

Abstract
The quaternary structure stability of proteins is typically studied under conditions that accelerate their aggregation/unfolding processes on convenient laboratory time scales. Such conditions include high temperature or pressure, chaotrope-mediated unfolding, or low or high pH. These approaches have the limitation of being nonphysiological and that the concentration of the protein in solution is changing as the reactions proceed. We describe a methodology to define the quaternary structure stability of the amyloidogenic homotetrameric protein transthyretin (TTR) under physiological conditions. This methodology expands from a described approach based on the measurement of the rate of subunit exchange of TTR with a tandem flag-tagged (FT2) TTR counterpart. We demonstrate that subunit exchange of TTR with FT2·TTR can be analyzed and quantified using a semi-native polyacrylamide gel electrophoresis technique. In addition, we biophysically characterized two FT2·TTR variants derived from wild-type and the amyloidogenic variant Val122Ile TTR, both of which are associated with cardiac amyloid deposition late in life. The FT2·TTR variants have similar amyloidogenic potential and similar thermodynamic and kinetic stabilities compared to those of their nontagged counterparts. We utilized the methodology to study the potential of the small molecule SOM0226, a repurposed drug under clinical development for the prevention and treatment of the TTR amyloidoses, to stabilize TTR. The results enabled us to characterize the binding energetics of SOM0226 to TTR. The described technique is well-suited to study the quaternary structure of other human aggregation-prone proteins under physiological conditions.
The amyloidoses are protein aggregation disorders characterized by the extracellular deposition of amorphous aggregates and amyloid fibrils in tissues, resulting in organ dysfunction and death.1,2 Transthyretin (TTR) is one of the nearly 30 human proteins known to aggregate in vivo. TTR is produced mainly in the liver and in the choroid plexus of the brain, and it circulates in the blood and the cerebrospinal fluid (CSF).2,3 The known functions of TTR are the transport of thyroxine (T4) in the blood and CSF and the transport of retinol through retinol-binding protein in blood only. It appears that the two T4 binding pockets of TTR can accommodate a large variety of small molecules, including peptides such as amyloid β peptide,4 suggesting that TTR’s function might also be that of a general detoxifier of circulating metabolites.3
Structurally, TTR is a homotetramer of 127 amino acid residue subunits with a molecular weight of ∼55 kDa. TTR aggregation results in a variety of morphologies including small oligomers, amorphous aggregates, protofibrils, and amyloid fibrils. Biophysical studies have shown that the mechanism of TTR aggregation requires the rate-limiting step of native tetramer disassembly into its corresponding monomer subunits. The released monomers misfold and aggregate in a downhill polymerization process.5 It is not yet clear whether the aggregation process is vectorial or whether diverging pathways are involved in the production of the several morphologies associated with the TTR deposits.
Wild-type (WT) and many mutant TTR variants are susceptible to aggregation and deposition, producing different clinical syndromes: In senile systemic amyloidosis (SSA), characterized by WT TTR deposition, and familial amyloid cardiomyopathy (FAC), characterized by mutant TTR deposition, the heart is the main affected organ, although other deposition sites have also been described.6,7 The most common TTR point mutation worldwide, Val122Ile (V122I) TTR, which is present in 4% of African Americans, produces late-onset FAC.8 The syndrome known as familial amyloid polyneuropathy (FAP) is characterized by mutant TTR deposition in peripheral nerve, heart, and other tissues.1
Until recently, the only means of therapy available for patients with TTR amyloidoses related to deposition of mutant TTR variants (FAP and FAC) was liver transplantation.9 Currently, two pharmacologic agents, tafamidis and the nonsteroidal anti-inflammatory drug diflunisal, are undergoing clinical trials in the U.S. for FAP therapy. Tafamidis is already approved in Europe and Japan for the treatment of early-stage FAP.10,11 These small molecules bind in the TTR T4 binding pocket and kinetically stabilize the tetramer, preventing its dissociation into monomers and thus aggregation and amyloid fibril formation.12,13
One of the limitations of the biophysical techniques employed to determine protein quaternary structure stability is the need to use conditions that are not physiological. For example, TTR aggregation capacity is often determined under mild acidic conditions (pH 4–4.5) or in methanol solutions to accelerate the aggregation process on a convenient laboratory time scale.14 Thermodynamic and kinetic stabilities of TTR have typically been measured with the use of chaotropes such as guanidinium chloride (GndCl), guanidinium thiocyanate (GndSCN), or urea. GndCl and GndSCN are very strong denaturants that do not require the disassembly of the native tetrameric TTR into its corresponding monomers before unfolding takes place. In both aggregation and denaturation studies, the conditions are not physiological, and the concentration of the substrate (tetrameric TTR) decreases as the reactions proceed (by aggregation or unfolding). Thus, determining biophysical constants in a continuously changing system is problematic. To overcome this limitation, the use of a tandem flag-tagged TTR variant (FT2·TTR) to study the stability of tetrameric TTR under physiological conditions was proposed.15 This protein has a short peptide (DYKDDDDKDYKDDDDK) appended to its N-terminus that significantly increases the total negative electric charge of TTR and adds 2024 units of molecular weight to each polypeptide chain. The TTR of interest is incubated with FT2·WT TTR under physiological conditions, and the rate of subunit exchange between the two proteins can be measured by ion exchange chromatography. Follow up studies demonstrated that the rate of subunit exchange is limited by the rate of tetramer dissociation;16 thus, the faster a TTR tetramer exchanges subunits with FT2·WT TTR, the less stable it is. In these reports, however, the TTR mixtures were analyzed by high-resolution ion exchange chromatography to detect the five different TTR tetramers that are generated upon subunit exchange, containing 0, 1, 2, 3, and 4 FT2-subunits. Most laboratories do not have such costly analytical instruments, hindering the broad applicability of this technique. Moreover, the biophysical characterization of the FT2·WT TTR protein is limited because GndSCN, a strong denaturant that does not require tetramer dissociation to unfold the protein, was used to study its thermodynamic stability.15 No data with respect to FT2·TTR kinetic stability was reported, an important parameter that significantly affects TTR amyloidogenicity.17
To address these issues, we focused on two TTR variants, WT and V122I TTR related to the senile forms of the TTR amyloidoses, SSA and FAC, and their FT2 counterparts (FT2·WT TTR and FT2·V122I TTR). We demonstrate that FT2·WT TTR and FT2·V122I TTR have an aggregation propensity similar to that of their non-FT2 counterparts. We have established that the FT2·TTR variants have thermodynamic stability similar (although slightly lower) to that of their non-FT2 counterparts in urea. On the other hand, while WT and FT2·WT kinetic stabilities appear to be similar, FT2·V122I TTR is kinetically less stable than its untagged counterpart.
Importantly, we have established a semi-native polyacrylamide gel electrophoresis (PAGE) system to analyze and quantify the rate and extent of subunit exchange, a method that can be utilized in most laboratories without a substantial investment in equipment. We demonstrate that, under semi-native PAGE conditions, TTR samples run as a tetramer and that WT and FT2·WT TTR can be clearly separated. We show that subunit exchange occurs faster at 4 °C than at 25 or 37 °C, consistent with the notion that tetramer stability is governed by hydrophobic interactions. We also report that the kinetically less stable V122I TTR exchanges subunits faster than WT TTR. Finally, we applied this methodology to examine the ability of SOM0226, a small molecule that binds in the T4 pocket of TTR and kinetically stabilizes the protein against dissociation, to stabilize TTR under physiological conditions. SOM0226 is a repurposed compound currently in Phase II proof-of-concept study in humans to treat any form of TTR amyloidosis.18 Our data show that SOM0226 is stronger than tafamidis at stabilizing WT and V122I TTR quaternary structure in vitro and that the developed methodology is robust enough to partially characterize the binding energetics of the small molecule to TTR.
Experimental Procedures
Recombinant Protein Preparation
All of the recombinant TTR variants were prepared in an Escherichia coli expression system and purified by chromatography as described elsewhere.15,19 The final purification step consisted of gel filtration chromatography on a Superdex 75 column (GE Healthcare). The mobile phase was 10 mM sodium phosphate buffer (pH 7.6), 100 mM KCl, and 1 mM EDTA (GF buffer). Only the fractions corresponding to tetrameric protein were pooled. For the F87M/L110M TTR, which is monomeric by design,20 only the fractions corresponding to the size of the monomer were collected. The identity of the proteins was confirmed by liquid chromatography–mass spectrometry (LC–MS). The molecular masses of the TTR variants were as follows: WT TTR, 13 892 (theoretical 13 893); V122I TTR, 13 906 (theoretical 13 907); FT2·WT TTR, 15 882 (theoretical 15 883); FT2·V122I TTR, 15 896 (theoretical 15 897); and F87M/L110M TTR, 13 895 (theoretical, 13 896). The proteins were stored in working size aliquots at −80 °C, at concentrations lower than 45 μM (2.5 mg/mL) to prevent aggregation.
