insect cell line. As in most animal genomes, fut8 is a single-copy gene organized in several exons. These properties and the high conservation of amino acids in FUT8 catalytic domain were used to retrieve unique fut8 orthologs from a large variety of metazoan genomes and to study their exon-intron structure evolution in several insect orders. The results of this analysis allow us to propose a model of fut8 evolution throughout the animal kingdom. Furthermore, fut8 evolutionary history could be used to measure the divergences among insect genomes highlighting the frequent intron losses and gains in arthropods.
Results
Cloning of fut8 cDNA from Sf9 cells
A partial cDNA sequence was obtained using a reverse transcription - polymerase chain reaction (RT-PCR) approach and degenerate primers (Table S1). This 860-bp (base pairs) sequence was compared to all non-redundant coding sequences (cds) in the GenBank database using the BlastX program [39] and was identified as belonging to a fut8 gene, with a high identity score (51% aa identity with human FUT8). This sequence was then used to design new exact-match primers for 3′- and 5′-RACE. The full-length cDNA sequence of Sf9 fut8 gene is available under the accession number KC538901.
Sequence analysis of this full-length cDNA sequence showed the presence of three in-frame ATG codons as potential initiation codons ( Figure 1 ). Inspection of the sequences immediately flanking the initiation codon of genes expressed in Sf9 cells (n = 168) [40] allowed us to propose a consensus sequence ( Table 1 ) for initiation of translation (most frequent for gene expression with high or low efficiency) that was in agreement with the start translation sites reported by Cavener and Ray [41] for invertebrates. Based on this consensus sequence, the third ATG codon ( Figure 1 , ATG3) seemed to have the most favorable environment as potential initiation codon ( Table 2 ). Sequence analysis showed that this cDNA encodes a type II protein of 561-aa with a short cytoplasmic tail of 8-aa at the N-terminus followed by a 17-aa (residues 9–25) transmembrane domain. The Golgi luminal part contains the catalytic C-terminal domain with the three highly conserved motif I (residues 336–348), motif II (residues 381–394) and motif III (residues 429–455) consensus sequences [4], [9]. All the critical aa residues (Arg-343, Asp-346, Lys-347, Glu-351, Tyr-360, Asp-387, Asp-431 and Ser-447) implicated in FUT8 enzymatic activity are also perfectly conserved in the Sf9 Fut8 protein ( Figure 1 ). The catalytic domain is preceded by a 78-aa proline-rich region (Pro-279 to Pro-336), like in mammalian FUT8 [15], [35], [36]. As for all known FUT8 sequences, Sf9 FUT8 has a 60-aa SH3 domain at its C-terminus (residues 484–543) [15], [16] ( Figure 1 ). All cysteine residues involved in the formation of disulfide bridges in human FUT8, are also perfectly conserved, except Cys-472 in motif III that is replaced by a glycine residue (G-450) ( Figure 1 and Figure S1). The amino acid identity of the Sf9 FUT8 aa sequences was 86.81%, and 79.89% with the lepidopteran Manduca sexta and Danaus Plexippus FUT8. Identity with dipteran FUT8 sequences ranged between 50.28% (Culex quinquefasciatus pipiens) and 46.90% (Drosophila melanogaster) and was about 44% with vertebrate FUT8 aa sequences (44.24% with human and 44.42% with bovine fut8).
Figure 1. Nucleotide and deduced aa sequences of the Sf9 α1,6-fucosyltransferase cDNA.

The three potential start codons are presented in boxes. ATG3 was considered as the start codon (see Table 2). A potential transmembrane domain (residues 9 to 25) is highlighted in grey. The conserved motifs I, II and III in the active site are underlined. The putative SH3 domain is in bold and underlined with a dashed line. The two potential N-glycosylation sites are underlined with a double line. Solid arrowheads indicate the position of introns. The numbers between brackets represent the position of critical aa conserved in human FUT8.
Table 1. Determination of the optimal sequence for initiation of translation in Sf9 cells.
| Sequences analyzed n = 168 | Position upstream of the start codon* | ||||||||||
| -10 | -9 | -8 | -7 | -6 | -5 | -4 | -3 | -2 | -1 | AUG | |
| A | 43 | 36 | 27 | 35 | 27 | 22 | 39 | 88 | 64 | 57 | |
| U | 26 | 34 | 29 | 27 | 31 | 41 | 11 | 0 | 13 | 6 | |
| G | 14 | 17 | 14 | 7 | 28 | 12 | 7 | 10 | 3 | 11 | |
| C | 17 | 13 | 30 | 31 | 14 | 25 | 43 | 2 | 20 | 26 | |
| Consensus sequence | A | A/U | A/U/C | A/U/C | A/U/G | U | A/C | A | A | A | AUG |
168 genes expressed in Sf9 cells [40] were included in this analysis.
*Nucleotide flanking Sf9 start codons, in percentage.
Table 2. Analysis of the sequences immediately flanking the three potential start codons (ATG1, ATG2 and ATG3 in Figure 1) identified in the fut8 cDNA.
To confirm that this cDNA encoded Sf9 FUT8, we evaluated the enzymatic activity of a recombinant protein produced by expressing the cloned cDNA in the baculovirus-Sf9 cell expression system. As shown in Table 3 , a characteristic fucosyltransferase activity was observed.
Table 3. Determination of α1–6-fucosyltransferase activity.
| Sample | α1,6-fucosyltransferase activity |
| 2D production | 1,229+/−80 |
| 3D production | 2,554+/−35 |
| Negative control | 62+/−1 |
Recombinant FLAG-tagged Sf9 FUT8 was produced using the baculovirus-Sf9 cell system and purified using anti-FLAG M2 affinity gel. The fucosyltransferase activity of the recombinant protein was tested with GDP-[14C]-L-fucose, as substrate, after two days (2D) and three days (3D) of production. The transfer of [14C]-fucose is expressed in cpm.
Restriction-enzyme digested genomic DNA from Sf9 cells hybridized to a single fragment, suggesting the presence of only one copy of the fut8 gene in the Sf9 genome ( Figure 2A ). Fut8 expression was then analyzed by Northern blotting using a specific mono-exonic (exon 3) fut8 probe. Only one transcript of the expected size was detected ( Figure 2B ).
Figure 2. Southern and Northern blot analyses.
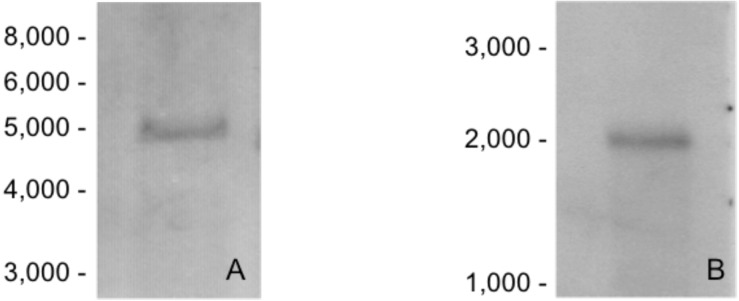
A. Ten µg/lane of EcoRI-digested genomic DNA from Sf9 cells was hybridized with a mono-exonic (exon 3, {E3l}) digoxigenin-labeled Fut8 probe prepared by PCR using the Forint10 and B24 primers (Table S1), as described in Materials and Methods. B. One µg/lane of total RNA was analyzed by Northern blotting. Hybridization was performed with a fut8 anti-sense RNA probe (pos. 825 to pos. 1686 on the cDNA, Figure 1). Molecular weight markers in bp are indicated on the left of each panel.
Phylogenomic analyses
We took advantage of the growing list of sequenced metazoan genomes to identify in silico several potential orthologs of the previously described FUT8 protein sequences. BLAST searches using the human FUT8 sequence and the previously described [4], [9] fucosyl motifs I, II and III, as hallmarks for ortholog identification, allowed the identification of 96 highly conserved FUT8-related sequences from early metazoan genomes to protostome (mainly arthropods) and deuterostome genomes (Table S2). In most of these genomes, one single-copy fut8 gene was identified with the notable exception of Danio rerio, Xenopus laevis and Saccoglossus kowalevskii in which two fut8 paralogs (named fut8A and fut8B) were found. Multiple sequence alignments using ClustalW ( Figure 3 ) highlighted the presence of the three conserved motifs (motifs I, II and III) and of a new “Cys-rich” peptide motif that is highly specific to FUT8 proteins.
Figure 3. Conserved aa and motifs found in all the α1,6-fucosyltransferases sequences.

