

Abstract
Apurinic/apyrimidinic endonuclease I (APE1) is an essential base excision repair enzyme that catalyzes a Mg2+-dependent reaction in which the phosphodiester backbone is cleaved 5′ of an abasic site in duplex DNA. This reaction has been proposed to involve either one or two metal ions bound to the active site. In the present study, we report crystal structures of Mg2+, Mn2+, and apo-APE1 determined at 1.4, 2.2, and 1.65 Å, respectively, representing two of the highest resolution structures yet reported for APE1. In our structures, a single well-ordered Mn2+ ion was observed coordinated by D70 and E96; the Mg2+ site exhibited disorder modeled as two closely positioned sites coordinated by D70 and E96 or E96 alone. Direct metal binding analysis of wild-type, D70A, and E96A APE1, as assessed by differential scanning fluorimetry, indicated a role for D70 and E96 in binding of Mg2+ or Mn2+ to APE1. Consistent with the disorder exhibited by Mg2+ bound to the active site, two different conformations of E96 were observed coordinated to Mg2+. A third conformation for E96 in the apo structure is similar to that observed in the APE1–DNA–Mg2+ complex structure. Thus, binding of Mg2+ in three different positions within the active site of APE1 in these crystal structures corresponds directly with three different conformations of E96. Taken together, our results are consistent with the initial capture of metal by D70 and E96 and repositioning of Mg2+ facilitated by the structural plasticity of E96 in the active site.
Apurinic/apyrimidinic endonuclease I (APE1) is an essential base excision repair enzyme that catalyzes a Mg2+-dependent reaction resulting in the cleavage of the phosphodiester backbone 5′ of an abasic site within double-stranded DNA.1,2 Pre-steady-state turnover rates for APE1’s endonuclease activity have been estimated to be greater than 7003 or 850 s–1,4 whereas steady-state turnover rates are approximately 2 s–1.5 Thus, substrate turnover is diffusion-limited, and the slow step of the reaction occurs after the chemistry.4 In fact, product release has been proposed to be the slow step of the reaction.6 The endonuclease reaction involves a one-step associative phosphoryl transfer mechanism, with water serving as the nucleophile7 and preference for the Rp stereoisomer during cleavage.2 However, the number of metal ions involved and their coordination in the enzyme remains controversial. A two metal ion mechanism was first proposed by Steitz and co-workers for Klenow fragment,8,9 related polymerases, and associated exonucleases.10 Since then, there has been a general assumption that most enzymes that cleave DNA, including APE1, will in fact use two metal ions.11 APE1 is most closely related in structure to Escherichia coli exonuclease III, which binds a single metal ion in the active site.12 The two metal ion assumption was challenged by Tainer and co-workers, who put forth a mechanism for APE1 involving only one metal ion based on structural and enzymatic characterizations of an APE1–DNA complex.6 In that work, they reported a 3.0 Å structure with one Mn2+ ion bound to E96 in the active site with cleaved DNA.
Crystal structures of APE1 with Pb2+ bound in the active site of the enzyme were then reported, and a two metal ion mechanism was proposed.13 In this mechanism, coordinating residues for the metal ions included D70 and E96 for one metal and H309, D210, and N212 for a second metal ion, despite the fact that Mg2+ strongly prefers coordinating oxygen ligands.14 The two metal ion binding sites then formed the basis of a proposed moving metal ion mechanism involving Mg2+ binding first to a site coordinated by D210 and N212 and then moving 5 Å to a site coordinated by D70, E96, and D308.15,16 Finally, a 25Mg solid-state NMR study reported that APE1 binds one mole equivalent of Mg2+, which is disordered due to its coordinating ligands, suggesting plasticity in the active site.17
Recently, a 2.4 Å resolution structure of an APE1–Mg2+–product complex was reported in which Mg2+ is coordinated solely to E96,18 as was shown previously for the Mn2+ complex. Although the effect of substituting E96 on the catalytic activity was characterized in this recent study,18 there is currently no solution data for direct metal binding by APE1. In the absence of substrate, Mg2+ is coordinated by D70 and E96 in crystal structures reported to date at moderate resolution.19
In this study, we present the highest resolution structure of APE1 as a complex with bound Mg2+ determined at 1.4 Å, the structure of APE1 bound to Mn2+, and the first apo-APE1 structure (i.e., without bound metal). Our motivation for this study was to establish a structural basis for metal binding in the absence of substrate and to determine the contributions of specific residues on metal binding and catalysis by APE1. Our results provide new insights on the initial capture of metal ion by APE1 involving remarkable plasticity of a metal-coordinating ligand within the active site.
Materials and Methods
Preparation of Human APE1 Proteins
For crystallization, a single amino acid substitution (C138A) was introduced within the N-terminally truncated protein lacking the first 40 amino acids (Δ40APE1). DNA encoding residues 40–318 of APE1 was inserted within the PET-28a vector by using the NheI and XhoI restriction sites. Site-directed mutagenesis was then used to introduce a C138A mutation, which was confirmed by DNA sequencing. This protein was expressed as an N-terminal hexa-His tagged protein and purified as previously described for the C65A and wild-type Δ40APE1 proteins.20 In brief, the cells were lysed by using a French press, and the crude extract was subjected to purification by Ni-NTA and SP-Sepharose ion-exchange chromatography. The affinity tag was then removed by treatment with thrombin, and the protein was further purified by SP-Sepharose ion-exchange chromatography.
For differential scanning fluorimetry assays, full-length human wild-type D70A, E96A, D70A/E96A, D210A, and D308A APE1 proteins were expressed as N-terminal-hexa His-SUMO fusions and prepared as previously described.21 Site-directed mutagenesis was used to introduce codon changes for D70A, E96A, D210A, and D308A using the QuikChange kit from Stratagene, Inc. Primers for D70A are 5′-gctcttggaatgtggctgggcttcgagcctg-3′ and 5′-caggctcgaagcccagccacattccaagagc-3′; for E96A, 5′-gttctctgaacatttggtcgcttgaaggcacagtatatc-3′ and 5′-gatatactgtgccttcaagcgaccaaatgttcagagaac-3′, for D210A, 5′-atgtgccacattgagggctccacacagcacaag-3′ and 5′-cttgtgctgtgtggagccctcaatgtggcacat-3′; and for D308A, 5′-tgataggacagtgagcactgccgagggcc-3′ and 5′-ggccctcggcagtgctcactgtcctatca-3′. In brief, the fusion proteins were first adhered to Ni-NTA affinity resin (Qiagen, Inc.). On-column cleavage with the SUMO-specific protease Ulp1 was used to cleave full-length APE1 encoding one extra N-terminal residue, Ser, followed by the native sequence. APE1 was eluted from Ni-NTA agarose resin (Qiagen, Inc.) and then applied to a tandem Q-Sepharose and SP-Sepharose. After loading the sample, the Q-Sepharose column, which binds any contaminating SUMO protein, was removed, and a linear NaCl gradient (10 column volumes from 50 mM to 1 M) was then applied to the SP-Sepharose column. Fractions containing APE1 protein were pooled and concentrated.