Acid-Mediated TTR Aggregation and Fibril Formation
Acid-mediated aggregation and fibril formation experiments were carried out as described elsewhere.21,22 Briefly, TTR variants in GF buffer (8 μM) were diluted 1:1 with acetate buffer (200 mM sodium acetate, pH 4.2, 100 mM KCl, 1 mM EDTA) to achieve a final pH of 4.4. The TTR solutions were incubated without agitation at 37 °C for up to 7 days. Blank samples consisted of a mixture of equal volumes of GF and acetate buffers. To minimize sample manipulation, the reactions that were used to measure aggregation by turbidity and thioflavin T fluorescence (see below) were incubated in cluster tubes (Genesee Scientific, San Diego, CA), whereas the reactions that were used to measure amounts of soluble and insoluble TTR were incubated in Eppendorf microcentrifuge tubes. In the experiments designed to quantify the capacity of the small molecule SOM0226 to inhibit TTR aggregation, 1000 μL of WT TTR or V122I TTR (8 μM, in GF buffer) was added to 1.6 μL of SOM0226 (10 mM or 5 mM dissolved in DMSO) to achieve TTR/SOM0226 ratios of 1:2 and 1:1. TTR solutions in the presence of DMSO only (vehicle) were prepared in parallel. The samples were then briefly vortexed and incubated at room temperature for 30 min to allow SOM0226 binding to TTR. Acid-mediated aggregation and fibril formation protocol were performed as detailed above. Blank samples consisted of GF/DMSO (1000:1.6) mixed with an equal volume of acetate buffer.
TTR Aggregation Measured by Turbidity
At the designated time points, the acid-mediated TTR aggregation reactions (above) were vortexed for 10 s and transferred into 1/2 area 96-well UV-transparent plates (Corning) in triplicate (50 μL/well). The turbidities of the solutions at 330 and 400 nm were recorded using a UV spectrophotometer (SpectramaxPlus, Molecular Devices). The average optical density of the blanks was subtracted from each experimental sample. The experiments were repeated at least twice in triplicate. The data presented correspond to the average values from one experiment; error bars represent standard deviations.
Measurement of Amounts of Soluble and Insoluble TTR
Four hundred microliters of aggregated TTR solutions incubated in Eppendorf tubes was directly centrifuged at 20 000g for 30 min at 4 °C. The supernatants were carefully separated from the pellets. The protein concentration in the supernatants was measured using 1/2 area 96-well UV-transparent plates in triplicate (50 μL/well). The amount of aggregated (insoluble) TTR was determined by adding 200 μL of 8 M GndCl to the protein pellets. The samples were then vortexed briefly and left at room temperature for 5 min to allow for the disassembly of the TTR aggregates. The total TTR in the solutions was then measured by UV spectrophotometry in 1/2 area 96-well UV-transparent plates in triplicate using an 8 M GndCl solution as blank. The percentages of total soluble and insoluble TTR were then calculated with respect to the total initial protein content (soluble) at time zero. The experiment was repeated twice. The data shown correspond to the average values from one experiment; the error bars correspond to standard deviations.
Thioflavin T (ThT) Binding Fluorescence
The acid-mediated TTR aggregation solutions described above were diluted 1:4 to a final TTR concentration of 1 μM (TTR tetramer equivalents) in 200 mM Tris pH 8.0, buffer with 150 mM NaCl. Two microliters of a ThT stock solution (2 mM in 200 mM Tris pH 8.0, 150 mM NaCl) was added to 400 μL of the diluted TTR aggregates. The samples were vortexed briefly and dispensed into black-wall, clear-bottom 96-well plates (Corning) in triplicate (100 μL/well). ThT fluorescence was recorded in a multiwell spectrofluorimeter (Tecan Safire 2, Austria) with excitation and emission wavelengths at 440 and 482 nm, respectively, and 5 nm bandwidths. The experiment was repeated twice. The data shown correspond to the average values from one experiment; the error bars are standard deviations.
Transmission Electron Microscopy (TEM)
Carbon-coated copper grids (400 mesh, Electron Microscopy Sciences, Hatfield, HA) were glow-discharged and inverted on 5 μL TTR samples subjected to acid-mediated aggregation (prepared as above) for 2 min. Excess sample was removed. The grids were then placed on a droplet of 0.1% ammonium acetate followed by a 2% uranyl acetate solution for 2 min. Excess stain was removed, and the grids were allowed to air dry. Grids were then examined on a Philips CM100 electron microscope (FEI, Hillsbrough, OR) at 80 kV, and images were collected using a Megaview III CCD camera (Olympus Soft Imaging Solutions, Lakewood, CO).
TTR Denaturation and Renaturation Curves
TTR denaturation curves were prepared by incubating TTR at 2 μM (∼0.1 mg/mL) with 0–8 M urea solutions in sodium phosphate buffer (50 mM sodium phosphate, 100 mM KCl, 1 mM EDTA, pH 7.6) for 4 days at room temperature. For the renaturation curves, TTR was first unfolded in 6.5 M GndCl at room temperature overnight. The next day, the proteins were buffer-exchanged into 9 M urea (in 50 mM sodium phosphate, 100 mM KCl, 1 mM EDTA, pH 7.6) and concentrated to 20 μM (∼1 mg/mL), conditions under which TTR remained completely unfolded. Dilutions were then prepared to yield a final TTR concentration of 2 μM over a wide range of urea concentrations (1–8 M). The renaturation solutions were incubated for 24 h at room temperature before measurement of the tertiary and quaternary structures.
Determination of the TTR Tertiary Structure by Tryptophan Fluorescence
The stability of the TTR tertiary structure was determined by measuring the intrinsic tryptophan fluorescence in the presence of urea over a concentration range of 0–8 M, as described previously.21 Briefly, the samples were vortexed and transferred into 1/2 area black 96-well plates (Corning) in triplicate (50 μL/well), and the tryptophan fluorescence was recorded with an excitation wavelength of 295 nm and emission wavelengths of 335 and 355 nm, with 10 nm bandwidth. The percentage of folded protein at each urea concentration was calculated from the fluorescence ratios (355/335 nm) with respect to TTR samples without urea (100% folded) or in 9 M urea (0% folded). The data were fit by sigmoidal curves with variable slope using GraphPad Prism (GraphPad, San Diego, CA), and the concentration of urea at which 50% of the TTR was folded (Cm) was calculated. For the determination of the dissociation constants (Kdiss), TTR samples (2 μM) in 4.5–6 M urea solutions in 0.5 M intervals (concentrations corresponding to the TTR unfolding post-transition zone) were dispensed into 1/2 area 96-well black plates in triplicate. The tryptophan fluorescence of the samples was recorded immediately and at several time-points over a 96 h period. Between readings, the plates were sealed and kept in an airtight moisturized plastic box to avoid sample evaporation, at room temperature. The experimental samples also included native TTR variants (0 M urea, 100% folded) and 100% unfolded TTR to serve as references. To obtain completely unfolded TTR, the proteins were first incubated in 6.5 M GndCl for 24 h and then buffer exchanged in 9 M urea as described previously.21 The raw data were plotted in GraphPad Prism, and they were fit to monoexponential curves to determine the dissociation rate constants (Kdiss). The natural logarithm of the obtained Kdiss at each urea concentration from two independent experiments was plotted against urea concentration, and a linear regression was used to obtain the Kdiss at 0 M urea by linear extrapolation (Table 2).
Table 2. Kinetic Stability of FT2·TTR Variants with Respect to That of Their Non-Flag-Tagged Counterparts Extrapolated to 0 M Urea.
| Kdissa (h–1) | mkina (M–1) | t50 range (h) | |
|---|---|---|---|
| WT | 0.0274 ± 0.00146 | 0.0875 ± 0.0360 | 30.1–44.1 |
| FT2·WT | 0.0253 ± 0.00178 | 0.0837 ± 0.0486 | 30.5–51.2 |
| V122I | 0.0447 ± 0.00195 | 0.1099 ± 0.0255 | 19.5–25.6 |
| FT2·V122I | 0.0627 ± 0.00155 | 0.07905 ± 0.01289 | 14.9–17.1 |
Kdiss is the dissociation rate constant for the tetramer; mkin is the dependence of Kdiss on urea concentration; t50 is the half time for dissociation. Data extrapolated from dissociation curves generated at several urea concentrations from two independent experiments (Figure 4).
Determination of the TTR Quaternary Structure by Resveratrol Binding Fluorescence
The TTR quaternary structure was determined using the small molecule resveratrol as described elsewhere.21,22 Resveratrol binds in the T4 binding pocket of tetrameric TTR, resulting in increased fluorescence quantum yield in a concentration-dependent manner.22 To determine the content of tetrameric TTR in the denaturation and renaturation solutions (above), 4 μL of 1 mM resveratrol stock (in DMSO) was added to 200 μL of each TTR sample and incubated for 30 min at room temperature. Aliquots of 50 μL were then transferred into 1/2 area black 96-well plates in triplicate, and the resveratrol fluorescence was recorded (ex/em 320/394 nm, 10 nm bandwidth). To calculate the percentage of tetrameric TTR at each urea concentration, the average of the resveratrol fluorescence recorded at low urea concentrations (1–2 M) and at high urea concentration (9 M) was regarded as 100% tetramer and 0% tetramer, respectively. The data were fit by sigmoidal curves with variable slopes using GraphPad Prism, and the concentration of urea at which 50% of TTR is tetrameric (Cm) was calculated.