Schematic representations of the FUT8 protein showing the cytoplasmic (C), transmembrane (T) and stem region (S) characteristic of α1,6-fucosyltransferases. The catalytic domain is in white and motifs I, II and III in grey. In addition, a region found only in α1,6-fucosyltransferase with conserved cysteine residues is indicated by dashed lines and was named “Cys-rich” domain. Conserved aa and those implicated in the enzymatic activity are highlighted with orange stars. The conserved peptide sequences used to generate the motif I, motif II and motif III sequence logos were extracted from multiple alignments of 96 α1,6-fucosyltransferase sequences identified in the databases (Table S2) and visualized at the Weblogos site at Berkeley, as described previously [63]. In the logos, aa are colored according to their chemical properties: polar aa (G, C, S, T, Y) are green, basic (K, R, H) are blue, acidic (D, E) are red, hydrophobic (A, V, L, I, P, W, F, M) are black and neutral polar aa (N, Q) are pink. The overall height of the stacks indicates the sequence conservation at a given position, while the height of the symbol within the stack indicates the relative frequency of each aa at that position. [69], [70].
We then assessed the evolutionary relationships of these animal sequences as described in the Materials and Methods section. To understand the orthology relationships of arthropods FUT8 sequences, we performed Maximum Likelihood (ML) ( Figure 4 ) and Neighbor Joining (NJ) reconstructions (Figure S2). The recovered ML topology was very similar to the one generated by the NJ method, showing high conservation in vertebrates and in each insect order. The newly described S. frugiperda fut8 sequence grouped with other early diverging lepidopteran FUT8 sequences. Both tree topologies tended to place nematode FUT8 sequences outside the protostome branch suggesting that these are rapidly evolving FUT8 sequences. We also investigated the extent of synteny and gene order conservation in the metazoan FUT8 sequences visualized at the Genomicus web site [42]. This preliminary analysis suggested microsynteny conservation in each insect order and no synteny conservation between insect orders (data not shown).
Figure 4. Molecular phylogenetic analysis using the Maximum Likelihood method.

Evolutionary analyses were conducted in MEGA5 [67] and the evolutionary history was inferred using the Maximum Likelihood method based on the Whelan and Goldman model [72]. This analysis involved 92 FUT8 aa sequences and the final dataset contained 336 positions (42% of 787). The bootstrap consensus tree inferred from 1050 replicates is taken to represent the evolutionary history of the analyzed taxa. The percentage of trees (only those >75%) in which the associated taxa clustered together is shown next to the branches [73]. Initial tree(s) for the heuristic search were obtained automatically as follows. When the number of common sites was <100 or less than one fourth of the total number of sites, the maximum parsimony method was used; otherwise the BIONJ method with the MCL distance matrix was used.
Intron-exon organization of the fut8 genes
Twelve exons were identified in Sf9 fut8, ranging from 75 bp to 216 bp in size. The intron-exon boundaries of Sf9 fut8 were in agreement with the consensus sequence for the splicing donor and acceptor sites (GT at position 1 and 2 at 5′ splice sites and AG at position -1 and -2 at 3′ splice sites) (not shown) [43], [44]. Comparative analysis of fut8 orthologs showed that the intron-exon organization was order-specific ( Figure 5 ). The structure of lepidopteran fut8 genes was overall well-conserved with only minor length disparities in exons 3, 9 and 12. Particularly, exon 3 contained 67 codons in Heliconius melpomene, 69 in D. plexippus and 71 codons in S. frugiperda and B. mori; exon 9 of H. melpomene fut8 included 112 codons as a result of exon 9 and 10 fusion.
Figure 5. Schematic illustration of the genomic organization of the fut8 genes identified in animal genomes.

Coding exons are represented by rectangles with their relative size in codons (in bold). Italic numbers represent the number of nucleotides belonging to codons that flank the intron insertion site and determine the intron insertion phase. Dashed lines represent conserved intron insertion sites. Conserved exons are in the same color. S. frugiperda and H. sapiens FUT8 proteins are in blue, specific domains are represented by rectangles with their relative size in aa (Cys-rich domain, in black and motifs I, II and III, in grey).
Despite the overall highly variable organization of fut8 genes ( Figure 5 ), some exons (such as exons 4 and 8) that encode domains involved in protein folding and catalytic activity were shared among arthropod fut8 genes. Exon 4 was also present in Pediculus humanus (phtiraptera) and Daphnia pulex (diplostraca) fut8, whereas exon 5 was found in P. humanus, Tribolium castaneum (coleoptera), Acyrthosiphon pisum and D. pulex fut8. Exon 8 was also found in Culicidae (e.g., C. pipiens and Anopheles gambiae). Lepidopteran exons 6 and 7 seemed to result from the splitting of an exon shared (2-78-2) by many arthropoda (phtiraptera, coleoptera, hemiptera and crustacea).
Sf9 fut8 contained 11 introns (noted i1l to i11l), a much higher number compared to other insect fut8 genes (between 2 and 9 introns) ( Table 4 ). Diptera had the lowest number of introns: 3 in Drosophilidae and 2 in Culicidae. Strikingly, although all the essential sequence motifs required for the enzymatic activity were conserved in the hymenoptera fut8 genes, the intron insertion sites identified in these genes (three in Apis mellifera, Bombus impatiens, Megachile rotundata and four in Atta cephalotes, Solenopsis invicta, Camponotus floridanus, Harpagnathos saltator) were hymenoptera-specific and were not shared by any of the other arthropod fut8 genes analyzed here. In addition, the intron position i3h, which splits the hymenopteran exon 3 in apidae and megachilidae, appeared to be formicidae-specific ( Table 5 , Figure 5 ).
Table 4. Analysis of intron positions shared with S. frugiperda fut8 (inumberl) in arthropoda and chordata orthologs fut8 genes.
| Protein domain | Stem | Cys-rich | Motif I | Motif II | Motif III | Total IS (bp) | Nb introns | |||||||||
| Intron insertion sites | i1l | i2l | i3l | i4l | i5l | i6l | i7l | i8l | i9l | i10l | i11l | |||||
| Arthropoda | Insecta | Lepidoptera | S. frugiperda | 2/3,459 | 2/344 | 2/1,645 | 0/8,467 | 1/3,290 | 2/733 | 2/1,097 | 0/989 | 0/494 | 0/601 | 0/952 | 22,071 | 11 |
| B. mori | 2/3,280 | 2/2,841 | 2/5,599 | 0/1,203 | 1/1,197 | 2/355 | 2/483 | 0/1,167 | 0/1,971 | 0/1,188 | 0/795 | 20,079 | 11 | |||
| D. plexippus | 2/4,756 | 2/312 | 2/1,263 | 0/2,173 | 1/512 | 2/299 | 2/1,177 | 0/1,393 | 0/468 | 0/590 | 0/71 | 13,014 | 11 | |||
| H. melpomene | 2/1,633 | 2/712 | 2/1,821 | 0/7,380 | 1/1111 | 2/204 | 2/146 | 0/795 | 0/129 | 0/248 | 14,179 | 10 | ||||
| Coleoptera | T. castaneum | 0/9,139 | 1/52 | 2/1,524 | 0/8,806 | 21,186 | 5 | |||||||||
| Hemiptera | A. pisum | 0/67 | 1/91 | 2/118 | 0/62 | 752 | 7 | |||||||||
| Phtiraptera | P. humanus | 2/342 | 0/82 | 1/215 | 2/295 | 3,507 | 9 | |||||||||
| Diptera | D. melanogaster | 0/109 | 2/65 | 582 | 3 | |||||||||||
| C. pipiens | 2/62 | 0/98 | 160 | 2 | ||||||||||||
| L. longipalpis* | 2/94 | 0/438 | ≥3,290 | ≥3 | ||||||||||||
| Arachnida | Mesostigmata | M. occidentallis | 0/210 | 1,581 | 8 | |||||||||||
| Trombidiformes | T. urticae | 1,173 | 6 | |||||||||||||
| Branchiopoda | Cladocera | D. pulex | 2/65 | 0/60 | 1/61 | 2/55 | 320 | 5 | ||||||||
| Maxilopoda | Siphonostomatoida | L. salmonis | 0 | 0 | ||||||||||||
| Chilopoda | Geoplilomorph | S. maritima | 2/292 | 0/56 | 2,591 | 6 | ||||||||||
| Chordata | Acidiacae | Enterogona | C. intestinalis A | 2/836 | 1/906 | 2/820 | 0/447 | 0/5,797 | 10,989 | 10 | ||||||
| Mammalia | Primate | H. sapiens | 2/13,118 | 1/52,295 | 2/2,125 | 0/8,711 | 130,359 | 8 | ||||||||
| Rodentia | R. norvegicus | 2/19,848 | 1/36,598 | 2/1,403 | 0/11,243 | 131,971 | 8 | |||||||||
| Artiodactyla | B. taurus | 2/14,139 | 1/40,145 | 2/4,795 | 0/11,832 | 173,134 | 8 | |||||||||
IS: intronic sequence, Nb: number. The intron phase was defined as follows: phase 0 introns are inserted between two codons; phase 1 introns are inserted after the first nucleotide of the codon, and phase 2 introns are inserted between the second and the third nucleotide of the codon.