Crystallization and Data Collection
Crystals were obtained by mixing equal parts (1 μL each) of microseeds with a precipitant solution containing 100 mM MES, pH 6.0, 200 mM NaCl, 18–21% PEG 4000, and C138A Δ40 hAPE1 (10 mg/mL) buffered in 10 mM HEPES, pH 7.5. The protein was diluted from a stock (120 mg/mL) stored in 50 mM MES, pH 6.0, 1 mM DTT, and 300 mM NaCl. Self-nucleated crystals of APE1 were obtained under similar conditions and used to produce microseeds.
Metal Complexes
Complexes with either MgCl2 or MnCl2 were obtained by cocrystallization under conditions similar to those described above with the exception that 1 mM MgCl2 or 5 mM MnCl2 was added directly to the protein prior to crystallization. Crystals were cryocooled in 100 mM MES, pH 6.0, 200 mM NaCl, 22% PEG 4000, 20–22% ethylene glycol, and either 1 mM MgCl2 or 5 mM MnCl2.
Data Collection
Data for the Mg2+ complex with C138A Δ40 APE1 were collected at beamline GM/CA 23ID-D at the Advanced Photon Source, Argonne National Laboratory, and processed with HKL2000.22 For the APE1–Mn2+ complex, anomalous data were collected on a Bruker X8 Prospector (Bruker Corporation, Billerica, MA) with Cu Kα radiation (1.5418 Å) at 100 K using an Oxford Cryosystem. Data were integrated using SAINT23 and scaled with SADABS.23 XPREP23 was used to determine the space group and to analyze the data. The apo-APE1 data were also collected on our home source instrument and processed as described for the MnCl2 complex. All of the crystals belong to space group P21212, with cell dimensions as shown in Table 1.
Table 1. Crystallographic Data.
| data set | apo | Mg2+ | Mn2+ |
|---|---|---|---|
| Data Statistics | |||
| a (Å) | 46.630 | 46.491 | 46.638 |
| b (Å) | 141.128 | 137.545 | 140.490 |
| c (Å) | 45.246 | 45.054 | 45.197 |
| space group | P21212 | P21212 | P21212 |
| resolution | 50–1.65 | 50–1.40 | 43–2.18 |
| completeness (%) | 98.8 (99.1) | 99.1 (96.8) | 99.4 (96.7) |
| Rmerge (%) | 6.0 (28.9) | 4.6 (28.7) | 9.9 (26.4) |
| I/σ | 13.2 (3.1) | 26.7 (4.9) | 10.3 (3.4) |
| Refinement Statistics | |||
| R value (%) | 20.0 | 17.1 | 19.6 |
| R free (%) | 23.8 | 19.3 | 25.6 |
| RMSD bonds (Å) | 0.005 | 0.006 | 0.005 |
| RMSD angles (deg) | 0.994 | 1.098 | 0.977 |
| average B-factor | 17.2 | 17.7a | 18.1 |
| Ramachandran Statistics | |||
| favored (%) | 97.8 | 98.2 | 97.8 |
| allowed (%) | 2.2 | 1.8 | 2.2 |
| outliers (%) | 0 | 0 | 0 |
All atoms included in this calculation rather than just protein atoms.
Structure Determination and Refinement
The apo and metal complex structures were phased by molecular replacement (MOLREP)24 using the coordinates of a previously refined structure of C138A Δ40 hAPE1 as the search model. Several rounds of initial refinement were carried out using REFMAC5,25 accompanied by iterative model building using COOT.26 Following placement of well-ordered water molecules, excluding those in the repair active site of the enzyme, bound metal was identified in difference Fourier maps. The position of Mn2+ was determined by anomalous difference Fourier analysis using the phases from the starting molecular replacement model prior to addition of Mn2+. The Mn2+ ion was identified as the highest peak (14.0σ) in the anomalous difference Fourier map as compared to the next highest peak at 5.4σ. For the Mg2+ structure, a single Mg2+ ion was placed in the highest positive peak in the Fo – Fc difference electron density map (11.55σ). The next highest peak in this difference map was 6.2σ. A distinctive feature of the Mg2+ difference peak was its somewhat elongated shape, suggesting potential disorder. Following refinement of a single Mg2+ placed in the middle of the difference electron density peak, positive peaks were observed on either side of its central location. The Mg2+ ion was then modeled as two partially occupied sites within the difference peak observed prior to its inclusion in the model, and the occupancy of each site was adjusted manually based on examination of difference electron density peaks leading to one site (site B) with an occupancy of 0.65 and the other (site A), 0.35. Similarly, there is evidence for disorder of one of the coordinating water ligands for the bound Mg2+ in the form of an elongated difference peak. Later stages of refinement were done using anisotropic temperature factors in REFMAC5.25 For the apo and Mn2+ structures, the final rounds of refinement were carried out by using PHENIX27 with isotropic temperature factor refinement. Statistics for refinements are compiled in Table 1. Coordinates have been deposited with the PDB under entries 4QH9, 4QHD, and 4QHE for Mn2+-bound, apo, and Mg2+-bound APE1 structures, respectively.
Differential Scanning Fluorimetry Assays
Differential scanning fluorimetry (DSF) assays28 were carried out in 100 mM HEPES, pH 7.0, 150 mM NaCl, and 4× SYPRO orange (Invitrogen). The final concentration of full-length (FL) APE1 used in the DSF assays was 2 μM, diluted from a 2.48 mM stock solution, buffered in 50 mM Tris, pH 8.0, and 100 mM NaCl. FL APE1 was incubated with 10 μM to 2.56 mM Mg2+ or Mn2+ for 15 min at room temperature prior to the addition of SYPRO orange dye. Reactions were then measured using a Roche Light Cycler 480 with an excitation wavelength of 483 nm and emission of 568 nm. The change in melting temperature (Tm) for each reaction was calculated by subtracting the Tm for the reaction containing no metal ion from that containing metal and plotted against metal ion concentration.
Enzyme Assays
APE1 endonuclease assays were performed as previously described using a fluorescently labeled substrate.5 Abasic DNA substrate used for the in vitro abasic DNA cleavage assay was annealed from 5′-(HEX)-AATTCACCGGTACC(THF)CCTAGAATTCG-3′ and its complementary strand, where HEX is a hexachlorofluoroscein tag at the 5′ end of the abasic site containing strand and THF is tetrahydrofuran. The oligonucleotides were purchased from Midland Certified Reagent. Reaction conditions were 50 mM HEPES (pH 7.5), 50 mM KCl, 1 mM MgCl2, 1 mM DTT, 10 mg/mL BSA, 20 nM DNA substrate, and varying concentrations of FL APE1 enzymes, as specified, in a final volume of 10 μL. Before use, FL APE1 enzyme stocks were diluted in a buffer containing 50 mM HEPES (pH 7.5), 50 mM KCl, 1 mM MgCl2, and 1 mM DTT to a concentration 2-fold higher than the final value.