TTR Subunit Exchange Reactions
The TTR proteins were dialyzed in Tris buffer (25 mM Tris-HCl, pH 7.4) and diluted to 8 μM. Equal volumes of non-flag-tagged TTR (WT or V122I) and flag-tagged TTR (FT2·WT or FT2·V122I) were mixed, and the reactions were incubated at 4, 25, and 37 °C for several periods of time (1–96 h). To determine the TTR stabilization effect of SOM0226 and tafamidis, the small molecules (dissolved in DMSO) were first preincubated for 30 min at room temperature with the desired TTR variants (8 μM) at a TTR/small molecule ratio of 1:2. For SOM0226 Kd determination, the TTR/small molecule ratio ranged from 1:0.25 to 1:2 mol equiv. Equal volumes of WT and FT2·WT were then mixed and incubated at 37 °C for up to 3 days. Aliquots of 50 μL were taken at multiple time points (0, 3, 6, 12, 24, 48, and 72 h) and stored at −80 °C until analyzed. For the studies in which the less stable V122I TTR variant was used, we noticed that upon sample storage at −80 °C the subunit exchange reactions were still proceeding, resulting in time 0 samples where subunit exchange was already occurring. To overcome this limitation, the experiments were performed in a reverse manner with the longer incubation time point reactions starting first. Thus, all of the samples were analyzed by semi-native PAGE without the need for storage at −80 °C.
Semi-Native PAGE
Subunit exchanged products were analyzed by semi-native PAGE as follows: we used in-house-cast 11% acrylamide/bis(acrylamide) (30:0.8 w/w), 0.1% SDS mini gels prepared in 395 mM TrisHCl buffer, pH 8.8, with a stacking layer containing 4% acrylamide/bis(acrylamide), 0.1% SDS in 375 mM TrisHCl buffer, pH 6.8. For sample analysis, 4 μL of 6× SDS loading buffer (350 mM TrisHCl, pH 6.8, 30% glycerol, 10% SDS, 0.6 M dithiothreitol, 0.012% bromophenol blue) was mixed with 20 μL of the subunit exchange reactions. Ten microliters was then directly loaded onto the gels in duplicate without boiling. The gels were run at a constant 40 V until the blue dye reached the bottom, and then they were stained with SYPRO Ruby following the manufacturer’s instructions (SYPRO Ruby Protein Gel Stain, Lonza). Briefly, the gels were fixed in 10% MeOH/7% AcOH (v/v) solution for 20 min at room temperature, and then SYPRO Ruby staining solution was added and incubated overnight. The gels were destained with 10% MeOH/7% AcOH solution for 1–3 h, and the protein bands were visualized and captured using a Gel Doc system (Bio-Rad). Particular attention was taken to make sure that the bands in the gels were not saturated. The band intensities were quantified using ImageJ software (NIH).
Quantification of Subunit Exchange
To quantitate the disappearance of the homotetramers with or without FT2, the band intensities at each time point were normalized to those at time 0. To quantitate the appearance of the heterotetramer composed of two FT2 subunits and two nontagged subunits, we measured the density of the middle band (2 FT2 subunits, band 3) of each sample and normalized it by the total density of the lane (total protein) to obtain the percent of band 3 or extent of exchange. For Kd determination of SOM0226 binding to WT TTR, we calculated the extent of exchange at each experimental time point. At saturation, the predicted stoichiometries for the five tetramers (containing 0, 1, 2, 3, or 4 FT2 subunits) are 1:4:6:4:1; thus, band 3 (2 FT2 subunits) represents 6/16 or 0.375 of the total band density. The fraction of exchange was calculated as (extent of exchange)/0.375. The reported rate constants of exchange were determined from plotting the fraction of exchange as a function of time and fitting the data by a first-order single-exponential kinetic equation, as implemented in the Mathematica software. The Kd values were then determined from the dependence of the exchange rate constants on the concentration of ligand as described previously.23,24
Glutaraldehyde Cross-Linking
The untagged TTR variants (WT TTR, V122I TTR) and tagged TTR (FT2·WT, FT2·V122I) were cross-linked with glutaraldehyde as previously described.19 Briefly, 40 μL of the TTR variants at 4 μM (in GF buffer) was treated with 4 μL of glutaraldehyde (25% solution, Sigma) and incubated at room temperature for 5 min. The reactions were quenched by addition of 4 μL of 7% NaBH4 (freshly prepared in 0.1 M NaOH).
In-Gel Staining of Tetrameric TTR by Resveratrol
We used resveratrol as a developing agent to detect tetrameric TTR on semi-native 15% PAGE. Resveratrol binds to tetrameric TTR, and the binding results in a sizable increase in fluorescence. Resveratrol does not bind or fluoresce with monomeric TTR. To stain the gels, we prepared 100 μM resveratrol solution in water from a 1 mM stock (in DMSO). The gels were first washed with Milli-Q water for 5 min, 3 times, and then incubated with 100 μM resveratrol solution for 30 min in the dark followed by another wash with Milli-Q water for 5 min. Resveratrol fluorescence was visualized and captured by a Gel Doc system (Bio-Rad) under UV light. The gels were then stained with Coomassie blue under standard conditions to visualize all of the proteins in the gel.
Results
Aggregation and Fibril Formation Propensity and Morphology of FT2·TTR Variants Are Similar to Those of Untagged TTR
TTR aggregation and amyloidogenesis can be induced in vitro by incubation under mildly acidic conditions.14,21 FT2·WT and FT2·V122I TTR together with their nontagged counterparts (WT and V122I) were incubated for 1, 3, and 7 days at 37 °C and pH 4.4 (the pH of maximum aggregation for both WT and V122I TTR). TTR aggregation was measured by quantification of total protein precipitated, turbidity at 330 nm, and by thioflavin T (ThT) binding fluorescence. The data show that FT2·TTR variants have similar, albeit slightly higher (5–10%), aggregation propensity as that of their nontagged counterparts when measured by total protein precipitated (Figure 1a). Measurement of aggregation by turbidity (Figure 1b) appears to result in an increase of 5–42% in the FT2-proteins compared to their nontagged counterparts. The differences are particularly pronounced for FT2·WT TTR with respect to WT (24–42% increase in turbidity). However, we have previously shown that turbidity measurements give an estimate of aggregation, but, due to the different morphologies of the aggregates, the values can substantially differ between different TTR variants, even though the total aggregated protein is the same.21 ThT fluorescence is an indicator that amyloid species are formed in the aggregation reactions.25 Both FT2·WT and FT2·V122I TTR display ThT fluorescence upon aggregation, suggesting that these proteins, as with their nontagged counterparts, form amyloid-like aggregates (Figure 1c). In all cases, the ThT signal for the FT2·TTR variants is lower than that of the nontagged WT and V122I TTR (Figure 1c). It is known that not all the diverse TTR aggregate morphologies bind ThT with the same efficiency.26 Our data suggest that FT2·WT and FT2·V122I TTR under acidic conditions might result in a distribution of aggregated species that is in a small degree different from that of WT and V122I TTR.
Figure 1.

Relative aggregation of FT2·WT TTR and FT2·V122I TTR variants with respect to their non-flag-tagged counterparts. TTR variants were incubated at pH 4.4 for up to 7 days at 37 °C. The extent of aggregation was measured by (a) percentage of insoluble protein at each time point with respect to initial total (soluble) protein, (b) turbidity at 330 nm, and (c) thioflavin T (ThT) fluorescence. WT TTR: solid squares, solid lines; FT2·WT TTR: solid circles, broken lines; V122I: open squares, solid lines; FT2·V122I: open circles, broken lines.
Transmission electron microscopy (TEM) images were acquired to evaluate the morphologies of the TTR species in the acid-mediated aggregation reactions (Figure 2). WT and V122I TTR samples incubated for 1 day at 37 °C (panels a and c, respectively) were characterized by the presence of elongated material (solid arrows) as well as abundant small spherical aggregates (broken circles). The samples also had thicker elongated deposits (arrow heads) and larger amorphous aggregates (solid circles) that were more abundant in the faster aggregating V122I TTR samples than in the WT TTR samples. For FT2·WT TTR (1 day incubation, panel b), the grids showed similar morphologies to those found in the WT TTR samples with apparently more abundant small amorphous spherical aggregates (broken circles). In contrast, the grids containing FT2·V122I TTR (1 day incubation, panel d) were rich in very thin elongated material (insert in panel d), but no thicker elongated deposits such as those structures shown in panel c (solid arrows and arrow heads) were observed. Samples incubated for 3 days at 37 °C (panels e–g) show more abundant and thicker elongated material (arrowheads) than the samples incubated for 1 day at 37 °C, indicating that the aggregation process was still developing. For the FT2·V122I TTR sample (panel h), we could see only the same structures as those found after 1 day of incubation: small spherical aggregates and very thin filaments (panel d). Only in the V122I TTR samples (panel g) did we find twisted fibrils 200–250 nm long (left) and large amorphous aggregates as those shown in panel g, right. We must note, however, that most of the material from the samples incubated for 3 and 7 days at 37 °C, especially those with V122I TTR variants, was characterized by the presence of large and dense deposits opaque to TEM (not shown).