(*) The gene is truncated and the 5′ exon is missing.
Table 5. Analysis of intron positions in fut8 orthologs in hymenoptera (inumberh).
| Protein domain | Stem | Cys-rich | |||||||
| Intron insertion sites | i1h | i2h | i3h | i4h | Total IS (bp) | Nb introns | |||
| Insecta | Hymenoptera | Apidae | A. mellifera | 2/49 | 2/85 | 0/109 | 243 | 3 | |
| B. impatiens | 2/101 | 2/84 | 0/84 | 269 | 3 | ||||
| Megachilidae | M. rotundata | 2/101 | 2/75 | 0/65 | 241 | 3 | |||
| Formicidae | A cephalotes | 0/92 | 2/80 | 2/96 | 0/84 | 352 | 4 | ||
| S. invicta | 2/725 | 2/84 | 2/130 | 0/71 | 1010 | 4 | |||
| C. floridanus | 2/116 | 2/77 | 2/137 | 0/75 | 405 | 4 | |||
| H. saltator | 2/145 | 2/89 | 2/97 | 0/271 | 457 | 4 | |||
The intron phase was defined as described in Table 4. IS: intronic sequence, Nb: number.
To understand the evolution of fut8 genomic organization, we analyzed the repartition of intronic insertion sites in 56 complete orthologous fut8 genes from organisms belonging to different animal phyla. As intron position analysis relies on consistent aa alignments, FUT8 sequences were aligned using Clustal Omega. The high aa identity observed in FUT8 catalytic domain (from exon 4 {E4l} to exon 11 {E11l}) allowed us to compare the intron-exon structure in this region. In contrast, the N-terminal region showed a very low level of aa conservation (Figure S1). As previously reported [44], when the insertion site was conserved, the intron phase was conserved as well. Several intron insertion sites were very well conserved ( Table 4 and Table 6 ), for instance i3l, i5l, i7l and i9l in chordata, hemichordata (S. kowalevskii) and echinodermata (Strongylocentrotus purpuratus). In nematoda, fut8 gene was interrupted by 9 or 7 intronic sequences, but only one in rhabditida (Caenorhabditis brenneri) and three in trichocephalida (Trichinella spiralis) (i.e. i9l and i6l, i7l and i9l, respectively) were shared with S. frugiperda fut8. The intron sites i7l and i9l were also conserved in placozoa (Trichoplax adhaerens), cnidaria (Hydra magnipapillata) and annelida (Capitella teleta) ( Table 6 ).
Table 6. Intron positions shared by S. frugiperda fut8 (inumberl) and with fut8 genes identified in chordata (inumberc).
| Protein domain | Stem | Cys-rich | Motif I | Motif II | Motif III | |||||||||||||
| Intron insertion sites in Sf9 fut8 gene | i1l | i2l | i3l | I4l | i5l | i6l | i7l | I8l | i9l | i10l | i11l | Total IS | Nb introns | |||||
| Specific introns insertion sites in Chordata fut8 gene | i4c | i7c | (bp) | |||||||||||||||
| Chordata | Mammalia | Primate | H. sapiens | X | + | X | X | + | X | 130,359 | 8 | |||||||
| Aves | Galliformes | G. gallus | X | + | X | X | + | X | 36,774 | 8 | ||||||||
| M. gallopavo | X | + | X | X | + | X | 37,003 | 8 | ||||||||||
| Passeriformes | T. guttata | X | + | X | X | + | X | 43,603 | 8 | |||||||||
| Reptilia | Squamata | A. carolinensis | X | + | X | X | + | X | 67,279 | 8 | ||||||||
| Amphibia | Anura | X. tropicalis | X | + | X | X | + | X | 72,102 | 8 | ||||||||
| Actinopterygii | Cypriniformes | D. rerio B | X | + | X | X | + | X | 88,524 | 8 | ||||||||
| Tetraodontiformes | T. rubipres | X | + | X | X | + | X | 23,935 | 8 | |||||||||
| Beloniformes | O. latipes | X | + | X | X | + | X | 44,674 | 8 | |||||||||
| Perciformes | O. niloticus | X | + | X | X | + | X | 45,900 | 8 | |||||||||
| Echinodermata | Echinoidea | Echinoida | S. purpuratus | X | + | X | X | + | X | 58,902 | 8 | |||||||
| Hemichordata | Enteropneusta | Enteropneusta | S. kowalevskii A | X | + | X | X | + | X | 21,376 | 8 | |||||||
| Nematoda | Secernantea | Trichocephalida | T spiralis | X | X | X | 1,122 | 7 | ||||||||||
| Rhabditida | C. brenneri | + | X | 652 | 9 | |||||||||||||
| Spirudida | L. loa | + | 3,347 | 9 | ||||||||||||||
| B. malayi | + | 2,417 | 9 | |||||||||||||||
| W. bancrofti | + | 2,954 | 10 | |||||||||||||||
| Ascaridida | A suum | + | 13,589 | 10 | ||||||||||||||
| Annelida | Polychaeta | C. teleta | X | + | X | + | X | 1,183 | 7 | |||||||||
| Placozoa | Tricoplacia | Tricoplaciformes | T. adhaerens* | X | + | X | ≥682 | ≥3 | ||||||||||
| Cnidaria | Hydroida | Anthomedusae | H. magnipapillata | X | + | X | 13,440 | 4 | ||||||||||
| Anthozoa | Actiniaria | N. vectensis | X | + | X | 2,492 | 4 | |||||||||||
IS: Intronic Sequence.
(X): Presence of lepidopteran intron insertion site.
(+): Presence of chordata intron insertion site.
(*) The gene is truncated and the 5′ exon is missing.
In the A. pisum (arthropoda) and chordata fut8 genes, exon 1-58-2 and exon 1-50-0 were flanked by the conserved intron sites i7l and i9l ( Figure 5 ). Comparison of these exons with the corresponding lepidopteran exon 8 (1-51-0) and exon 9 (0-58-0) led us to hypothesize that these two exons derived from a common ancestral exon (1-109-0) that was found in T. castaneum. Similarly, the exon pairs 1-55-0/0-62-1 (E4l and E5l) in lepidoptera and 1-38-0/0-78-1 in chordata (E4c and E5c), which were flanked by the conserved intron sites i3l and i5l, could derive from a common ancestral exon.
Discussion
FUT8 can be considered part of the GT-23 family within the CAZy classification, which groups enzymes acting on carbohydrates. This classification is based on the structural (presence of conserved peptide motifs) and mechanistic features (α1,6-fucosyltransferase) of these glycosyltransferases. GT-23 is a single-copy nuclear gene family, whereas most GT families are polygenic. fut8 cDNAs have been cloned essentially from a few mammalian species (bovine, rat, pig, murine, and human, [15], [34], [35], [37], [45] and from two invertebrates, Caenorhabditis elegans and Drosophila [38]. In this work, we report the identification and molecular cloning of a fut8 ortholog in the lepidoptera S. frugiperda and the analysis of the molecular function and genomic organization of this new lepidopteran gene.