Reactions were assembled on ice, and enzymes were added to initiate the reaction. Reaction mixtures were incubated at 37 °C for 15 min and then terminated by the addition of 10 μL of 99.5% formamide. Denatured DNA products were separated on a 20% denaturing polyacrylamide gel containing 7 M urea. All gels were run in 1× TBE buffer (90 mM Tris, 90 mM boric acid, 2 mM EDTA) at 200 V for 1 h at room temperature. Following electrophoresis, the HEX labeled DNA bands were visualized using an SHG 532 nm green laser and LPG filter on a Fujifilm FLA-5100 system. Fluorescence was quantified using Multi Gauge V3.1 software. For metal titration assays, 20 nM DNA substrate was incubated with 0.6 nM FL APE1 (WT) in a final volume of 10 μL for 15 min at 37 °C in buffer as above except that the MgCl2 or MnCl2 concentrations were 0, 0.25, 0.5, 1.0, 2.0, 4.0, or 8.0 mM. Specific activities were determined for concentrations of enzyme within the linear range of the assay and assigned values relative to the wild-type enzyme.
Results
Crystals of C138A Δ40 APE1 Diffract to High Resolution
To date, the crystal structures reported for APE1 all contain bound metal, Mg2+, Pb2+, or Sm3+ ions,13,19,29 which appear to be required for crystallization of wild-type or N-terminally truncated versions of APE1. In this work, we sought to analyze the effects of metal binding in APE1 at high resolution. Ideally, one would crystallize APE1 in the presence and absence of metal ions in the same crystal form. To accomplish this, we substituted a surface residue with the goal of changing the packing interactions within the lattice and obtaining APE1 crystals under conditions that would not require metal ions for crystallization. We selected C138 for substitution due to its solvent accessibility and tendency to form a disulfide bond with C138 from a neighboring molecule in the samarium acetate crystal form.29 Substitution of Cys 138 with Ala does not affect APE1’s endonuclease activity.30,31 Thus, we initially prepared C138S and C138A Δ40 APE1, referring to an N-terminally truncated form lacking 39 amino acids from the N-terminus. The C138S Δ40 APE1 was even more soluble than the wild-type sequence and did not crystallize in initial screening experiments. However, following an incomplete factorial screen, crystals of the C138A Δ40 APE1 were obtained from a sodium chloride and PEG 4000 containing precipitant solution without the use of divalent metal ions. We refer to this structure as the apo structure of APE1 crystallized in the absence of metal bound to the active site. We also cocrystallized APE1 in the same lattice with either Mg2+ or Mn2+. Interestingly, this is the same lattice that we obtained for C65A APE1,20 but, in that case, the crystallization conditions contained Sm3+. These crystals diffract to high resolution with complete data collected in the best case to 1.4 Å for the Mg2+ complex (Table 1). Data for the apo and Mn2+ structures were collected to 1.65 and 2.2 Å, respectively, on our home source instrument (see Materials and Methods). The lower resolution of the Mn2+ data set results from a strategy designed to collect relatively complete anomalous data that would allow unambiguous identification of metal binding sites in this structure. Previously, the highest resolution structures reported were a 1.92 Å for the wild-type–Mg2+ complex and 1.9 Å for the C65A APE1. Thus, our crystals exhibit a marked improvement in resolution, allowing us to analyze alternate side chain conformations within the active site for the Mg2+ and apo structures. The structures were determined by molecular replacement and refined as described in the Materials and Methods.
Mn2+ Binds to D70 and E96 in the Active Site of APE1
It has been previously shown that Mn2+ will effectively substitute for Mg2+ to promote APE1 catalysis, albeit with a pre-steady-state catalytic rate reduced by a factor of ∼1.5-fold, as determined by rapid quench flow techniques.3 Structurally, Mn2+ has the advantage of having an anomalous signal that can be measured using Cu Kα X-radiation. We determined a 2.2 Å crystal structure for an APE1–Mn2+ complex (Table 1) and confirmed the site of bound Mn2+ by anomalous difference Fourier analysis (Figure 1A). A 14σ anomalous difference peak corresponds to the refined Mn2+ position in our final model for this complex, with D70 and E96 serving as coordinating ligands. There is no anomalous peak consistent with binding of Mn2+ to a second proposed metal site involving H309, D210, and N212 as coordinating ligands. Thus, Mn2+ does not appear to bind to this second site despite a much higher propensity to accept coordinating N atoms as ligands than Mg2+. In the APE1–Mn2+ structure, Mn2+ is coordinated by one oxygen atom from D70 and one from E96 with coordination distances of 2.3 and 2.1 Å, respectively, and the four remaining ligands are water molecules with coordinating distances of 2.2–2.4 Å (Figure 1B). The six coordinating ligands exhibit octahedral coordination geometry for the Mn2+ bound to APE1. D308 is too far away to coordinate Mn2+ directly but is hydrogen-bonded to two of the coordinating water molecules at distances of 2.6 and 2.7 Å, respectively. The only other residue within hydrogen-bonding distance of a metal coordinating water molecule is K98.
Figure 1.

Metal binding sites are shown for Mn2+, Mg2+, and apo APE1 crystals structures. In each panel, the protein model is shown as a gray cartoon rendering, with D70, E96, and D308 shown as stick renderings with carbons in yellow and oxygens in red. (A) The anomalous difference Fourier map (magenta) contoured at 6σ (magenta mesh) and 2Fo – Fc difference electron density map contoured at 1.2σ (blue mesh) are shown for the Mn2+–APE1 complex structure. Both D70 and E96 directly coordinate the Mn2+ ion. (B) A cartoon and stick rendering of the metal binding site as in panel A is shown with coordination distances for protein and water ligands bound to Mn2+. (C) Initial Fo – Fc difference electron density map contoured at 3σ (green mesh) and final 2Fo – Fc difference electron density map contoured at 1.2σ (blue mesh) are shown for the Mg2+–APE1 complex structure. Mg2+ atoms (pink spheres) and one water molecule (red spheres) are shown modeled in the elongated Fo – Fc difference peaks. The Mg2+ site denoted as the B site is shown in (D) and the A site in (E) along with coordinating ligands and distances. (F) The final 2Fo – Fc electron density map is shown for the metal binding site of apo APE1 structure (i.e., without bound metal). Red spheres are water molecules. (G) The metal binding site in the apo structure is shown with a water molecule (red sphere) coordinated to D70 and E96.