Figure 2.

Representative transmission electron microscopy (TEM) images of WT (a, e), FT2·WT TTR (b, f), V122I TTR (c, g), and FT2·V122I TTR (d, h) subjected to acid-mediated aggregation conditions for 1 (a–d) and 3 days (e–h) at 37 °C. All images were taken at 92 000× magnification. Scale bars in panels a–f and h represent 200 nm; scale bars in panel g (left and right images), 100 nm. Scale bar in the inset of panel d represents 40 nm.
FT2·TTR Variants Have Similar Thermodynamic Stability Compared to That of Their Nontagged Counterparts
Urea-mediated denaturation/renaturation curves were obtained to determine the tetramer thermodynamic stability of FT2·WT and FT2·V122I TTR with respect to that of WT and V122I TTR. Previous studies have shown that urea cannot directly unfold tetrameric TTR;27 the unfolding process, like the aggregation process, requires rate-limiting tetramer dissociation into its corresponding monomers, which is followed by monomer unfolding. Thus, urea is a suitable chaotrope to estimate tetrameric TTR stability.
The loss of TTR quaternary structure was measured by resveratrol fluorescence,22 which is known to bind to and fluoresce only in the presence of tetrameric TTR and not in the presence of monomeric TTR.21,22 Under the conditions of the assay, resveratrol minimally perturbs the tetramer–monomer equilibrium; thus, it is an appropriate probe to quantify the loss of TTR quaternary structure upon incubation with urea.
The urea-induced TTR tertiary structural changes (i.e., unfolding) were measured by intrinsic tryptophan fluorescence, which allows the quantification of the fraction of folded protein at any urea concentration.21,22,28 Given that in urea the limiting step in the unfolding process is the tetramer disassembly, the rate and extent of unfolding are measures of tetramer stability as well.14
Figure 3 shows the unfolding and refolding curves for the WT and V122I isoforms in urea. The data were fitted by sigmoidal dose–response curves with variable slope. The midpoint of the transitions between folded/unfolded (measured by tryptophan fluorescence) or tetramer/nontetramer (measured by resveratrol binding fluorescence) Cm (in molar units) was calculated and are summarized in Table 1.
Figure 3.

Thermodynamic stability curves of FT2·TTR and TTR variants in urea. WT TTR and FT2·WT TTR (a–d) and V122I TTR and FT2·V122I TTR (e–h) were incubated in the presence of various concentrations of urea. The curves were generated in the denaturation direction (a, c, e, g) starting with folded proteins and in the renaturation direction (b, d, f, h) starting with unfolded proteins. The amount of folded TTR at any given urea concentration was measured by tryptophan fluorescence (a, b, e, f), and the amount of tetrameric TTR at any given urea concentration was measured by resveratrol fluorescence (c, d, g, h). WT TTR: solid squares, solid lines; FT2·WT TTR: solid circles, broken lines; V122I TTR: open squares, solid lines; FT2·V122I TTR: open circles, broken lines. Data shown represents the average of two independent experiments performed in triplicate (mean ± SD).
Table 1. Midpoint Transition Values (Cm) of TTR Denaturation–Renaturation Curves in Urea.
| refolding |
tetramer
reassembly |
unfolding |
tetramer disassembly |
|||||
|---|---|---|---|---|---|---|---|---|
| Cma | pb | Cm | p | Cm | p | Cm | p | |
| WT | 3.5 ± 0.01 | 3.3 ± 0.03 | 3.6 ± 0.04 | 3.2 ± 0.04 | ||||
| FT2·WT | 3.3 ± 0.01 | ** | 3.1 ± 0.03 | * | 3.4 ± 0.03 | * | 3.0 ± 0.05 | ns |
| V122I | 3.4 ± 0.02 | 3.0 ± 0.03 | 3.5 ± 0.03 | 2.9 ± 0.04 | ||||
| FT2·V122I | 3.2 ± 0.02 | ** | 2.6 ± 0.03 | ** | 3.2 ± 0.02 | ** | 2.5 ± 0.03 | ** |
Cm values in molar (M) units. TTR unfolding and refolding were measured by tryptophan fluorescence; tetramer disassembly and reassembly were measured by resveratrol-binding fluorescence.
p values measure the significant changes in Cm for the flag-tagged proteins with respect to their non-flag-tagged counterparts. Unpaired Student’s t test: ns (not significant), p > 0.05, * p ≤ 0.05, ** p ≤ 0.01.
For all proteins, the Cm values for tetramer disassembly and reassembly are slightly lower than the Cm values for unfolding and refolding, consistent with the notion that the tetramer must first dissociate before it can unfold. In urea, tetramer disassembly and unfolding are processes that take days and can vary substantially for different TTR variants.17 The refolding and tetramer reassembly processes are much faster and are mostly complete in a matter of seconds.22 Thus, the approach to thermodynamic equilibrium is more easily reached from the unfolded state in the refolding/reassembly reactions. This fact is reflected in Figure 3, where, in the denaturation direction, folded and tetrameric TTR can still be detected at the time of measurement (96 h incubation time). Longer incubation times in urea can result in urea-mediated protein modification and are not desirable.29 Although the Cm values for the denaturation and renaturation processes are similar, for the sake of clarity, we will focus on the renaturation processes where thermodynamic equilibrium has been reached.
The data show that the FT2·TTR variants have slightly lower Cm values than their nontagged counterparts (Table 1), suggesting that appending FT2 at the N-terminus of the protein decreases, to some degree, its thermodynamic stability. The effect appears to be more pronounced for the V122I variant, for which the tetramer reassembly Cm values for V122I and FT2·V122I TTR are 3.0 and 2.6 M, respectively.
FT2·TTR Variants Have Similar Kinetic Stabilities to Those of Their Nontagged Counterparts
We next studied the kinetic stability of the FT2·TTR variants by measuring the rates of denaturation. In our experience, measuring the rate of unfolding by tryptophan fluorescence gives more robust data than measuring the rate of tetramer dissociation by resveratrol fluorescence. Given that tetramer dissociation is the limiting step of the overall unfolding process, the rate of unfolding can be linked to the rate of tetramer dissociation, as long as the urea concentration is high enough to guarantee complete unfolding. Hammarström et al. validated this notion by determining that monomeric TTR unfolding rate is ∼5 × 105 times faster than that of tetrameric TTR.14 Thus, we measured the rate of unfolding of the FT2·TTR variants side-by-side with their nontagged counterparts. We used urea concentrations that are in the post-transition region for tertiary structural changes (i.e., >4.0 M urea), to be able to link tetramer disassembly and monomer unfolding (Figure 4). The dissociation time courses fit best to a single-exponential curve over the range of urea concentrations described by the equation I355/335= IN355/335 + A (1 – e–Kdiss.t), were IN355/335 is the native protein fluorescence intensity ratio (355/335 nm), A is the amplitude change, Kdiss is the tetramer dissociation rate constant, and t is the time in hours. Figure 4a shows the denaturation kinetic curves at 6 M urea. The natural logarithm of the dissociation constants (Kdiss) change linearly with urea concentration14 (Figure 4b). This observation allows the extrapolation of Kdiss to the more physiologic 0 M urea concentration (Table 2). The data, which represents the average of two independent experiments, show that FT2·WT TTR has the same kinetic stability as that of WT TTR within error, whereas FT2·V122I TTR appears to be kinetically less stable than V122I TTR. The estimated half time for dissociation (1/Kdiss) is in the range of 30–51 h for WT and FT2·WT TTR, 19–25 h for V122I, and 15–17 h for FT2·V122I TTR (Table 2).
Figure 4.

Kinetics of TTR dissociation measured by unfolding rates. (a) Unfolding kinetics at 6 M urea measured by tryptophan fluorescence; data fitted to one-phase association reactions. (b) Natural logarithm of the rate of tetramer dissociation (ln Kdiss, h–1) plotted as a function of urea concentration. The plots are linear, allowing the extrapolation of Kdiss to 0 M urea (Table 2). The data correspond to the average of two independent experiments. WT TTR: solid squares, solid lines; FT2·WT TTR: solid circles, broken lines; V122I TTR: open squares, solid lines; FT2·V122I TTR: open circles, broken lines.