All conserved motifs (I, II and III) and all essential aa residues implicated in FUT8 enzymatic activity were identified in the Sf9 FUT8 sequence. However, analysis of the cysteine residues involved in the disulfide bridges formation in most FUT8 proteins shows that one cysteine residue, which corresponds to Cys-472 in human FUT8, is missing in the lepidopteran protein. This change is accompanied by the presence of a new cysteine residue (Cys-438 in S. frugiperda FUT8) that is also conserved in many FUT8 sequences (tricocephalida, spirudida and ascaridia, Ciona intestinalis, Nematostella vectensis and in the hymenoptera FUT8 sequences analyzed in this study) ( Figure 1 and Figure S1). The high enzymatic activity measured with recombinant Sf9 FUT8 suggests that the disulfide bridge detected between Cys-465 and Cys-472 in most species can probably be equally established between Cys-465 with Cys-438 in S. frugiperda FUT8 without affecting its enzymatic activity or protein stability. Two potential N-glycosylation sites (Asn-167 and Asn-325) ( Figure 1 ) are also present in the Sf9 sequence, but these sites are not conserved in agreement with the fact that the enzyme activity is not dependent on the presence of N-glycans [15].
In an attempt to trace back the origin and evolutionary relationships of fut8 genes, we identified fut8 orthologs in a large panel of metazoan genomes and determined their gene organization.
Most GT families have significantly expanded in early vertebrates through whole genome duplications, and differential loss or retention of duplicated genes has contributed to the functional divergence of these GT families (e.g., sialyltransferases families) [46], [47]. GT-23 represents a unique case of an evolutionary ancient GT family that did not diverge during metazoan evolution. We have chosen to study its gene structure as a way to characterize the evolutionary relationships of these genes.
Comparative analysis of the fut8 gene in various animal genomes provided few insights into the evolution of this single-copy gene in metazoans. In contrast with genes encoding α1,3-, α1,3/4- and α1,2-FucTs, all the characterized fut8 genes presented a common poly-exon organization. Metazoan fut8 genes contained a highly variable number of exons (from 12 exons in lepidoptera to 2 or 3 in diptera and no introns in chilopoda Lepeophtheirus salmonis), suggesting a complex evolutionary history with many intron gain and loss events. Analysis of the structure and evolution of the fut8 genes in arthropods showed that the high evolutionary rates found for instance in the coleoptera and diptera branches (see branch length differences between Drosophila and Aedes aegypti in Figure 4 ) corresponded to differences in gene organization as well.
The total size of the intronic sequences inserted in fut8 genes varied greatly among insects with more than 10,000 bp in lepidoptera and only 160 bp in diptera (culicidae), with the notable exception of A. aegypti, which presented 13,978 bp of intronic sequences. This observation is in agreement with the analysis of the average intron size of the A. aegypti genome [48] that shows a 4-fold increase due to intron infiltration by transposable elements compared to other diptera species, such as A. gambiae and D. melanogaster. Similarly, the intronic sequences identified in D. plexippus fut8 were about two times smaller than in B. mori and S. frugiperda, possibly because D. plexippus has the smallest genome among lepidoptera [49]. The same diversity was observed in other phyla. For instance, in nematoda, fut8 has either 7 or 10 introns with relatively short intronic sequences (from 652 bp in C. brenneri to 3,676 bp for Caenorhabditis japonica). In placozoa and cnidaria, only 3 or 4 introns were found for a total of 682 bp of intronic sequences in T. adhaerens fut8 and 13,440 bp in H. magnipapillata fut8. In vertebrates, the intron number appeared to be constant (8 introns). The main difference, compared to other phyla, was the very large total size of intronic sequences (about 130,000 bp in Homo sapiens and Rattus norvegicus 173,134 bp in Bos taurus) ( Table 4 and Table 6 ).
Intron insertion sites in phase 0 were over-represented in gene sequences encoding the most conserved part of the FUT8 proteins: i4l and i4c in the cysteine-rich domain, and i8l (motif II), i9l (motif III), i10l (SH3) and i11l (SH3) in the catalytic domain. Moreover, in human FUT8 structure [9], introns i4c and i8c/i9l are located at the C- and N- terminal domain of alpha helix 3 and alpha helix 11 respectively. Such features are proposed to be characteristic of ancient conserved genes [50].
Many exons were shared by different arthropoda orders, such as phtiraptera, coleoptera, hemiptera, diptera, lepidoptera and even crustacea ( Figure 5 ). Remarkably, despite the high conservation of lepidoptera FUT8 sequence with vertebrates and hymenoptera (S. frugiperda FUT8 sequence shares 44.24% and 45.93% aa identity with H. sapiens and A. mellifera respectively), the exon-intron organization of hymenoptera fut8 genes is order-specific with no shared exons. This is particularly surprising because when the analysis is extended to orthologous fut8 genes of other phyla, many Spodoptera intron insertion sites are still conserved throughout the animal kingdom. Particularly, intron positions i7l and i9l are common to most of the fut8 genes analyzed in this work ( Table 4 and Table 6 ). As these introns are located in a gene portion encoding motif I and III respectively (highly conserved regions of FUT8), a high selection pressure could be exerted on these parts, such as for instance the presence of regulatory elements that are important for gene expression. The i8l insertion site is only found in lepidoptera and some diptera, such as culicidae and psychodoidea (Lutzomyia longipalpis). This site is very close to the chordate site i7c with only 24 nucleotides between these two sites (Figure S1) forming a “near intron pair” as defined by [51]. In contrast to i8l, i7c is conserved in many arthropods (A. pisum, P. humanus, D. pulex, Strigamia maritima), cnidarians (N. vectensis and H. magnipapillata) and nematodes (Brugia malayi and C. brenneri). Thus, position i7c appeared before i8l and we hypothesize that the i8l site might have resulted from i7c sliding. Similarly, the chordate insertion site i4c is found in the arthropod S. maritima (Figure S1). Comparison of the lepidoptera and chordata exon pairs 1-55-0/0-62-1 and 1-38-0/0-78-1, located between the conserved sites i3l and i5l, suggests the existence of a common ancestral exon. As hypothesized for i8l, position i4l could have derived from the sliding of the ancestral position i4c (51 nt between i4l and i4c). However, unlike i7c, i4c is not well conserved outside chordates ( Figure 5 ).
To conclude, we propose a model of fut8 gene evolution in animals ( Figure 6 ). In this model, i7c and i9l are considered as ancestral intron insertion sites. Until the arthropoda-chordata split, intron gain seems to have been the most favored event with gain of i7l about 855MYA (Million years ago) and of i3l, i5l, i4c about 783 MYA [52]. After this divergence, intron loss seems to have become rather more common with the consecutive loss in arthropods of i3l (372 MYA), i7c (300 MYA), i5l and i9l (265 MYA). These intron losses could be accompanied by specific intron site gains (for instance, gain of i6l, i10l and i11l in lepidoptera) ( Figure 6 , compare lane 1 and 2) or not (for instance in D. pulex). It has to be noted that the intron site gains and losses identified in arthropods are not observed in chordates. Indeed, insertions of spliceosomal introns are rarely observed during evolution of vertebrates [53]. As fut8 intron-exon organization is order-specific, these intron losses and gains may be linked to evolutionary innovations, such as appearance of new orders.
Figure 6. Schematic illustration of the correlation between animal fut8 gene phylogeny, intron gain/loss and intron position (gene organization).

This analysis was carried out by considering only the intron insertion sites in the fut8 gene sequences encoding the conserved region of FUT8 proteins, between i3l and the stop codon. On the right, column 1 shows the total number of intron insertion sites (IS) identified in the different fut8 genes, and column 2 shows the number of order- or family-specific intron insertion sites. Intron gains and losses are highlighted (grey and white boxes, respectively) as well as putative intron sliding in “near-intron-pairs” (light grey boxes). Putative ancestral introns are in a dark grey box.
Finally, the gain of some intron sites, such as i8l in some diptera (culicidae, C. pipiens and psychodidae, L. longipalpis), i10l in C. intestinalis and i6l in T. spiralis ( Figure 6 , Figure S1, Table 4 and Table 6 ), may be explained by convergent/parallel intron gains, as recently described by [54] for hymenopteran paralogs, suggesting the presence of intron insertion hot spots [55].
Unlike other well studied glycosyltransferases [46], [47], we found that this single-copy gene characterized by highly conserved motifs is present from the very first metazoans to vertebrates. This could be explained by FUT8 important function. Evolution of fut8 gene organization is also very specific and may accommodate different way of regulating its expression for instance in response to the environment of these insects.
Materials and Methods
Cells and viruses
The Sf9 subclone of the S. frugiperda Sf21 cell line [56] was maintained at 28°C in TC100 medium (GIBCO) containing 5% heat-inactivated fetal calf serum. Cells were infected with baculoviruses AcMNPV clone 1.2 [57] at a multiplicity of infection (MOI) of 2 plaque-forming units (PFU) per cell. After 1 h incubation at room temperature with the viral suspension, fresh culture medium was added and cells were incubated at 28°C for 5 days. The viral titers were determined by plaque assay [58].