Mg2+ Exhibits Disorder within the Active Site of APE1
In the crystal structure of the APE1–Mg2+ complex determined at 1.4 Å, a single elongated 11.55σ Fo – Fc peak was identified as the Mg2+ binding site (Figure 1C). There is no evidence for Mg2+ bound to another site in the Fo – Fc map. As described in the Materials and Methods, refinement efforts led to a model with two partially occupied Mg2+ sites that are 0.7 Å apart: one involving coordination primarily with E96 (A site, 0.35 occupancy) and the second involving coordination with both D70 and E96 (B site, 0.65 occupancy) (Figure 1D,E). The designations of A and B in this context refer to alternate conformations for the Mg2+ in our crystal structure and should not be confused with previous reports in which a second proposed binding site involving coordination with D210, N212, and H309 was referred to as the B site.13 As noted above, there is no evidence in our structures for binding to this second site. The A site for Mg2+ in our structure is more closely associated with the A conformation of E96 at a distance of 2.0 Å between the coordinating oxygen and Mg2+. This same E96 coordinating oxygen is 2.4 Å from the B site for Mg2+. Similarly, the coordinating oxygens from D70 and the B conformation of E96 are 1.9 and 2.0 Å, respectively, from Mg2+ in the B site, whereas that same D70 oxygen is 2.5 Å from Mg2+ in the A site (Figure 1D,E). Although three of the coordinating waters are in common, there is evidence for one water ligand that is unique to each of the two Mg2+ sites, providing further evidence for modeling of two different Mg2+ positions. Coordinating waters are 1.9–2.5 Å from the metal positions. As was true in the Mn2+ structure, D308 is 4.0 Å from the closest Mg2+ site and therefore not close enough to coordinate the metal directly. However, D308 is hydrogen-bonded to two water molecules that coordinate Mg2+ in the structure with distances of 2.7 and 2.8 Å. In this structure, K98 is too far away, but Y171 is hydrogen-bonded to one of the two coordinating water molecule conformations associated with the disordered Mg2+ at a distance of 2.8 Å.
Glu 96 Adopts Three Different Conformations in APE1 Structures
In the apo structure, there is only one conformation observed for E96 (Figure 1F), and in place of bound metal, there is a water molecule hydrogen-bonded to D70 (2.66 Å) and E96 (2.45 Å) (Figure 1G). Although the apo protein and metal complexes reported here were crystallized in the same lattice, there is some variation in b, the longest cell parameter. In the Mg2+ complex structure, b is 137.5 Å, whereas in the other two structures, apo and Mn2+ complex, b is approximately 141 Å (Table 1). Structural differences in the backbone atom positions that correlate with the difference in this cell parameter include residues 77–86, 98–102, and 106–113, which comprise surface loop and helical elements in the structure (Figure 2A). Thus, the difference observed in the position of K98 in the Mn2+ vs Mg2+ structure likely results from this difference in the b cell parameter.
Figure 2.
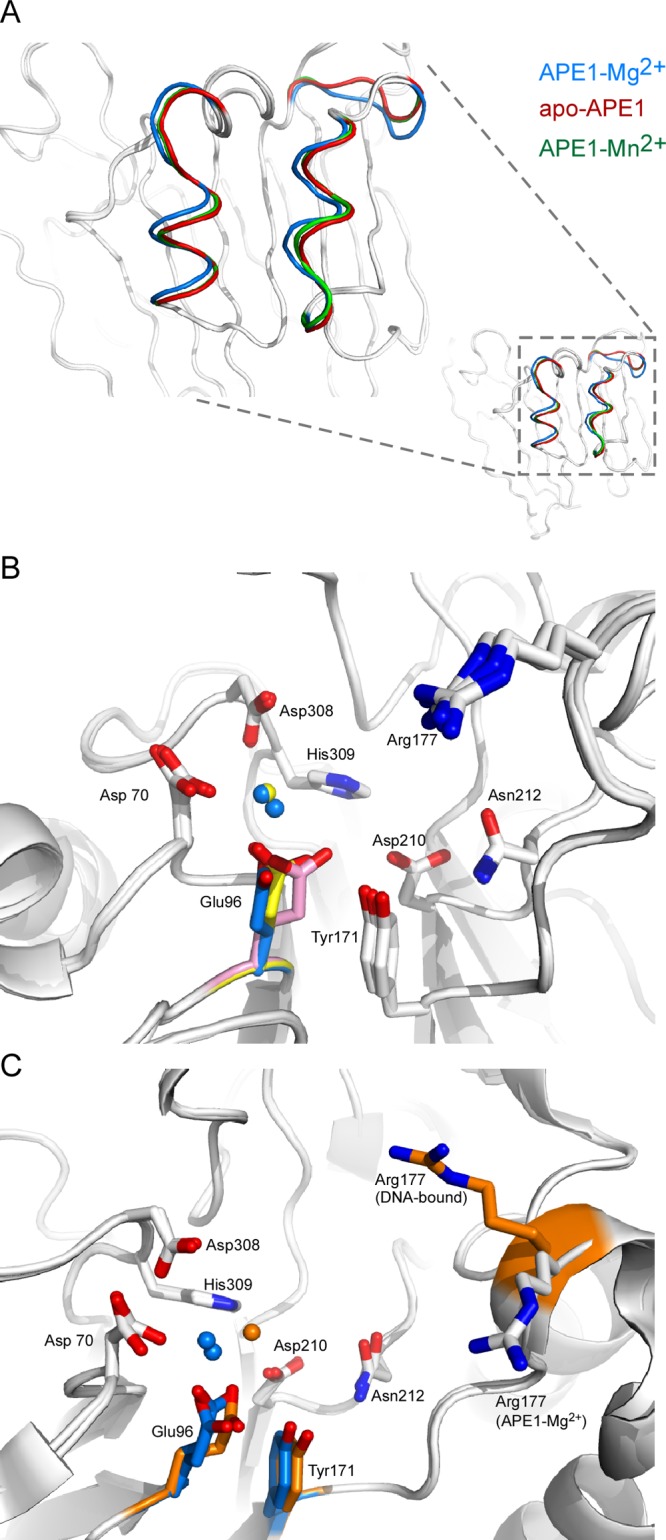
Structural comparisons are shown for different APE1 structures. (A) Differences between the Mg2+-bound APE1 (blue ribbon rendering) and apo-APE1 (red) or Mn2+-bound APE1 (green) structures are shown close-up and in the context of the full structure (inset) for the surface loop (amino acids 98–102) and two alpha helices (amino acids 77–86 and 106–113, respectively). Structural differences appear to correlate with the length of the b cell axis (Table 1), which is shorter in the Mg2+-bound APE1 structure. (B) Active site residues are shown in stick renderings with carbons in gray, oxygens in red, and nitrogens in blue for superimposed structures of apo-APE1, APE1–Mg2+, and APE1–Mn2+. Overall, active site residues in each of the structures have a single conserved conformation including D210, N212, and H309 in all three structures. Small differences in the conformation for Y171 were observed. Remarkably, three different conformations for E96 were found in apo-APE1 (pink), APE1–Mg2+ (marine blue), and APE1–Mn2+ (yellow). Mg2+ (marine blue) and Mn2+ (yellow) are depicted as spheres in the active sites. (C) A comparison of our Mg2+-bound APE1 structure with that of a ternary complex with DNA and Mg2+ bound to APE1 (PDB ID: 4IEM) reveals a single conformation for D210, N212, and H309, as was true for our three structures. Differences are observed in the conformations for E96 and R177 in the DNA-bound complex (orange) due to intercalation in the DNA. Bound Mg2+ ions are shown as blue spheres for our structure and orange for the DNA complex.