The fact that FT2-labeled proteins are somewhat less stable than their nontagged counterparts implies that there will always be FT2·TTR subunits available to exchange with a nontagged protein. The FT2-labeled proteins will not be limiting the rate and extent at which the reactions take place, thus affording bona fide measurement of the stability of the TTR variant under study.
TTR Quaternary Structure Is Preserved under Semi-Native PAGE Conditions
WT TTR and FT2·WT TTR in Laemmli sample buffer with or without a boiling step were analyzed in an 11% PAGE system. In order to determine the size of the TTR species under these analytical conditions, we also cross-linked one set of TTR samples with glutaraldehyde. The cross-linked samples, with or without boiling, were analyzed by PAGE in parallel lanes. Glutaraldehyde cross-linking followed by PAGE is a well-established method to determine protein quaternary structure.23,30 After electrophoretic separation, the gels were developed by Sypro Ruby staining (Figure 5a). Boiled native TTR runs near the 15 kDa molecular weight maker, consistent with the size of the monomer; boiled cross-linked TTR runs mainly as a wide band near the 55–75 kDa markers, consistent with the size of a tetramer. Bands of lower intensity with the size of dimer and monomer can also be observed and probably reflect the fact that glutaraldehyde cross-linking is not 100% efficient. When the boiling step was omitted, TTR appeared near the 35 kDa marker for WT TTR and near the 40 kDa marker for FT2·WT TTR whether they were cross-linked or not, suggesting that the cross-linked and non-cross-linked TTR species were of the same size. Glutaraldehyde cross-linked TTR coelutes with native TTR in a calibrated gel filtration Superdex 75 column (Figure 5b), indicating that cross-linking preserves the TTR tetramer (and does not generate larger species).
Figure 5.

Analyses of TTR species by (a) PAGE and (b) size-exclusion chromatography. (a) WT TTR and FT2·WT TTR were analyzed on an 11% PAGE gel under several conditions, including with or without denaturing (boiling) before electrophoresis and with or without quaternary structure fixation by cross-linking with glutaraldehyde before analysis. Denatured (boiled) TTR runs near the 15 kDa marker (monomer size), cross-linked and boiled TTR runs near 55–75KDa (tetramer size), and nondenatured (nonboiled) TTR (cross-linked or not) runs near the 30–40 kDa marker. (b) Size-exclusion chromatography on a calibrated Superdex 75 analytical column demonstrates that both WT TTR (solid lines) and cross-linked WT TTR (dotted lines) have elution volumes consistent with the size of a tetramer and no higher molecular weight species are generated upon cross-linking.
Under standard PAGE conditions, the electrophoretic mobility of the proteins depends primarily on their molecular mass, whereas under our semi-native PAGE conditions (unboiled samples), the mobility of the proteins depends also on their shape and charge. This principle may explain why the unboiled TTR samples (cross-linked or not) run much faster through the gel than what is predicted from their molecular weight (observed 35 kDa vs predicted 56 kDa). In these samples, TTR is still folded and thus more compact compared to that in the boiled TTR samples, which are denatured. The compact proteins move more freely through the acrylamide mesh than the unfolded proteins, resulting in bands with apparent lower molecular weight. Overall, the data demonstrate that nonboiled native TTR runs as a tetramer in SDS-PAGE. We named these conditions semi-native PAGE.
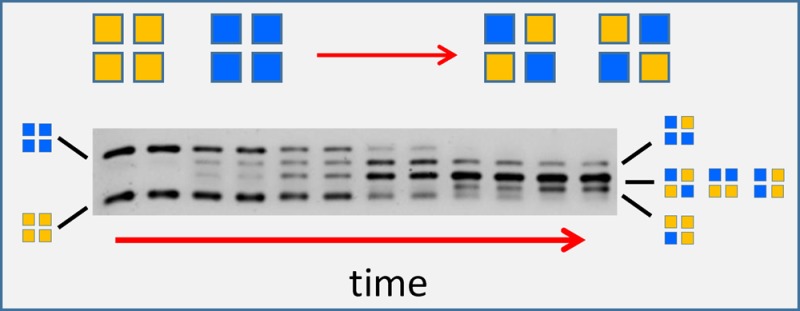
To further validate the notion that under semi-native PAGE conditions TTR runs as a tetramer, we used the small molecule resveratrol that, by virtue of binding in the T4 binding pocket present only in the native tetramer, becomes fluorescent. As mentioned above, resveratrol does not bind to or fluoresce with monomeric TTR.22 The TTR samples analyzed by semi-native PAGE were as follows: WT TTR, FT2·WT TTR, and an equimolar mixture of WT and FT2·WT TTR that had been first unfolded in 6.5 M GndCl and then refolded to allow the formation of mixed heterotetramers. A well-characterized TTR variant that is monomeric was also analyzed as a control.20 After electrophoretic separation, the gels were first stained with a 100 μM solution of resveratrol (Figure 6, left panel) as detailed in the Experimental Procedures and then with Coomassie blue to reveal all of the proteins in the gel (Figure 6, right panel). Figure 6 (resveratrol staining) shows that the TTR variants run as tetramer under these conditions because they are fluorescent; in contrast, the engineered monomeric TTR variant (M-TTR) does not show resveratrol fluorescence. Control gels in which the resveratrol solution was substituted by DMSO (vehicle) display no fluorescence (not shown). We also measured resveratrol fluorescence in the presence of WT TTR, S112I TTR (a recombinant dimeric TTR variant),31 and the monomeric M-TTR at concentrations ranging from 0.01 to 0.1 mg/mL. Only resveratrol in the presence of WT tetrameric TTR displays fluorescence (not shown). These results support the notion that in semi-native PAGE TTR runs as a tetramer. Furthermore, it is clear from the gels shown in Figures 5 and 6 that FT2·TTR variants have much lower mobility than that of their nontagged TTR counterparts. Samples consisting of unfolded/refolded equimolar mixtures of WT TTR with FT2·TTR contain at least 3 resolved bands, in agreement with the presence of TTR heterotetramers. Staining with the more sensitive Sypro Ruby dye clearly shows that there are 5 bands forming upon subunit exchange (Figure 7), consistent with the formation of heterotetramers containing 0, 1, 2, 3, and 4 FT2-TTR subunits.
Figure 6.

TTR species analyzed by semi-native PAGE (nonboiled samples) and visualized consecutively by resveratrol fluorescence staining (left panel) and Coomassie staining (right panel). The samples analyzed are WT TTR, FT2·WT TTR, an equimolar mixture of WT TTR and FT2·WT TTR that had previously been unfolded and refolded to allow the formation of mixed heterotetramers (labeled WT/FT2·WT), and a well characterized engineered monomeric TTR variant, M-TTR.20 When resveratrol is bound to tetrameric TTR species, it is fluorescent, demonstrating that semi-native PAGE conserves TTR tetrameric quaternary structure.
Figure 7.

Subunit exchange analysis by semi-native PAGE between WT TTR and FT2·WT TTR incubated at 4, 25, and 37 °C for different time periods (0–72 h). (a) Gels stained with Sypro Ruby (arrowhead points to band 3). (b) Quantitation of the upper bands in each gel corresponding to FT2·WT TTR. (c) Quantitation of lower bands in each gel corresponding to WT TTR. (d) Quantitation of the TTR heterotetramer composed of two FT2·WT subunits and two WT TTR subunits (band 3). Data representing quantitation of the homotetramers (b, c) were normalized to band intensities at time 0; the data were fitted by one-phase decay curves. Data representing quantitation of band 3 (d) were not normalized; the data were fitted by one-phase association curves. Symbols: circles, 4 °C; squares, 25 °C; triangles, 37 °C.
All of our FT2·WT and FT2·V122I TTR preparations have a small impurity whose mobility is consistent with that of a homotetramer composed of 3 FT2-subunits and one untagged subunit. We attribute it to a proteolysis event during synthesis in E. coli. The presence of this same impurity was also noticed in a recent paper describing the use of TTR subunit exchange coupled to ultraperformance liquid chromatography,32 where it was demonstrated that it does not modify the kinetics of the subunit exchange reactions.
Semi-native PAGE Can Be Used To Determine the Rate of TTR Subunit Exchange with FT2·TTR under Physiological Conditions
Equal amounts of FT2·WT and WT TTR were mixed and incubated over time at 4, 25, and 37 °C in buffer at pH 7.4. The samples were then analyzed in duplicate by semi-native PAGE, and the gels were stained with Sypro Ruby (Figure 7). We quantified the disappearance of the bands corresponding to FT2·WT homotetramer (top band, panel b) and WT homotetramer (lower band, panel c) and the appearance of the most abundant heterotetramer (middle band) consisting of 2 FT2·WT subunits and 2 WT subunits (band 3, panel d). The data were fitted by decreasing (for FT2·WT and WT TTR disappearance) or increasing (for heterotetramer formation, band 3) monoexponential equations; the r2 values for the fits were >0.9. The subunit exchange is faster at 4 °C than at 25 or 37 °C. This observation is consistent with previous studies in which similar TTR mixtures were analyzed by high-resolution ion exchange chromatography15 and is also in agreement with the notion that there is a significant contribution from the hydrophobic effect on TTR tetramer stability.14 We attribute the faster disappearance of FT2·WT homotetramer compared to that of the WT homotetramer at all temperatures to the fact that the former is less stable than the latter (Figure 3 and Table 1).