DNA and RNA purification
High molecular weight genomic DNA was extracted from Sf9 cells (15×106 cells) using the Genomic-Tip Kit (QIAGEN) as recommended by the manufacturer. Total cytoplasmic RNA was extracted from 2×107 Sf9 cells using a method previously described [59]. After precipitation, RNA pellets were washed with a cold solution containing 75% ethanol and 25% sodium acetate and resuspended in 50 µl of distilled water. Fifty µg of RNA were then incubated with 15 units of RNase-free DNase I (New England Biolabs) at 37°C for 20 min and then the DNase was inactivated with 5 mM EDTA at 75°C for 10 min. RNA samples were loaded on an RNeasy column (QIAGEN) as recommended by the manufacturer. Purified RNA was immediately stored at −80°C.
Cloning of the Sf9 α1,6-FucT cDNA
Sf9 fut8 cDNA was cloned by RT-PCR using degenerate primers [60]. The two ForFut1deg and BacFut2deg degenerate primers were designed based on the conserved aa sequence found in five species in which fut8 cDNA had been already isolated and characterized: H. sapiens (AJ536056), B. taurus (BC104555), D. melanogaster (AF441264), Drosophila yakuba (AJ830719) and Drosophila pseudoobscura (AJ830720) (Table S1, for primer sequences) (Eurogentech, Belgium). RT was performed as follows: a mixture (final volume of 20 µl) including 1 µg of total RNA from Sf9 cells, 4 µl of 10 X RT buffer (QIAGEN), 4 µl of 5 mM of each dNTP (QIAGEN), 4 µl of 10 pmoles/ml BacFut2deg, 20 units of RNase inhibitor (Roche) and 8 units of Omniscript reverse transcriptase (QIAGEN) was incubated at 37°C for 1 hour. The reverse transcriptase was inactivated at 93°C for 5 min.
Polymerase chain reaction (PCR) amplification was then carried out in a final volume of 20 µl containing 2 µl of 10 X Vent DNA polymerase (New England Biolabs), 2 µl of 10 mM each dNTP (Biolabs), 20 pmoles of each degenerate primer, 1.5 µl of 25 mM MgSO4, 1 unit of Vent Polymerase and 0.5 µl of the RT mixture. Thirty-five cycles of amplification were performed on a Mastercycler apparatus (Eppendorf): denaturation at 94°C for 30 sec, annealing for 1 min at a temperature depending on the primer used, extension at 72°C for 1 min and a final extension of 10 min at 72°C.
PCR products were separated on low melting agarose gels (FMC, Nusieve GTG) and purified with the QIAquick Gel Extraction Kit (QIAGEN). The 5′-ends were adenylated by incubating the DNA with 1 unit of Taq DNA Polymerase (Biolabs) and 5 µl of 10X ThermoPol buffer (20 mM Tris-HCl pH 8.8, 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4, 0.1% Triton X-100) and 200 µM dATP (final volume: 50 µl) at 72°C for 20 min. The PCR fragments were purified with the QIAquick PCR Purification Kit (QIAGEN) before ligation in the pGEM-T Easy vector system (Promega) for sequencing.
The 5′- and 3′- ends of the cDNA were amplified using the 5′/3′-RACE Kit, 2nd generation (Roche), following the manufacturer’s protocol, and the exact-match primers Bac5′RACEFut8 and For3′RACEFut8 (Table S1), designed based on the fut8 cDNA sequence obtained with the degenerate primers. The AMV reverse transcriptase and total RNA from Sf9 cells, as before, were used for the RT step. Briefly, 3′-RACE uses the polyA stretch generally present at the 3′-end of each mRNA as hybridization site for the oligo(dT) anchor primer. To specifically amplify the 3′ fut8 cDNA end, RNA was thus reverse transcribed with the oligo(dT) anchor primer and then the RT product was PCR amplified with For3′RACEFut8 and the oligo(dT) primers. For the 5′-RACE, the specific Bac5′RACEFut8 primer was used in the RT step. Then, to specifically amplify the 5′-end of the fut8 cDNA, a polyA stretch was added at the 5′-end of the cDNA by a terminal transferase and used as a hybridization site for the oligo(dT) anchor primer. The entire 1734 bp ORF was reconstituted in the pGEM-T Easy plasmid, giving the final pGEMFUT8Sf construct.
Genomic DNA analysis
Sf9 cell genomic DNA (10 µg) was digested with EcoRI or HindIII restriction endonucleases. DNA fragments were then separated by electrophoresis on 0.9% agarose gels (SeaKem GTG, Lonza) and transferred to positively charged nylon membranes (Roche). Membranes were pre-hybridized at 68°C in 5X SSC, 0.1% N-laurylsarcosine, 0.02% (w/v) SDS, 1% blocking reagent (Roche) and 100 µg/ml calf thymus DNA for 3 hours. Overnight hybridization was performed at 68°C in the same buffer with a 204bp fut8 specific probe (nucleotide 217–420 of the fut8 cDNA cloned in pGEM-T Easy, Figure 1 ) labeled with digoxigenin using the PCR DIG Probe Synthesis Kit (Roche). After hybridization, filters were washed twice at room temperature with 2X SSC, 0.1% SDS for 5min and then twice with 0.1X SSC, 01% SDS at 68°C for 15minutes. Probe-target hybrids were revealed by incubating the membranes with an alkaline-phosphatase conjugated anti-digoxigenin antibody (Roche) and by a chemoluminescent reaction using CSPD as substrate (Roche). Membranes were exposed to Kodak films for 20min.
RNA analysis
Total RNA from Sf9 cells was resolved (1 µg per lane) on 1% agarose gels and transferred to positively charged nylon membranes (Roche). A digoxigenin-labeled riboprobe was synthesized using the DIG Northern Starter Kit (Roche) using as a template the complete FUT8 coding sequence (pos. -1 to pos. 1686 in the fut8 cDNA, Figure 1 ) cloned in pGEM-T Easy (pGEMFUT8Sf). Hybridization and revelation were performed as described for Southern blotting.
PCR analysis of the genomic structure of the Sf9 α1,6-FucT gene
PCR amplifications were performed using Sf9 genomic DNA as a template. DNA was isolated as described above using the Genomic-Tip Kit (QIAGEN) and resuspended in water. PCR amplifications were carried out in a final volume of 50 µl containing 10 µl of 5X Phusion DNA polymerase buffer (HF buffer) (Finnzymes, Finland), 1 µl of 10 mM each dNTP (Biolabs), 0.5 µmoles of each primer (Table S1; Cloning of introns), 1 unit of Phusion DNA Polymerase and 200 ng of Sf9 genomic DNA. After denaturation at 98°C for 2 min, a two-step protocol was used with (i) seven cycles of amplification in the following conditions: denaturation at 98°C for 15 sec, annealing and extension for 3 minutes at 72°C and (ii) thirty cycles of amplification with denaturation at 98°C for 15 sec, annealing and extension for 5 min at 68°C. PCR products were isolated on agarose gels (SeaKem GTG, Lonza) and purified with the QIAquick Gel Extraction Kit (QIAGEN). PCR fragments were then adenylated with Taq Polymerase as described above and cloned in pGEM-T Easy (Promega) for sequencing analysis (Operon Eurofins MWG, Germany).
Construction of the recombinant baculovirus expressing soluble and tagged Sf9 FUT8
To produce soluble recombinant FUT8, the 5′-end of the cDNA encoding the N-terminal domain of FUT8 (from aa 1 to aa 30 at the end of the putative transmembrane domain) was deleted and replaced by the signal peptide sequence of the baculovirus ecdysteroid glycosyltransferase (EGT) [61] followed by a sequence encoding the FLAG epitope (Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys) to produce a recombinant protein that can be easily purified and that can be differentiated from the endogenous protein. To this aim, a HpaI-BglII DNA fragment encoding the FLAG epitope and aa 31 to aa 40 of Sf9 FUT8 was synthesized using overlapping oligonucleotides and inserted in frame with the EGT signal peptide present in the pUC PS-EGT vector (pUC PS-EGT-FLAG-NterFut8Sf construct) [62]. Then, the C-terminal domain of fut8 cDNA was isolated from the pGEMFut8Sf plasmid by EcoRI-BamHI digestion and inserted in the pUC PS-EGT-FLAG-NterFut8Sf construct to obtain the pUC PS-EGT-FLAG-Fut8Sf construct. After BamHI and HindIII digestion, the 1722 bp fragment containing the EGT-FLAG-Fut8 cDNA was isolated by agarose gel electrophoresis (Nusieve GTG, Lonza), purified using the QIAGEN Gel Extraction Kit and ligated in the p119 baculovirus transfer vector digested with BglII and HindIII to produce the p119-EGT-FLAG-Fut8Sf construct.