Of relevance to the present study is an analysis of residues within these three structures that vary in side chain conformation. Within these three structures, 24 residues have side chain conformations that differ significantly from one another. Most of these residues are surface residues, with the most prevalent being Glu, Lys, and Arg residues; see Table 2 for a complete list of residues with varying side chain conformations. While some variations in side chain conformation for surface residues may result from poorly resolved density, residues within the active site, with few exceptions, have well-ordered electron density. Overall, the structures of the active site residues are remarkably similar in all of our structures. Notably, important active site residues including D210, N212, and H309 along with surrounding residues show no differences in conformation in the three structures (Figure 2B). In the mechanism proposed by Tainer et al., N212 and H309 play critical roles in positioning the scissile P–O3′ bond for catalytis, whereas D210 activates a nucleophilic water that attacks the bond.6 Small differences in the position of Y171 are apparent in the three structures, but the only residue in the active site that exhibits markedly different conformations in the metal-bound versus apo structure is E96. The conformation of E96 observed in the apo structure is clearly distinct from the E96 conformation observed in the Mg2+- and Mn2+-bound structures (Figure 2B).
Table 2. Residues with Multiple Side Chain Conformations in Mg2+, Mn2+, and Apo APE1 Structuresa.
| Glu | Lys | Arg | Trp | Ser | Val | Thr | Asn | Gln | Cys | Met |
|---|---|---|---|---|---|---|---|---|---|---|
| 46 | 125 | 73 | 83 | 143 | 166 | 266 | 272 | 245 | 99 | 270 |
| 87 | 224 | 187 | 180 | 271 | ||||||
| 96 | 228 | 193 | ||||||||
| 110 | 276 | 202 | ||||||||
| 183 | 303 |
Amino acids with multiple side chain conformations are shown with specific residue numbers for each amino acid residue type.
Conformational Plasticity of E96 Correlates with Metal Binding Sites Observed in APE1
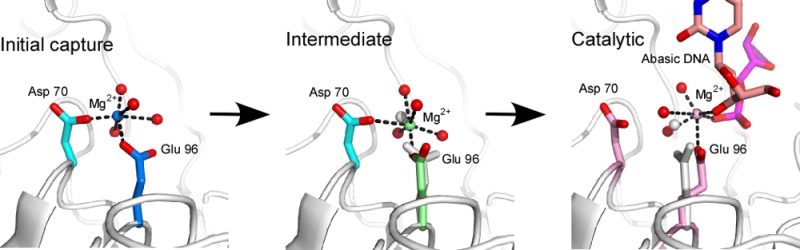
The structures of the active sites of APE1 with Mg2+ bound in the presence and absence of abasic site containing DNA also show remarkably few differences. R177 in the product-bound complex intercalates at the abasic site adopting a significantly different conformation than in the complex without DNA, and Y171 adopts a slightly different conformation in the two structures (Figure 2C). The only other residue that adopts a significantly different conformation is E96. In the DNA-bound complex, E96 serves as the sole protein ligand for bound Mg2+ and adopts a conformation similar to that observed in our apo structure (Figure 3D). Thus, collectively, there are three different binding sites identified for Mg2+ and three different conformations for E96 in these two Mg2+-bound structures (Figures 2C). The first conformation of E96 is also observed in the APE1–Mn2+ structure, with D70 and E96 ligands bound to the B site observed in the Mg2+ structure (Figure 3A). A second E96 conformation is observed for coordination to the A site in the Mg2+ structure (Figure 3B), and a third conformation of E96 observed in both the product complex and the apo structure positions of E96 to coordinate metal and product DNA in the APE1–Mg2+–DNA complex (Figure 3C,D). This metal binding site is 2.1 Å from the Mg2+ site A in our structure that is coordinated primarily by E96. Collectively, these different metal binding sites and conformations of E96 may represent different sites of occupancy for the bound metal ion and the ability to move from an initial capture site to a site involving interaction with bound substrate.
Figure 3.

Structural models suggest movement of Mg2+ between an initial capture site and a final DNA-bound position within the active site of APE1. A gray cartoon rendering of the APE1 structure is shown with stick models (carbons in specified colors; oxygens in red) for D70 and E96. (A) Mg2+ (blue sphere) is bound in the B site with E96-B conformation (blue stick) and coordinating waters (red spheres) representing initial capture of the metal in the active site. (B) Mg2+ shifts to bind in the A site to the E96-A conformation (light green stick) and coordinating waters (red spheres), possibly in an intermediate binding mode. The gray sphere indicates the position of the B site Mg2+ metal. (C) In the presence of DNA, Mg2+ (pink sphere) bound to E96 shifts to interact with substrate DNA in this catalytically competent mode. The gray stick rendering is the A conformation of E96. The DNA-bound model is from the APE1–DNA complex structure (PBD ID: 4IEM). (D) Mg2+ bound to E96 in 4IEM structure is superimposed on the apo APE1 structure. The conformation of E96 within these two structures is similar. Thus, the conformational plasticity of E96 may facilitate initial capture, an intermediate binding mode, and a catalytically relevant binding mode for Mg2+.
D70, E96, and D308 Are Involved in Metal Binding by APE1
To examine metal binding by APE1, we employed differential scanning fluorimetry (DSF) as a measure of metal binding through determination of the melting temperatures. In this assay, the dye SYPRO orange was used to monitor unfolding of the protein with increasing temperature; the inflection point of the transition curve (Tm) is calculated from plotting the fluorescence intensity against the temperature.28 Full-length APE1 was used for these experiments. The melting temperature of APE1, as measured by DSF, is ∼48–50 °C. Addition of increasing concentrations of MgCl2 or MnCl2 increased the melting temperature of APE1 by a maximum of 4.5–6.4 °C, respectively (Figure 4), consistent with stabilization of the structure upon binding of metal.
Figure 4.
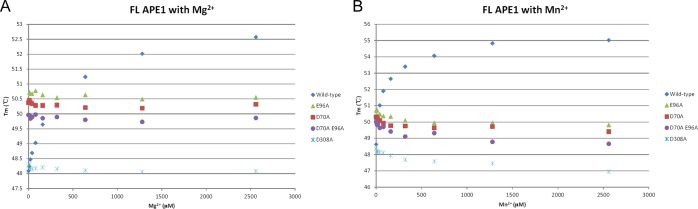
Effect of metal binding on the stability of wild-type, D70A, E96A, D70A/E96A, D308A, and full-length APE1. Differential scanning fluorimetry was used to measure the change in melting temperature, which is plotted versus the concentration of Mg2+ (A) or Mn2+ (B).