We also compared the subunit exchange rates between WT TTR and FT2·WT TTR and the kinetically less stable V122I and FT2·V122I TTR. As expected, V122I/FT2·V122I TTR exchanged subunits much faster than WT/FT2·WT TTR (Supporting Information Figure S1). The half time for the disappearance of the untagged tetramers was 10.2 ± 3.5 and 2.3 ± 0.4 h for WT and V122I TTR, respectively (Table 3). In agreement with this kinetic data, the half time for the formation of the mixed heterotetramers (band 3, Supporting Information Figure S1) was 12.5 ± 4.7 and 2.8 ± 0.6 h for the WT/FT2·WT and V122I/FT2·V122I heterotetramers, respectively (Table 3). In further analyses, we focused on the appearance of band 3 (heterotetramer composed of 2 FT2 subunits and 2 untagged subunits) because, in general, it gave us less variability than the quantification of the disappearance of the homotetramers. This observation is probably due to the fact that it is difficult to quantitate with precision very faint bands like those of the homotetramers when the subunit exchange reactions have reached completion.
Table 3. Subunit Exchange Kinetic Analysis between WT and FT2·WT TTR and between V122I and FT2·V122I TTRa.
| FT2-homotetramer disappearanceb | nontagged TTR disappearanceb | 2 FT2/2 TTR (heterotetramer) formationb | |
|---|---|---|---|
| WT/FT2·WT TTR | 6.49 ± 0.42 | 10.21 ± 3.46 | 12.54 ± 4.71 |
| V122I/FT2·V122I TTR | 1.92 ± 0.14 | 2.28 ± 0.37 | 2.84 ± 0.62 |
Calculated half-time (t50, h) for the disappearance of the homotetramers FT2 and untagged homotetramers and the appearance of the heterotetramer composed of 2 FT2 subunits and 2 untagged subunits.
Small Molecule-Mediated TTR Kinetic Stabilization Can Be Determined by Semi-Native PAGE
Semi-native PAGE was used to quantify tetrameric TTR stability in the presence of small molecules known to bind in the T4 binding pocket and kinetically stabilize tetrameric TTR against dissociation. We used this methodology to evaluate the potency of SOM0226, a repurposed drug intended to be used as a TTR kinetic stabilizer.18 First, we showed that SOM0226 is able to prevent TTR aggregation in vitro under acid-mediated aggregation and in a fibril formation standard assay (Figure 8). SOM0226 was able to prevent WT TTR and V122I TTR fibril formation by 75 and 66%, respectively, when tested at 1 mol equiv with respect to TTR and by 98% for both WT and V122I TTR when tested at 2 mol equiv. These data demonstrate that SOM0226 is a very potent inhibitor of TTR aggregation and fibril formation in vitro.
Figure 8.

Inhibition of TTR aggregation and fibril formation by SOM0226. WT TTR (a) and V122I TTR (b) at 4 μM were incubated at pH 4.4 for 3 days in the absence (DMSO, vehicle) or presence of SOM0226 (4 and 8 μM). Aggregation was measured by quantification of precipitated protein (insoluble) and normalized to total soluble protein at time 0. Percentage of fibril formation inhibition by SOM0226 is shown on the figure. Statistical significance was calculated by Student’s t test: **** p < 0.0001.
We then evaluated the capacity of SOM0226 to prevent WT and V122I TTR subunit exchange in the semi-native PAGE system under physiological conditions. At the same time, we tested tafamidis, an excellent inhibitor of TTR fibril formation in vitro, which is currently being used to treat TTR amyloidosis. We performed the experiments in two settings: one in which only WT TTR was stabilized with the small molecules before mixing it with nonstabilized FT2·WT TTR (asymmetrical) and the other in which both WT and FT2·WT TTR were preincubated with the small molecules prior to subunit exchange (symmetrical). We quantified the formation of band 3 (the tetramer containing 2 WT subunits and 2 FT2·WT subunits, Figure 9). The same experiments were done for V122I TTR/FT2·V122I TTR pairs (Supporting Information Figure S2).
Figure 9.

Stabilization of WT TTR and FT2·WT TTR by small molecules measured by subunit exchange. WT TTR and FT2·WT TTR were preincubated with SOM0226, tafamidis, or DMSO (vehicle control). Mixtures of WT TTR and FT2·WT TTR with or without small molecules were prepared and incubated for up to 48 h at 37 °C. Subunit exchange rates were measured by quantifying the formation of mixed heterotetramers composed of 2 WT and 2 FT2·WT subunits (band 3, arrowhead). (a, c) Asymmetric mode where only WT TTR was preincubated with small molecules; (b, d) symmetric mode where both WT and FT2·WT TTR were preincubated with small molecules. Symbols: DMSO, diamonds; SOM0226, circles; tafamidis, triangles.
As expected, when both WT and FT2·WT (or V122I and FT2·V122I TTR) are stabilized with the drugs (symmetrical setting, right panel), there is less subunit exchange than when only the non-flag-tagged proteins were stabilized with the small molecule (asymmetrical setting, left panel). These analyses also show that the asymmetrical setting appears to be more sensitive to discern the stabilization potency among small molecules. While in the symmetrical setting the stabilization differences between SOM0226 and tafamidis are distinguishable but small, it is clear in the asymmetrical setting that SOM0226 is a more effective stabilizer than tafamidis.
Subunit exchange and semi-native PAGE were used to determine the binding constants of SOM0226 with WT TTR and to evaluate cooperativity between the two TTR T4 binding sites. WT TTR and FT2·WT TTR were preincubated with SOM0226 at 0, 0.25, 0.5, 1, 1.5, and 2 mol equiv, and subunit exchange was followed for 72 h incubation at 25 °C (Figure 10). We quantified the appearance of band 3 (tetramer composed of 2 FT2 and 2 nontagged subunits), calculated the fraction of exchange as detailed in the Experimental Procedures, and fit the data as in Penchala et al.23 and Bulawa et al.24 The best-fit exchange rate constant is Kex = 0.28 ± 0.04 h–1; the ΔG1 for SOM0226 binding to the first binding site is −10.1 ± 0.4 kcal/mol, which corresponds to a Kd1 = 41 nM, and the ΔG2 for binding to the second binding site is −7.3 ± 1.0 kcal/mol, which corresponds to a Kd2 = 4.3 μM. The data are consistent with a negative cooperative mode of binding of SOM0226 to WT TTR, which has also been described for tafamidis, diflunisal, AG10 (another small molecule that binds and stabilizes TTR)23 and many other ligands.
Figure 10.

Analysis of subunit exchange between WT TTR and FT2·WT TTR in the presence of 0 (DMSO) to 2 equiv of SOM0226 incubated for up to 72 h at 25 °C. (a) Semi-native PAGE images (arrowhead points to band 3). (b) Quantification of the appearance of band 3 with time expressed as fraction exchanged (see Experimental Procedures). Each symbol represents a different stoichiometry of SOM0226 to TTR: solid circles, 0:1; solid squares, 0.25:1; solid triangles, 0.5:1; open squares 1:1; open circles 1.5:1; solid diamonds, 2:1.
Discussion
In the transthyretin amyloidoses the dissociation of the native TTR tetramer is required and is the rate-limiting step in the aggregation cascade.33−35 Kinetic stabilization of native tetrameric TTR is an effective means to prevent amyloidogenesis in vitro and in vivo. TTR stabilization has been achieved in vivo genetically and pharmacologically. Genetically, it has been observed that humans heterozygous for both the amyloidogenic V30M TTR variant and the kinetically stable T119M TTR variant do not develop FAP or manifest very mild symptoms.36 Their secreted TTR consists of heterotetramers with mixed V30M and T119M subunits. Biophysical in vitro studies have clearly demonstrated that the presence of a single T119M subunit in the TTR heterotetramer prevents V30M amyloidogenicity by 50% and that the aggregation propensity of a TTR heterotetramer composed of two T119M subunits and two V30M TTR is completely abrogated.37 These observations underscore the importance of studying quaternary protein structure to understand disease etiology. Moreover, TTR isolated from plasma of V30M/T119M TTR heterozygotes is more stable than that of V30M/WT TTR carriers.38 Pharmacologically, there are two small molecules, tafamidis and the nonsteroidal anti-inflammatory drug diflunisal, that have undergone clinical trials as possible therapeutics to treat FAP. Tafamidis is already approved in Europe and Japan for early-stage FAP treatment. These small molecules bind in the T4 pocket of TTR, selectively decreasing the energetic ground state of the tetramer and increasing thus the energetic barrier for tetramer dissociation.