Sf9 cells (4×106 cells) were seeded in 25 cm2 flasks and co-transfected with 500 ng of purified AcSLP10 viral DNA [59] and 5 µg of p119-EGT-FLAG-Fut8Sf by lipofection [63] with DOTAP (Roche). The p119 transfer vector is designed for recombination in the P10 locus. AcSLP10 is derived from wild type AcMNPV 1.2 [57] and has only one strong promoter (P10) to drive the expression of a polyhedrin gene [59]. After 5 days of incubation at 28°C, recombinant (i.e., polyhedron-negative) viruses were purified by plaque assay [58]. Viral stocks were generated by propagating viruses in Sf9 cells (75 cm2 flasks) and titrated using plaque assays.
Production and purification of soluble FUT8
Sf9 cells seeded in roller bottles at a density of 400,000 cells/mL in serum-free medium were infected at a MOI of 2 PFU per cell. After 3-day incubation at 28°C, supernatants were collected and stored at −80°C before use. Supernatants were then concentrated and diafiltered against TS buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl) using Centromate cassettes (Pall, 0.1 m2/30 kDa) before loading on a 5 ml column of anti-FLAG M2 Affinity gel (Sigma) equilibrated with TS buffer. The FLAG-tagged protein was eluted with 30 ml of 100 µg/ml FLAG peptide (in TS buffer). Fractions containing the purified protein were concentrated (AMICON Ultra 50 k, Millipore) and analyzed by PAGE and western blotting.
FUT8 assays
After 2 hours of dialysis against 50 mM sodium acetate buffer (pH 7.5), 5 mg of human apotransferrin (Sigma) were incubated with 120 mU Arthrobacter urefasciens neuraminidase (Sigma) and 50 mU Escherichia coli β-galactosidase (Sigma) in a final volume of 10 ml at 37°C for 24 hours. After 4 hours dialysis against water, asialo-agalacto apotransferrin was freeze-dried. The absence of galactose and sialic acid residues was verified by gas chromatography-mass spectrometry (GC-MS). To test the enzymatic activity of recombinant Sf9 FUT8, assays were performed in 50 µl total volume that included 20 µl of purified recombinant protein solution (140 µg/ml), 70 mM cacodylate buffer (pH 7.2) 10 mM L-fucose, 6 mM GDP-[14C]-L-fucose (299 mCi/mMol, Amersham), 10 mM GDP-fucose and 100 µg asialo-agalacto apotransferrin as acceptor, as previously described [34]. After incubation at 30°C for 4 hours, the reaction was stopped with 150 µl of water and samples were precipitated with 1 ml of 5% PTA and processed for scintillation counting. The transfer of [14C]-fucose is expressed in cpm.
In silico α1,6-FucT sequences retrieval and phylogenetic analysis
Only eukaryotic core α1,6-FucT sequences were considered for this study. Homologous fut8 sequences were searched by querying all genomic and expressed sequence tag (EST) divisions of the National Center for Biotechnology Information (NCBI database, Washington, DC, USA), as described previously for sialyltransferases [22], [64] (Table S2). DNA and aa sequences were analyzed using DNA Strider [65]. PSORT II (http://expasy.org) was used for the prediction of protein localization sites in cells. Homology searches were performed using the BLAST program [39]. Multiple sequence alignments of the 86 deduced aa sequences homologous to α1,6-fucosyltranferases were generated using ClustalW 2.0.8, Clustal Omega 1.1.0 or MAFFT, which are multiple sequence alignment programs available at http://www.ebi.ac.uk. Gblocks (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) [66] was used to select the 336/785 informative sites. Evolutionary analyses were conducted using the Neighbor Joining (NJ) and Maximum Likelihood (ML) methods implemented in MEGA 5.05 [67]. The branch robustness was tested with 1050 bootstrap replicates. Conservation of synteny and gene order in the metazoan were visualized at the Genomicus web site (version 19.01) (http://www.genomicus.biologie.ens.fr/genomicus-metazoa-19.01/cgi-bin/search.pl) [42].
Supporting Information
Amino acid sequences of α1,6-fucosyltranferases from different phyla were aligned using Clustal Omega 1.1.0 ( www.ebi.ac.uk). Letters on grey background indicate the position of intron insertion in the genes. Numbers indicate the intron phase. When the insertion phase is 1 or 2, the aa corresponding to the split codon is in highlighted in grey, when the insertion phase is 0, the two flanking aa are in grey. Putative transmembrane domains determined using http://wolfpsort.org are underlined. Conserved cysteine residues are highlighted in yellow.
(PDF)
The evolutionary history was inferred using the Neighbor-Joining method [68] and 86 amino acid sequences aligned with MAFFT (EBI). All positions containing gaps and missing data were eliminated. The final dataset contained 461 positions selected in 17 blocks by GBlocks [66] (58% of the original 785 positions). The Dictyostelium discoideum sequence was used as outgroup. The optimal tree with a branch length sum = 6.93435487 is shown. The percentage of replicated trees (>75%) in which the associated taxa clustered together in the bootstrap test (1050 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths (next to the branches) in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method [69] and refers to the number of amino acid differences per site.
(PDF)
Primers used in this study.
(PDF)
Nomenclature, name and GenBank accession number of the fut8 gene sequences used in this study. (*) Nucleotide sequence data available in the Third Party Annotation Section of the DDBJ/EMBL/GenBank databases. (**) Ensembl accession number.
(PDF)
Acknowledgments
We thank the anonymous reviewers for their very interesting comments that helped us improving the manuscript.
Data Availability
The authors confirm that, for approved reasons, some access restrictions apply to the data underlying the findings. Nucleotide sequence data are available in the Third Party Annotation Section of the DDBJ/EMBL/GenBank databases with accession numbers BK008802, BK008805, BK008806, BK008807, BK008808, BK008809, BK008810, BK008811, BK008812, BK008813, BK008814.