To determine the role of E96, D70, and D308 in binding metal ions, we prepared substituted full-length APE1 proteins D70A, E96A, D70A/E96A, and D308A and measured binding of MgCl2 or MnCl2 at concentrations up to 2.6 mM using DSF. We elected to substitute these acidic residues with Ala in order to ensure that no metal binding through the side chain atoms would be possible. As a control to assess metal binding in solution to the proposed site involving D210, N212, and H309, we also prepared and analyzed D210A APE1. Melting temperatures of D70, E96, D70/E96, and D308 were 50.3, 50.6, 50.0, and 48.2 °C, respectively and thus were similar to the Tm of the wild-type protein, 48.4 °C. However, the D210A enzyme exhibited a significantly lower melting temperature of 38.4 °C, suggesting that this substitution was destabilizing and cannot be compared directly to the other enzymes. The melting temperature of D210A APE1 did increase by 2.5 °C maximally upon addition of increasing concentrations of Mg2+. As shown in Figure 4, addition of metal to D70A, E96A, D70A/E96A, or D308A proteins has no effect on the melting temperature, consistent with loss of stabilization by metal. A slight decrease of −0.9 °C in the Tm was observed upon addition of either Mg2+ or Mn2+ to the D70A APE1 protein. Similar results were obtained for addition of Mg2+ or Mn2+ to E96A or the doubly substituted D70A/E96A APE1 proteins with decreases of Tm of −0.9 and −1.35 °C, respectively. The Tm for the D308A protein was unaffected by addition of either Mg2+ or Mn2+. Thus, in the absence of bound substrate, we conclude that D70, E96, and D308 play a role in metal binding, resulting in stabilization of APE1. This result is consistent with our crystallographic results for Mg2+- and Mn2+-bound complexes with APE1 in which both D70 and E96 directly coordinate metal and D308 coordinates metal through a water molecule (Figure 1).
D70A/E96A APE1 Shows Significantly Impaired Enzymatic Activity
Having established a role for D70, E96, and D308 in the binding of metal as assessed by DSF, we next sought to determine the effects of substituting these residues on catalytic activity. Enzymatic activity for the endonuclease activity of wild-type, D70A, E96A, D70A/E96A, D308A, and D210A enzymes was measured as previously described.5 The D70A and E96A enzymes exhibited modest decreases of 8.7- vs 8.4-fold, respectively, in activity compared to that of the wild-type protein, as shown in Table 3. However, the D70A/E96A enzyme had significantly lower activity than either single substitution, with an approximately 300-fold decrease relative to that of the wild-type protein. In contrast, D308A has slightly higher than wild-type activity, suggesting that, although it contributes to capture of metal under the conditions used for DSF experiments, it plays a much less significant role under the conditions of catalysis where the ratio of metal to enzyme is at least 3 orders of magnitude higher than that in the DSF assay. Thus, the relative contribution of D308 in binding metal is smaller during catalysis. The D210A enzyme had no observable activity under the conditions assayed.
Table 3. Comparison of Relative Specific Activities for APE1 Enzymes.
| APE1 enzyme | relative activity | fold-decrease vs wild type |
|---|---|---|
| wild type | 100 | 1 |
| D70A | 11.8 | 8.7 |
| E96A | 11.9 | 8.4 |
| D70A/E96A | 0.35 | 287 |
| D308A | 125 | |
| D210A | 0 |
Mg2+-Catalyzed APE1 Activity Is Higher than That of Mn2+
Both Mg2+ and Mn2+ are coordinated by D70 and E96 in the absence of bound substrate and coordinated by E96 alone in the presence of DNA product in crystal structures. Thus, it was of interest to compare steady-state activity of full-length APE1 catalyzed by Mg2+ or Mn2+ and determine optimal concentrations supporting activity. The concentration of either Mg2+ or Mn2+ was varied from 0.25 to 8 mM for reactions containing 0.6 nM APE1, a concentration within the linear response range of the assay. Under the conditions of our assay, the cleavage rates for Mg2+ at low concentrations of metal were approximately 1.7-fold higher than those for Mn2+ at the same concentrations, especially at the lower concentrations used, as shown in Figure 5. However, each showed optimal activity between 4 and 8 mM.
Figure 5.
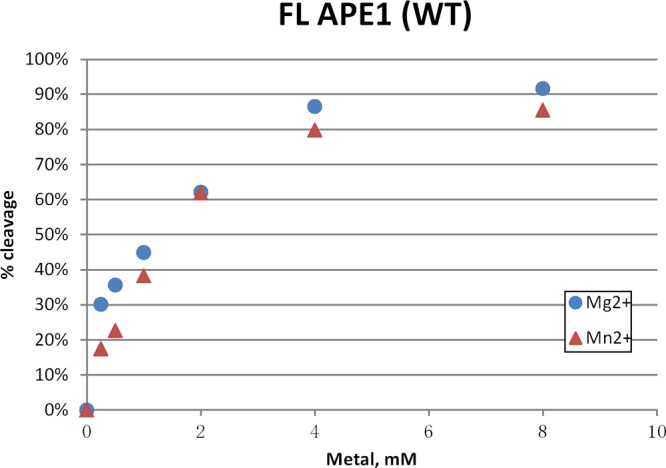
Comparison of catalytic activity supported by varying concentrations of Mg2+ or Mn2+. In this experiment, 0.6 nM full-length APE1 (WT) was used in the reaction mixture containing 20 nM HEX labeled DNA substrate and increasing concentrations of MgCl2 or MnCl2 (i.e., 0.25, 0.5, 1.0, 2.0, 4.0, 8.0 mM). The percentage of cleaved product is represented as a function of metal concentration. For all concentrations, activity catalyzed by Mn2+ is lower than that of Mg2+.
Discussion
Collectively, our studies support a role for a single preformed metal binding site in APE1 involving D70, E96, and, to a lesser extent, D308 in facilitating endonuclease activity. All three of these residues appear to be essential for stabilization of the enzyme by Mg2+ or Mn2+ measured by DSF, as substitution with Ala for any of these residues results in loss of stabilization. These findings are supported by our structural results and those of others19,32 in which D70 and E96 directly coordinate the bound metal and D308 is hydrogen-bonded to coordinating water ligands. No experimental evidence was found for binding to the previously proposed B site involving coordination by D210, N212, and H309,13 consistent with results reported from the solid state 25Mg NMR study.17
To determine what role a preformed metal binding site might play in catalysis, we determined the enzymatic activity of D70A, E96A, and D308A enzymes. Modest reductions in specific activity, 8.4- to 8.7-fold, were observed for D96A or E70A enzymes, respectively, and slightly increased activity was observed for D308A (Table 3). In previous reports for enzymatic analysis of E96A APE1, reductions in activity range from 15-fold18 to 105-fold,33 with several groups reporting 400- to 600-fold reductions.34−36 Thus, we felt it was important to compare all of the substituted APE1 enzymes prepared in the same way and to the same level of purity in our system. Our results for E96A are in fairly close agreement with the most recently reported 15-fold reduction in activity for E96A.18 Our 8.7-fold reduction in D70A APE1 activity is in close agreement with a previous report of a 6.7-fold reduction in activity.37 Finally, we find that D308A is slightly more active than that of wild-type enzyme, while a previous study reported a modest 1.4-fold reduction in activity.37 These results suggest that substitution of one of the three residues, D70, E96, or D308, involved in coordinating metal has relatively modest effects on enzymatic activity. However, the D70A/E96A enzyme had a much more profound effect and is ∼300-fold less active than the wild-type enzyme and ∼30-fold less active than the singly substituted enzymes, D70A and D96A APE1. These decreases in activity are not comparable to those observed for residues involved directly in the chemistry of the reaction such as D210 or H309, which have reductions in activity of 30,000 and 25,000-fold, respectively.36,38−40 However, the decrease in activity observed for the D70A/E96A APE1 enzyme is sufficiently large to suggest an important role in the overall reaction. The proposed role for the single Mg2+ ion is to help position the substrate and stabilize the transition state and 3′-OH leaving group.6 Although the substrate would likely have bound metal ions associated with it, these metal ions would not be strategically located to facilitate the reaction. Thus, potentially, the advantage of a preformed metal binding site for the enzyme is the increase in effective metal concentration at the desired site to promote catalysis.