Most of the studies of TTR aggregation in the absence or presence of small molecules are done under nonphysiological conditions such as acidic buffers or in the presence of methanol.14,34 In other instances, highly amyloidogenic TTR variants, not related to clinical diseases, that can aggregate at physiological pH have been generated to study the process of aggregation.39 Moreover, determination of TTR thermodynamic and kinetic stabilities are typically established using chaotropes such as urea, GndCl, or GndSCN to accelerate protein unfolding rates on convenient laboratory time scales.14,15,21,22 In both fibrillogenesis and denaturation assays, depletion of the substrate is occurring during the course of the experiment because TTR either aggregates or dissociates/unfolds. Furthermore, these nonphysiological conditions might have an impact on how small molecules bind and stabilize the TTR tetramer or on how the monomeric polypeptide chains interact to form the native tetramer. For these reasons, it is difficult to predict the precise stability of TTR under physiological conditions, either by itself or in the presence of small molecules that kinetically stabilize the tetramer.
The use of subunit exchange methodology to study TTR stability circumvents these limitations.15 The concentration of TTR in solution is constant over time because there is no precipitation or denaturation occurring during the course of the experiment. As the TTR of interest exchanges subunits with FT2·TTR, five different tetramers are generated containing 0, 1, 2, 3, and 4 FT2 subunits.15 To a first approximation, the faster a TTR molecule exchanges subunits with FT2·TTR, the less stable it is. The implementation of this methodology, however, has not been widespread, most likely because of the need for high-resolution ion exchange chromatography systems to quantify the subunit exchange reactions, which are not readily available in most laboratories. Follow up applications of the methodology suffer from the same drawback.16,23,32 Coupling subunit exchange reactions to semi-native PAGE analysis overcomes this limitation.
With respect to the biophysical studies, our data show that FT2·TTR variants have a similar (although slightly higher) aggregation propensity to that of their non-flag-tagged counterparts. TEM imaging show the same type of morphologies for WT and FT2·WT TTR, although the relative proportions of each appeared to be different in the grids examined. FT2·V122I TTR differed from V122I TTR in that the elongated material deposited in the grids was very thin. These morphological differences could explain the higher turbidity observed in the FT2·WT samples compared to that in WT TTR and the lower ThT fluorescence signal of the two FT2 proteins compared to that of their native counterparts. However, it is difficult to assign any specific effect to any particular morphology because of the heterogeneous nature of the samples. It is also noteworthy that the morphologies of the TTR aggregates obtained in vitro under acidic conditions appear to be very poor in amyloid fibers. In our samples, only the faster aggregating V122I TTR showed twisted 150–250 nm long structures that might represent the so-called protofibrils (structures 4–11 nm in diameter and ∼200 nm long). It is possible that longer periods of time might be required to obtain TTR amyloid morphologies, but it could also be the case that under acidic conditions the formation of fibrils per se is not favorable. More exhaustive and long-term studies on aggregate morphology of TTR in vitro are consistent with our observations.40 In these studies, amyloid TTR fibrils from ex vivo tissues were also visualized by TEM and shown to be several micrometers long. It is known that in both tissues of patients with TTR amyloidosis and transgenic mice overexpressing human WT TTR, amyloid fibrils and amorphous aggregates are present in the deposits.41,42
Consistent with the aggregation studies, the thermodynamic stability of the FT2·WT TTR is comparable (albeit somewhat lower) to that of WT TTR (Table 1). In previous studies using GndSCN, FT2·WT TTR thermodynamic stability was found to be identical to that of WT TTR.15 The thermodynamic stability of FT2·V122I TTR is also lower than that of V122I TTR (Table 1). The kinetic stabilities of the FT2-labeled proteins at 6 M urea appear to be lower than those of their unlabeled counterparts. However, extrapolation of the kinetic data obtained at several urea concentrations to 0 M urea shows that FT2·WT TTR has the same kinetic stability as that of WT TTR, whereas FT2·V122I TTR is kinetically less stable than V122I TTR (Figure 4 and Table 2). That the FT2·TTR variants are of similar but lower stability to that of their nontagged counterparts is, in fact, a desirable trait to perform subunit exchange experiments since the tagged variants will not be the limiting factor for the reactions to proceed. Our data highlights the importance of using mild chaotropes such as urea, which mimic the TTR amyloidogenic process (i.e., disassembly of the tetramer before aggregation), to perform TTR stability studies.
We show that subjecting TTR to semi-native PAGE conditions (nonboiled samples) keeps the native TTR quaternary structure intact (Figures 5 and 6). A new methodology developed herein for the purpose of defining the TTR quaternary structure is the staining of the gels with resveratrol, a small molecule that is fluorescent when it binds to the T4 binding pocket of tetrameric TTR (Figure 6). We demonstrate that semi-native PAGE can be used as a quantitative tool to define TTR stability under physiological pH values. Our studies show that TTR is less stable at 4 °C than at 25 or 37 °C, at pH 7.4 (Figure 7), which is consistent with previous studies15 and compatible with the notion that TTR stability is governed by hydrophobic interactions, which are weaker at lower temperatures.27 Our data also show that V122I TTR exchanges subunits much faster than does WT TTR (Supporting Information Figure S1), which is in agreement with its reported lower kinetic stability,43 as illustrated in Figure 4 and Table 2. These experiments demonstrate that subunit exchange coupled with semi-native PAGE can be used to assess TTR quaternary structure stability under physiological conditions.
We applied the described methodology to quantify the TTR stabilization effect of small molecules under physiological conditions. For that we chose SOM0226, a repurposed drug candidate for the treatment of FAP,18 and tafamidis. Subunit exchange inhibition between non-flag-tagged TTR and FT2·TTR was more efficient when both TTR variants were stabilized with the small molecules (symmetrical setting) than when only the non-flag-tagged protein was stabilized with the small molecules (asymmetrical setting) before the subunit exchange reactions took place (Figure 9). The data also show that the asymmetrical setting appears to be more sensitive at discriminating the TTR binding potency of small molecules.
TTR has two T4 binding sites located at the dimer–dimer interface. Each TTR tetramer can, in principle, accommodate two small molecules in its T4 sites. To study the affinity and the mode of binding of small molecules to the TTR tetramer, techniques such as isothermal titration calorimetry (ITC), surface plasmon resonance (SPR), change in fluorescence polarization of a fluorescently labeled TTR competing with a small molecule, or subunit exchange reactions coupled to high-resolution ion exchange chromatography systems have been employed.23,24,44 All of these techniques require expensive instrumentation, which limits their broad applicability. We show that subunit exchange reactions coupled with semi-native PAGE can be used to measure such interactions and that the dissociation constants for binding of the small molecules to TTR can be calculated. Our data obtained at 25 °C is consistent with a negative cooperativity mode of binding of SOM0226 to WT TTR, with a dissociation constant for the first binding site Kd1 = 41 nM and a dissociation constant for the second binding site Kd2 = 4.3 μM. The calculated Kd1 for tafamidis using subunit exchange coupled to high-resolution ion exchange chromatography (UPLC) is about 1 order of magnitude below that of SOM0226 (4 nM), whereas the Kd2 value is ∼1 μM.23 The binding of SOM0226 and tafamidis to WT TTR are highly uncooperative. The large difference between Kd1 and Kd2 in practical terms means that these small molecules will mostly be bound to one single T4 site of TTR rather than binding to both sites, resulting in more TTR molecules being stabilized at a given concentration of the ligand. Because only a single small molecule per tetramer is necessary to efficiently stabilize TTR,45 this mode of binding allows lower dosage for treatment of patients with TTR amyloidoses than if a small molecule with cooperative binding was used. SOM0226 appears to be a stronger stabilizer of WT TTR (and V122I TTR) than is tafamidis (Figure 9 and Supporting Information Figure S2), although its Kd1 to WT TTR is higher than that of tafamidis. This effect could be due to an unusually high off rate of SOM0226 for the first T4 binding site. If this is correct, once SOM0226 binds in the TTR T4 pocket it will remain tightly bound to it and it will hardly come off, resulting in a more efficiently stabilized WT TTR tetramer.
There is substantial interest in developing small molecules that can prevent protein aggregation for a variety of protein misfolding disorders by modulating the kinetics of the process.46 We believe that subunit exchange reactions coupled to semi-native PAGE, such as that demonstrated for TTR, can be a simple and versatile methodology to study oligomeric protein stability under physiological conditions as well as for determining the potency of small molecules to stabilize the native state. For that purpose, it will be necessary to generate a tagged protein with similar stability to that of the protein of interest but with different PAGE mobility. Although we used the highly charged FT2 for our studies, we predict that a noncharged tag that simply increases the molecular weight of the complex might be sufficient to obtain the desired separation. PAGE in the absence of SDS might be desirable if the oligomeric protein of interest is not very stable. A protein such as the homodimeric superoxide dismutase 1 (SOD1), whose misfolding and aggregation is related to amyotrophic lateral sclerosis (ALS),47 would be a perfect candidate to adapt this methodology, particularly because SOD1, like TTR, is thermally very stable and will probably maintain its quaternary structure under semi-native PAGE conditions.48 Other protein misfolding scenarios in which heterotypic interactions stabilize the native complex might also be candidate systems to test this approach, provided that appropriate tags that do not modify the interaction and the native stability of the proteins can be developed.