Funding Statement
This work was supported by Centre National de la Recherche Scientifique to MD-C, AH-L, FM, MLV, Institut National de la Recherche Agronomique to PC, SJ, AO, Laboratoire Français du Fractionnement et des Biotechnologies de Lille to BC, and PPF Bioinformatique Lille1 to AH-L. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, et al. (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 37: D233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Becker DJ, Lowe JB (2003) Fucose: biosynthesis and biological function in mammals. Glycobiology 13: 41R–53R. [DOI] [PubMed] [Google Scholar]
- 3. Zhao Y, Sato Y, Isaji T. Fukuda T. Matsumoto A, et al. (2008) Branched N-glycans regulate the biological functions of integrins and cadherins. FEBS J 275: 1939–1948. [DOI] [PubMed] [Google Scholar]
- 4. Oriol R, Mollicone R, Cailleau A, Balanzino L, Breton C (1999) Divergent evolution of fucosyltransferase genes from vertebrates, invertebrates, and bacteria. Glycobiology 9: 323–334. [DOI] [PubMed] [Google Scholar]
- 5. Mollicone R, Cailleau A, Oriol R (1995) Molecular genetics of H, Se, Lewis and other fucosyltransferase genes. Transfus Clin Biol 2: 235–242. [DOI] [PubMed] [Google Scholar]
- 6. Liu YC1, Yen HY, Chen CY, Chen CH, Cheng PF, et al. (2011) Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc Natl Acad Sci U S A 108: 11332–11337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shields RL, Lai J, Keck R, O’Connell LY, Hong K, et al. (2002) Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fc gamma RIII and antibody-dependent cellular toxicity. J Biol Chem 277: 26733–26740. [DOI] [PubMed] [Google Scholar]
- 8. Wang X, Inoue S, Gu J, Miyoshi E, Noda K, et al. (2005) Dysregulation of TGF-β1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc Natl Acad Sci USA 102: 15791–15796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ihara H, Ikeda Y, Toma S, Wang X, Suzuki T, et al. (2007) Crystal structure of mammalian alpha1,6-fucosyltransferase, FUT8. Glycobiology 17: 455–466. [DOI] [PubMed] [Google Scholar]
- 10. Rao ST, Rossmann MG (1973) Comparison of super-secondary structures in proteins. Journal of Molecular Biology. 76: 241–250. [DOI] [PubMed] [Google Scholar]
- 11. Breton C, Oriol R, Imberty A (1998) Conserved structural features in eukaryotic and prokaryotic fucosyltransferases. Glycobiology 8: 87–94. [DOI] [PubMed] [Google Scholar]
- 12. Takahashi T, Ikeda Y, Tateishi A, Yamaguchi Y, Ishikawa M, et al. (2000) A sequence motif involved in the donor substrate binding by alpha1,6-fucosyltransferase: the role of the conserved arginine residues. Glycobiology 10: 503–510. [DOI] [PubMed] [Google Scholar]
- 13. Martinez-Duncker I, Mollicone R, Candelier JJ, Breton C, Oriol R (2003) A new superfamily of protein-O-fucosyltransferases, alpha 2-fucosyltransferases, and alpha 6-fucosyltransferases: phylogeny and identification of conserved peptide motifs. Glycobiology 13: 1C–5C. [DOI] [PubMed] [Google Scholar]
- 14. Okajima T, Xu A, Lei L, Irvine KD (2005) Chaperone activity of protein O-fucosyltransferase 1 promotes notch receptor folding. Science 307: 1599–1603. [DOI] [PubMed] [Google Scholar]
- 15. Javaud C, Dupuy F, Maftah A, Michalski JC, Oriol R, et al. (2000) Ancestral exonic organization of the FUT8, the gene encoding the α6-fucosyltransferase, reveals successive peptide domains which suggest a particular three dimensional core structure for the α6-fucosyltransferase family. Mol Biol Evol 17: 1661–1672. [DOI] [PubMed] [Google Scholar]
- 16. Javaud C, Dupuy F, Maftah A, Julien R, Petit JM (2003) The fucosyltransferasegene family: an amazing summary on the underlying mechanisms of gene evolution. Genetica 118: 157–170. [PubMed] [Google Scholar]
- 17. Tomiya N, Narang S, Lee YC, Betenbaugh MJ (2004) Comparing N-glycan processing in mammalian cell lines to native and engineered lepidopteran insect cell lines. Glycoconj J 21: 343–360. [DOI] [PubMed] [Google Scholar]
- 18. Marchal I, Mir AM, Kmiécik D, Verbert A, Cacan R (1999) Use of inhibitors to characterize intermediates in the processing of N-glycans synthesized by insect cells: a metabolic study with Sf9 cell line. Glycobiology 9: 645–654. [DOI] [PubMed] [Google Scholar]
- 19. Juliant S, Lévêque M, Cérutti P, Ozil A, Choblet S, et al. (2013) Engineering the baculovirus genome to produce galactosylated antibodies in lepidopteran cells. Methods Mol Biol 988: 59–77. [DOI] [PubMed] [Google Scholar]
- 20. Lopez M, Coddeville B, Langridge J, Lemoine J, Plancke H, et al. (1997) Microheterogeneity of the oligosaccharides carried by the N-glycosylation sites of the bovine lactotransferrin expressed in Mamestra brassicae using a baculovirus vector. Glycobiology 7: 635–651. [DOI] [PubMed] [Google Scholar]
- 21. Hooker AD, Green NH, Baines AJ, Bull AT, Jenkins N, et al. (1999) Constraints on the transport and glycosylation of recombinant IFN-gamma in Chinese hamster ovary and insect cells. Biotechnol Bioeng 63: 559–672. [DOI] [PubMed] [Google Scholar]
- 22. Petit D, Mir AM, Petit JM, Thisse C, Delannoy P, et al. (2010) Molecular phylogeny and functional genomics of β-galactoside α2,6-sialyltransferases to explain ubiquitous expression of st6gal1 in Amniotes. J Biol Chem 285: 38399–38414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marchal I, Jarvis DL, Cacan R, Verbert A (2001) Glycoproteins from insect cells: sialylated or not? Biol Chem 382: 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Altmann F, Kornfeld G, Dalik T, Staudacher E, GlössI J (1993) Processing of the asparagine-linked oligosaccharides in insect cells. N-acetylglucosaminyltransferase I and II activities in cultured lepidopteran cells. Glycobiology 3: 619–625. [DOI] [PubMed] [Google Scholar]
- 25. Velardo MA, Bretthauer RK, Boutaud A, Reinhold B, Reinhold VN, et al. (1993) The presence of UDP-N-acetylglucosamine:alpha-3-D-mannoside beta 1,2-N-acetylglucosaminyltransferase I activity in Spodoptera frugiperda cells (IPLB-SF-21AE) and its enhancement as a results of baculovirus infection. J Biol Chem 268: 17902–17907. [PubMed] [Google Scholar]
- 26. Geisler C, Jarvis DL (2012) Substrate specificities and intracellular distributions of three N-glycan processing enzymes functioning at a key branch point in the insect N-glycosylation pathway. J Biol Chem 287: 7084–7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Altmann F, Schwihla H, Staudacher E, Glössl J, März L (1995) Insect cells contain an unusual, membrane bound (beta)-N-acetylglucosaminidase probably involved in the processing of proteins N-glycans. J Biol Chem 270: 17344–17349. [DOI] [PubMed] [Google Scholar]
- 28. Wagner R, Liedtke S, Kretzschmar E, Geyer H, Klenk HD (1996) Elongation of the N-glycans of the fowl-plague virus hemaglutinin expressed in Spodoptera frugiperda (Sf9) cells by coexpression of the human β1,2-N-acetylglucosaminidase I. Glycobiology. 6: 165–175. [DOI] [PubMed] [Google Scholar]
- 29. Fabini G, Freilinger A, Altmann F, Wilson IB (2001) Identification of core alpha 1,3-fucosylated glycans and cloning of the requisite fucosyltransferase cDNA from Drosophila melanogaster. Potential basis of the neural anti-horseadish peroxidase epitope. J Biol Chem 276: 28058–28067. [DOI] [PubMed] [Google Scholar]
- 30. Staudacher E, Kubelka V, März L (1992) Distinct N-glycan fucosylation potentials of three lepidopteran cell lines. Eur J Biochem 207: 987–993. [DOI] [PubMed] [Google Scholar]
- 31. Kubelka V, Altmann F, Kornfeld G, März L (1994) Structures of the N-linked oligosaccharides of the membrane glycoproteins from three lepidopteran cell lines (Sf-21, IZD-Mb-0503, Bm-N). Arch Biochem Biophys 308: 148–157. [DOI] [PubMed] [Google Scholar]
- 32. Hancock K, Narang S, Pattabhi S, Yushak ML, Khan A, et al. (2007) False positive reactivity of recombinant, diagnostic, glycoproteins produced in High Five insect cells: Effect of glycosylation. J Immunol Methods 330: 130–136. [DOI] [PubMed] [Google Scholar]
- 33. Voss T, Ergülen E, Ahorn H, Kubelka V, Sugiyama K, et al. (1993) Expression of interferon w1 in Sf9 cells. No evidence for complex-type N-linked glycosylation or sialylation. Eur J Biochem. 217: 913–919. [DOI] [PubMed] [Google Scholar]
- 34. Teylaert B, Meurice E, Bobowski M, Harduin-Lepers A, Gaucher C, et al. (2011) Molecular cloning, characterization, genomic organization and promoter analysis of the α1,6-fucosyltransferase gene (Fut8) expressed in the rat hybridoma cell line YB2/0. BMC Biotechnol 11: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Uozumi N, Yanagidani S, Miyoshi E, Ihara Y, Sakuma C, et al. (1996) Purification and cDNA cloning of porcine brain GDP-L-Fuc: N-acetyl-β-D-glucosaminide α1,6-fucosyltransferase. J Biol Chem 271: 27810–27817. [DOI] [PubMed] [Google Scholar]
- 36. Yanagidani S, Uozumi N, Ihara Y, Miyoshi E, Yamaguchi N, et al. (1997) Purification and cDNA cloning of GDP-L-Fuc: N-acetyl-beta-D-glucosaminide: alpha1,6-fucosyltransferase (alpha1,6-FucT) from human gastric cancer MKN45 cells. J Biochem 121: 626–632. [DOI] [PubMed] [Google Scholar]
- 37. Hayashi H1, Yoneda A, Asada M, Ikekita M, Imamura T (2000) Molecular cloning of mouse alpha-1,6-fucosyltransferase and expression of its mRNA in the developing cerebrum. DNA Seq 11: 91–6. [DOI] [PubMed] [Google Scholar]
- 38. Paschinger K, Staudacher E, Stemmer U, Fabini G, Wilson IB (2005) Fucosyltransferase substrate specificity and the order of fucosylation in invertebrates. Glycobiology 15: 463–474. [DOI] [PubMed] [Google Scholar]
- 39. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Landais I, Ogliastro M, Mita K, Nohata J, Lopez-Ferber M, et al. (2003) Annotation pattern of ESTs from Spodoptera frugiperda Sf9 cells and analysis of the ribosomal protein genes reveal insect-specific features and unexpectedly low codon usage bias. Bioinformatics 19: 2343–2350. [DOI] [PubMed] [Google Scholar]
- 41. Cavener DR Ray SC (1991) Eukaryotic start and stop translation sites. Nucl Acids Res 19: 3185–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Louis A, Muffato M, Roest Crollius H (2013) Genomicus: five genome browsers for comparative genomics in eukaryota. Nucleic Acids Research 41: D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Senapathy P, Shapiro MB, Harris NL (1990) Splice junctions, branch point sites and exons: sequence statistics, identification and applications to genome project. Methods in enzymology 183: 252–278. [DOI] [PubMed] [Google Scholar]
- 44. Long M, Deutsch M (1999) Association of intron phases with conservation at splice site sequences and evolution of spliceosomal introns. Mol Biol Evol 16: 1528–1534. [DOI] [PubMed] [Google Scholar]
- 45. Yamaguchi Y, Fujii J, Inoue S, Uozumi N, Yanagidani S, et al. (1999) Mapping of the alpha-1,6-fucosyltransferase gene, FUT8, to human chromosome 14q24.3. Cytogenet Cell Genet 84: 58–60. [DOI] [PubMed] [Google Scholar]
- 46. Harduin-Lepers A, Mollicone R, Delannoy P, Oriol R (2005) The animal sialyltransferases and sialyltransferase-related genes: a phylogenetic approach. Glycobiology 15: 805–817. [DOI] [PubMed] [Google Scholar]
- 47. Harduin-Lepers A (2010) Comprehensive Analysis of Sialyltransferases in Vertebrate Genomes Glycobiology Insights. 2: 29–61. [Google Scholar]
- 48. Nene V, Wortman JR, Lawson D, Haas B, Kodira C, et al. (2007) Genome sequence of Aedes aegypti, a major arbovirus vector. Science 316: 1718–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gregory TR (2007) Animal genome size data. http://www.genomesize.com.