Of particular interest was our finding that Mg2+ is disordered in our 1.4 Å structure and exhibits two partially occupied binding sites involving coordination by D70 and E96. Two independent lines of evidence support this finding. In two other lower resolution crystal structures available for APE1 with bound Mg2+ (PDB IDs: 4LND and 3U8U), the position of Mg2+ corresponds to one or the other of the two binding modes that we identified (Figure 6). In solid-state 25Mg NMR studies, a single mole equivalent of Mg2+ was bound to APE1 in both the presence and absence of bound THF containing oligonucleotide substrate with two distinct resonances for Mg2+. This result is consistent with disorder of the Mg2+ due to differences in coordinating ligands and thus plasticity in the active site.17 Our results now provide a structural context for the observed disorder of bound Mg2+ in the absence of bound DNA and implicate E96 as the residue conferring plasticity to the active site. These results set the stage for the ability of Mg2+ to move in the enzyme.
Figure 6.

A comparison of coordinating ligands and distances for Mg2+-bound APE1 structures. Two other structures from the PDB, 4LND (A) and 3U8U (B), are shown superimposed on our Mg2+–APE1 structure. A cartoon rendering of our APE1 structure is shown in lime green. (A) The B conformation of E96 from our Mg2+ structure (marine blue) is in a similar conformation to that of E96 in 4LND (pink, oxygens in red). The A conformation of E96 is shown in gray. (B) The A conformation of E96 in our Mg2+ structure shown in lime green is in a similar conformation to that of E96 in 3U8U shown in orange; oxgyens are shown in red. The B conformation of E96 is shown in gray. D70 is shown in a stick rendering with carbons in blue and oxygens in red for our structure, with carbons in pink for 4LND (A) and with carbons in orange for 3U8U (B). Bound Mg2+ for our structure is shown as a blue sphere in (A) and a green sphere in (B). Mg2+ in 4LND is shown as a pink sphere in (A) and in 3U8U is shown as an orange sphere in (B).
As observed in pre-steady-state measurements of APE1’s endonuclease activity,3 under steady-state conditions as shown in Figure 5, the rate of reaction for Mn2+ is also slower than that for Mg2+ at lower concentrations, but it is more similar at higher concentrations. In crystal structures of APE1–DNA–Mn2+ and APE1–DNA–Mg2+ complexes, Mn2+ and Mg2+ are both coordinated by E96 and the DNA product, and in APE1–Mg2+ and APE1–Mn2+ structures, the metals are coordinated by D70 and E96. However, we did not observe disorder in Mn2+ binding in our APE1–Mn2+ structure as was observed for Mg2+. On the basis of structural analysis, we cannot determine whether this is a property of Mn2+ vs Mg2+ or whether the much lower resolution of the Mn2+ structure masks this type of disorder. The fact that the Mn2+-catalyzed endonuclease reaction is slower in both pre-steady-state3 and steady-state experiments suggests that there may be differences in the reaction catalyzed by Mn2+ vs Mg2+. One possibility is that the larger size of Mn2+ affects its ability to move similarly to Mg2+ within the active site and thereby affects the rate of the reaction, as previously suggested.17
In conclusion, our studies provide evidence for initial capture of metal ion in the active site of APE1 by D70 and E96 and suggest that this capture is important for catalysis. Our structures provide evidence for structural plasticity within the active site involving E96, which directly coordinates Mg2+ and is captured in three different conformations. This novel plasticity observed for E96 provides a possible mechanism for binding of Mg2+ in three different positions all involving coordination with E96, two observed in our APE1–Mg2+ structure and the third in the product DNA complex structure (4IEM).18
Acknowledgments
We thank the staff at the Advanced Photon Source GM/CA beamline for assistance with our data collection efforts. We also thank our colleague, Tom Hurley, for helpful discussions related to this manuscript.
Glossary
Abbreviations
- hAPE1
human apurinic/apyrimidinic endonuclease
- Mg2+
magnesium ion
- Mn2+
manganese ion
- Pb2+
lead ion
- Arg (R)
arginine
- Lys (K)
lysine
- Glu (E)
glutamate
- Asp (D)
aspartate
- His (H)
histidine
- Asn (N)
asparagine
- Cys (C)
cysteine
- Ser (S)
serine
- DSF
differential scanning fluorimetry
- MES
2-(N-morpholino)ethanesulfonic acid
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- Å
angstroms
Author Contributions
§ H.H. and Q.C. contributed equally to this work.
This work was supported by a grant from the National Institutes of Health, CA14571 to M.M.G. Data for this work were collected at GM/CA at the Advanced Photon Source, which has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract no. DE-AC02-06CH11357.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Demple B.; Herman T.; Chen D. S. (1991) Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc. Natl. Acad. Sci. U.S.A. 88, 11450–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D. M. III; Takeshita M.; Grollman A. P.; Demple B. (1995) Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J. Biol. Chem. 270, 16002–16007. [DOI] [PubMed] [Google Scholar]
- Schermerhorn K. M.; Delaney S. (2013) Transient-state kinetics of apurinic/apyrimidinic (AP) endonuclease 1 acting on an authentic AP site and commonly used substrate analogs: the effect of diverse metal ions and base mismatches. Biochemistry 52, 7669–7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher R. L.; Bloom L. B. (2007) Pre-steady-state kinetic characterization of the AP endonuclease activity of human AP endonuclease 1. J. Biol. Chem. 282, 30577–30585. [DOI] [PubMed] [Google Scholar]
- Kreklau E. L.; Limp-Foster M.; Liu N.; Xu Y.; Kelley M. R.; Erickson L. C. (2001) A novel fluorometric oligonucleotide assay to measure O6-methylguanine DNA methyltransferase, methylpurine DNA glycosylase, 8-oxoguanine DNA glycosylase and abasic endonuclease activities: DNA repair status in human breast carcinoma cells overexpressing methylpurine DNA glycosylase. Nucleic Acids Res. 29, 2558–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol C. D.; Izumi T.; Mitra S.; Tainer J. A. (2000) DNA-bound structure and mutants reveal abasic DNA binding by APE1 DNA repair and coordination. Nature 403, 451–456. [DOI] [PubMed] [Google Scholar]
- Mundle S. T.; Delaney J. C.; Essigmann J. M.; Strauss P. R. (2009) Enzymatic mechanism of human apurinic/apyrimidinic endonuclease against a THF AP site model substrate. Biochemistry 48, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beese L. S.; Derbyshire V.; Steitz T. A. (1993) Structure of DNA polymerase I Klenow fragment bound to duplex DNA. Science 260, 352–355. [DOI] [PubMed] [Google Scholar]