- 50. de Souza SJ, Long M, Klein RJ, Roy S, Lin S, Gilbert W (1998) Toward a resolution of the introns early/late debate: only phase zero introns are correlated with the structure of ancient proteins. Proc Natl Acad Sci USA 95: 5094–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Krauss V, Pecyna M, Kurz K, Sass H (2005) Phylogenetic mapping of intron positions: a case study of translation initiation factor eIF2gamma. Mol Biol Evol 22: 74–84. [DOI] [PubMed] [Google Scholar]
- 52. Hedges SB, Dudley J, Kumar S (2006) TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22: 2971–2972. [DOI] [PubMed] [Google Scholar]
- 53. Kumar A, Bhandari A, Sinha R, Goyal P, Grapputo A (2011) Spliceosomal intron insertions in genome compacted ray-finned fishes as evident from phylogeny of MC receptors, also supported by a few other GPCRs. PLoS One 6: e22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Klopfstein S, Ronquist F (2013) Convergent intron gains in hymenopteran elongation factor-1α. Mol Phylogenet Evol 67: 266–276. [DOI] [PubMed] [Google Scholar]
- 55. Li W, Tucker AE, Sung W, Thomas WK, Lynch M (2009) Extensive, recent intron gains in Daphnia populations. Science 326: 1260–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vaughn JL, Goodwin RH, Tompkins GJ, McCawley P (1977) The establishment of two cell lines from the insect Spodoptera frugiperda (Lepidoptera: Noctuidae). In Vitro 13: 213–217. [DOI] [PubMed] [Google Scholar]
- 57. Croizier G, Croizier L, Quiot JM, Lereclus D (1988) Recombinaison of Autographa californica and Rachiplusia ou nuclear polyhedrosis viruses in Galleria mellonella L. J Gen Virol. 69: 177–185. [Google Scholar]
- 58.Summers MD, Smith GE (1987) A manual of methods for baculovirus vectors and insect cell culture procedures. Tex Agr Exp Stat Bull N°1555.
- 59. Chaabihi H, Ogliastro MH, Martin M, Giraud C, Devauchelle G, et al. (1993) Competition between baculovirus polyhedrin and p10 gene expression during infection of insect cells. J Virol 67: 2664–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, et al. (1985) Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230: 1350–1354. [DOI] [PubMed] [Google Scholar]
- 61. O’Reilly DR, Miller LK (1989) A baculovirus blocks insect molting by producing ecdysteroid UDP-glucosyl transferase. Science 245: 1110–1112. [DOI] [PubMed] [Google Scholar]
- 62. Marchal I, Cérutti M, Mir AM, Juliant S, Devauchelle G, et al. (2001) Expression of a membrane-bound form of Trypanosoma cruzei trans-sialidase in baculovirus-infected cells: a potential tool for sialylation of glycoproteins produced in the baculovirus-insect cells system. Glycobiology 11: 593–603. [DOI] [PubMed] [Google Scholar]
- 63.Felgner PL, Ringold GM (1989) Cationic liposome-mediated transfection. Nature 337, 387–388. [DOI] [PubMed]
- 64. Petit D, Teppa RE, Petit JM, Harduin-Lepers A (2013) A practical approach to reconstruct evolutionary history of animal sialyltransferases and gain insights into the sequence-function relationships of Golgi-glycosyltransferases. Methods Mol Biol 1022: 73–97. [DOI] [PubMed] [Google Scholar]
- 65. Marck C (1988) ‘DNA Strider’: a ‘C’ program for the fast analysis of DNA and protein sequences on the Apple Macintosh family of computers. Nucleic Acids Res 16: 1829–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution 17: 540–552. [DOI] [PubMed] [Google Scholar]
- 67. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425. [DOI] [PubMed] [Google Scholar]
- 69.Nei M, Kumar S (1992) Molecular Evolution and Phylogenetics. Oxford University Press. 2000.
- 70. Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schneider TD, Stephens RR (1990) Sequence Logos: A New Way to Display Consensus Sequences. Nucl Ac Res 18: 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Whelan S, Goldman N (2001) A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Molecular Biology and Evolution 18: 691–699. [DOI] [PubMed] [Google Scholar]
- 73. Felsenstein J (1985) Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39: 783–791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Amino acid sequences of α1,6-fucosyltranferases from different phyla were aligned using Clustal Omega 1.1.0 ( www.ebi.ac.uk). Letters on grey background indicate the position of intron insertion in the genes. Numbers indicate the intron phase. When the insertion phase is 1 or 2, the aa corresponding to the split codon is in highlighted in grey, when the insertion phase is 0, the two flanking aa are in grey. Putative transmembrane domains determined using http://wolfpsort.org are underlined. Conserved cysteine residues are highlighted in yellow.
(PDF)
The evolutionary history was inferred using the Neighbor-Joining method [68] and 86 amino acid sequences aligned with MAFFT (EBI). All positions containing gaps and missing data were eliminated. The final dataset contained 461 positions selected in 17 blocks by GBlocks [66] (58% of the original 785 positions). The Dictyostelium discoideum sequence was used as outgroup. The optimal tree with a branch length sum = 6.93435487 is shown. The percentage of replicated trees (>75%) in which the associated taxa clustered together in the bootstrap test (1050 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths (next to the branches) in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method [69] and refers to the number of amino acid differences per site.
(PDF)
Primers used in this study.
(PDF)
Nomenclature, name and GenBank accession number of the fut8 gene sequences used in this study. (*) Nucleotide sequence data available in the Third Party Annotation Section of the DDBJ/EMBL/GenBank databases. (**) Ensembl accession number.
(PDF)
Data Availability Statement
The authors confirm that, for approved reasons, some access restrictions apply to the data underlying the findings. Nucleotide sequence data are available in the Third Party Annotation Section of the DDBJ/EMBL/GenBank databases with accession numbers BK008802, BK008805, BK008806, BK008807, BK008808, BK008809, BK008810, BK008811, BK008812, BK008813, BK008814.