- Beese L. S.; Friedman J. M.; Steitz T. A. (1993) Crystal structures of the Klenow fragment of DNA polymerase I complexed with deoxynucleoside triphosphate and pyrophosphate. Biochemistry 32, 14095–14101. [DOI] [PubMed] [Google Scholar]
- Beese L. S.; Steitz T. A. (1991) Structural basis for the 3′–5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 10, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W. (2011) Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys. 44, 1–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol C. D.; Kuo C. F.; Thayer M. M.; Cunningham R. P.; Tainer J. A. (1995) Structure and function of the multifunctional DNA-repair enzyme exonuclease III. Nature 374, 381–386. [DOI] [PubMed] [Google Scholar]
- Beernink P. T.; Segelke B. W.; Hadi M. Z.; Erzberger J. P.; Wilson D. M. III; Rupp B. (2001) Two divalent metal ions in the active site of a new crystal form of human apurinic/apyrimidinic endonuclease, Ape1: implications for the catalytic mechanism. J. Mol. Biol. 307, 1023–1034. [DOI] [PubMed] [Google Scholar]
- Cowan J. A. (1998) Metal activation of enzymes in nucleic acid biochemistry. Chem. Rev. 98, 1067–1088. [DOI] [PubMed] [Google Scholar]
- Oezguen N.; Schein C. H.; Peddi S. R.; Power T. D.; Izumi T.; Braun W. (2007) A “moving metal mechanism” for substrate cleavage by the DNA repair endonuclease APE-1. Proteins 68, 313–323. [DOI] [PubMed] [Google Scholar]
- Oezguen N.; Mantha A. K.; Izumi T.; Schein C. H.; Mitra S.; Braun W. (2011) MD simulation and experimental evidence for Mg2+ binding at the B site in human AP endonuclease 1. Bioinformation 7, 184–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton A. S.; Heck R. W.; Primak S.; McNeill D. R.; Wilson D. M. III; Ellis P. D. (2008) Characterization of Mg2+ binding to the DNA repair protein apurinic/apyrimidic endonuclease 1 via solid-state 25Mg NMR spectroscopy. J. Am. Chem. Soc. 130, 9332–9341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa S. E.; Shin D. S.; Mol C. D.; Izumi T.; Arvai A. S.; Mantha A. K.; Szczesny B.; Ivanov I. N.; Hosfield D. J.; Maiti B.; Pique M. E.; Frankel K. A.; Hitomi K.; Cunningham R. P.; Mitra S.; Tainer J. A. (2013) Conserved structural chemistry for incision activity in structurally non-homologous apurinic/apyrimidinic endonuclease APE1 and endonuclease IV DNA repair enzymes. J. Biol. Chem. 288, 8445–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manvilla B. A.; Pozharski E.; Toth E. A.; Drohat A. C. (2013) Structure of human apurinic/apyrimidinic endonuclease 1 with the essential Mg2+ cofactor. Acta Crystallogr., Sect. D: Biol. Crystallogr. 69, 2555–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiadis M.; Luo M.; Gaur R.; Delaplane S.; Li X.; Kelley M. (2008) Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat. Res. 643, 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Luo M.; Marasco D.; Logsdon D.; LaFavers K. A.; Chen Q.; Reed A.; Kelley M. R.; Gross M. L.; Georgiadis M. M. (2013) Inhibition of apurinic/apyrimidinic endonuclease I’s redox activity revisited. Biochemistry 52, 2955–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (2007) SAINT, SADABS, and XPREP, Bruker AXS Inc., Madison, WI.
- Vagin A.; Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 22–25. [DOI] [PubMed] [Google Scholar]
- (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- Gorman M. A.; Morera S.; Rothwell D. G.; La Fortelle E.; Mol C. D.; Tainer J. A.; Hickson I. D.; Freemont P. S. (1997) The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 16, 6548–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M.; Zhang J.; He H.; Su D.; Chen Q.; Gross M. L.; Kelley M. R.; Georgiadis M. M. (2012) Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry 51, 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. J.; Kim D.; Illuzzi J. L.; Delaplane S.; Su D.; Bernier M.; Gross M. L.; Georgiadis M. M.; Wilson D. M. III. (2011) S-Glutathionylation of cysteine 99 in the APE1 protein impairs abasic endonuclease activity. J. Mol. Biol. 414, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L. H.; Barsky D.; Erzberger J. P.; Wilson D. M. III. (2000) Mapping the protein–DNA interface and the metal-binding site of the major human apurinic/apyrimidinic endonuclease. J. Mol. Biol. 298, 447–459. [DOI] [PubMed] [Google Scholar]
- Chou K. M.; Cheng Y. C. (2003) The exonuclease activity of human apurinic/apyrimidinic endonuclease (APE1). Biochemical properties and inhibition by the natural dinucleotide Gp4G. J. Biol. Chem. 278, 18289–18296. [DOI] [PubMed] [Google Scholar]
- Barzilay G.; Walker L. J.; Robson C. N.; Hickson I. D. (1995) Site-directed mutagenesis of the human DNA repair enzyme HAP1: identification of residues important for AP endonuclease and RNase H activity. Nucleic Acids Res. 23, 1544–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilay G.; Mol C. D.; Robson C. N.; Walker L. J.; Cunningham R. P.; Tainer J. A.; Hickson I. D. (1995) Identification of critical active-site residues in the multifunctional human DNA repair enzyme HAP1. Nat. Struct. Biol. 2, 561–568. [DOI] [PubMed] [Google Scholar]
- Erzberger J. P.; Wilson D. M. III. (1999) The role of Mg2+ and specific amino acid residues in the catalytic reaction of the major human abasic endonuclease: new insights from EDTA-resistant incision of acyclic abasic site analogs and site-directed mutagenesis. J. Mol. Biol. 290, 447–457. [DOI] [PubMed] [Google Scholar]
- Kim W. C.; Berquist B. R.; Chohan M.; Uy C.; Wilson D. M. III; Lee C. H. (2011) Characterization of the endoribonuclease active site of human apurinic/apyrimidinic endonuclease 1. J. Mol. Biol. 411, 960–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry D. F.; Hoyt D. W.; Khazi F. A.; Bagu J.; Lindsey A. G.; Wilson D. M. III. (2003) Investigation of the role of the histidine-aspartate pair in the human exonuclease III-like abasic endonuclease, Ape1. J. Mol. Biol. 329, 311–322. [DOI] [PubMed] [Google Scholar]
- Lucas J. A.; Masuda Y.; Bennett R. A.; Strauss N. S.; Strauss P. R. (1999) Single-turnover analysis of mutant human apurinic/apyrimidinic endonuclease. Biochemistry 38, 4958–4964. [DOI] [PubMed] [Google Scholar]
- Rothwell D. G.; Hang B.; Gorman M. A.; Freemont P. S.; Singer B.; Hickson I. D. (2000) Substitution of Asp-210 in HAP1 (APE/Ref-1) eliminates endonuclease activity but stabilises substrate binding. Nucleic Acids Res. 28, 2207–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]