In summary, we present a new methodology to study TTR stability and define the effect of small molecules as TTR kinetic stabilizers as well as their mode of binding under physiological conditions. Our results provide insights into the protein quaternary structure stabilization by small molecules and therefore open new avenues for therapeutic intervention in human amyloidogenic diseases. We suggest its application to the study of other misfolding-prone proteins with quaternary structure as well as heterotypic protein–protein interactions such as those occurring in chaperone–client protein processes like folding, refolding, assembly, or unfolding.
Acknowledgments
We thank Prof. Evan T. Powers for the calculation of Kd values for SOM0226 to WT TTR and for fruitful discussions and Dr. Malcolm Wood (TSRI) for the acquisition of the TEM images. We also thank Dr. Jaime Pascual and Prof. Joel N. Buxbaum (TSRI) for helpful discussions and careful editing of the manuscript. We thank Prof. Jeffery Kelly (TSRI) and the Kelly lab for providing plasmids for FT2·WT TTR and FT2·V122I TTR; Dr. Raul Insa, Dr. Marc Centellas, and Dr. Núria Reig from SOM Biotech (Barcelona, Spain) for providing SOM0226; Dr. Lorena Saelices (UCLA) for providing recombinant S112I TTR; and Jeanine Witkowski for preparing and purifying some of the recombinant protein used for these studies.
Glossary
Abbreviations
- CSF
cerebrospinal fluid
- FAC
familial amyloid cardiomyopathy
- FT2
tandem flag-tagged
- GndCl
guanidinium chloride
- GndSCN
guanidinium thiocyanate
- GF buffer
10 mM sodium phosphate, pH 7.6, 100 mM KCl, 1 mM EDTA
- ITC
isothermal titration calorimetry
- PAGE
polyacrylamide gel electrophoresis
- SPR
surface plasmon resonance
- SSA
senile systemic amyloidosis
- T4
thyroxine
- TEM
transmission electron microscopy
- ThT
thioflavin T
- TTR
transthyretin
- V122I TTR
Val122IIe TTR
- WT TTR
wild-type TTR.
Supporting Information Available
Analysis and quantification of the rate of subunit exchange reactions between WT and FT2·WT TTR and between V122I and FT2·V122I TTR (Figure S1); stabilization of V122I TTR and FT2·V122I TTR by small molecules measured by subunit exchange (Figure S2). This material is available free of charge via the Internet at http://pubs.acs.org.
This research was supported by the National Institutes of Health (NIA, AG032285 to N.R.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Buxbaum J. N. (2004) The systemic amyloidoses. Curr. Opin. Rheumatol. 16, 67–75. [DOI] [PubMed] [Google Scholar]
- Buxbaum J. N. (2008) in Rheumatology (Hochberg M. C., Silman A. J., Smolen J. S., Weinblatt M. E., and Weisman M. H., Eds.) pp 1671–1681, Elsevier, London. [Google Scholar]
- Buxbaum J. N.; Reixach N. (2009) Transthyretin: the servant of many masters. Cell. Mol. Life Sci. 66, 3095–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Masliah E.; Reixach N.; Buxbaum J. N. (2011) Neuronal production of transthyretin in human and murine Alzheimer’s disease: is it protective?. J. Neurosci. 31, 12483–12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurshman A. R.; White J. T.; Powers E. T.; Kelly J. W. (2004) Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 43, 7365–7381. [DOI] [PubMed] [Google Scholar]
- Cornwell G. G. III; Murdoch W. L.; Kyle R. A.; Westermark P.; Pitkanen P. (1983) Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am. J. Med. 75, 618–623. [DOI] [PubMed] [Google Scholar]
- Tanskanen M.; Peuralinna T.; Polvikoski T.; Notkola I. L.; Sulkava R.; Hardy J.; Singleton A.; Kiuru-Enari S.; Paetau A.; Tienari P. J.; Myllykangas L. (2008) Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann. Med. 40, 232–239. [DOI] [PubMed] [Google Scholar]
- Jacobson D. R.; Pastore R. D.; Yaghoubian R.; Kane I.; Gallo G.; Buck F. S.; Buxbaum J. N. (1997) Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N. Engl. J. Med. 336, 466–473. [DOI] [PubMed] [Google Scholar]
- Herlenius G.; Wilczek H. E.; Larsson M.; Ericzon B. G. (2004) Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial Amyloidotic Polyneuropathy World Transplant Registry. Transplantation 77, 64–71. [DOI] [PubMed] [Google Scholar]
- Adams D.; Theaudin M.; Cauquil C.; Algalarrondo V.; Slama M. (2014) FAP neuropathy and emerging treatments. Curr. Neurol. Neurosci. Rep. 14, 435. [DOI] [PubMed] [Google Scholar]
- Sekijima Y. (2014) Recent progress in the understanding and treatment of transthyretin amyloidosis. J. Clin. Pharm. Ther. 39, 225–233. [DOI] [PubMed] [Google Scholar]
- Coelho T.; Maia L. F.; Martins da S. A.; Waddington C. M.; Plante-Bordeneuve V.; Lozeron P.; Suhr O. B.; Campistol J. M.; Conceicao I. M.; Schmidt H. H.; Trigo P.; Kelly J. W.; Labaudiniere R.; Chan J.; Packman J.; Wilson A.; Grogan D. R. (2012) Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 79, 785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk J. L.; Suhr O. B.; Sekijima Y.; Yamashita T.; Heneghan M.; Zeldenrust S. R.; Ando Y.; Ikeda S.; Gorevic P.; Merlini G.; Kelly J. W.; Skinner M.; Bisbee A. B.; Dyck P. J.; Obici L. (2012) The diflunisal trial: study accrual and drug tolerance. Amyloid 19, 37–38. [DOI] [PubMed] [Google Scholar]
- Hammarström P.; Jiang X.; Hurshman A. R.; Powers E. T.; Kelly J. W. (2002) Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc. Natl. Acad. Sci. U.S.A. 99, 16427–16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider F.; Hammarstrom P.; Kelly J. W. (2001) Transthyretin slowly exchanges subunits under physiological conditions: a convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein Sci. 10, 1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman R. L.; Green N. S.; Kelly J. W. (2005) Kinetic stabilization of an oligomeric protein under physiological conditions demonstrated by a lack of subunit exchange: implications for transthyretin amyloidosis. Biochemistry 44, 9265–9274. [DOI] [PubMed] [Google Scholar]
- Sekijima Y.; Wiseman R. L.; Matteson J.; Hammarstrom P.; Miller S. R.; Sawkar A. R.; Balch W. E.; Kelly J. W. (2005) The biological and chemical basis for tissue-selective amyloid disease. Cell 121, 73–85. [DOI] [PubMed] [Google Scholar]
- Centellas M., Planas A., Reig N., Gavalda N., and Insa R. (2013) SOM0226: a reprofiled drug intended for the prevention and treatment of familial transthyretin amyloidosis, in Proceedings of the XIIIth International Symposium on Amyloidosis: From Misfolded Proteins to Well-Designed Treatment (Hazenberg B. P. C., and Bijzet J., Eds.) pp 451–453, May 6–10, Groningen, The Netherlands.
- Reixach N.; Deechongkit S.; Jiang X.; Kelly J. W.; Buxbaum J. N. (2004) Tissue damage in the amyloidoses: transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. U.S.A. 101, 2817–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Smith C. S.; Petrassi H. M.; Hammarstrom P.; White J. T.; Sacchettini J. C.; Kelly J. W. (2001) An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry 40, 11442–11452. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Buxbaum J. N.; Reixach N. (2013) Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry 52, 1913–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reixach N.; Foss T. R.; Santelli E.; Pascual J.; Kelly J. W.; Buxbaum J. N. (2007) Human–murine transthyretin heterotetramers are kinetically stable and non-amyloidogenic: a lesson in the generation of transgenic models of diseases involving oligomeric proteins. J. Biol. Chem. 283, 2098–2107. [DOI] [PubMed] [Google Scholar]
- Penchala S. C.; Connelly S.; Wang Y.; Park M. S.; Zhao L.; Baranczak A.; Rappley I.; Vogel H.; Liedtke M.; Witteles R. M.; Powers E. T.; Reixach N.; Chan W. K.; Wilson I. A.; Kelly J. W.; Graef I. A.; Alhamadsheh M. M. (2013) AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc. Natl. Acad. Sci. U.S.A. 110, 9992–9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulawa C. E.; Connelly S.; Devit M.; Wang L.; Weigel C.; Fleming J. A.; Packman J.; Powers E. T.; Wiseman R. L.; Foss T. R.; Wilson I. A.; Kelly J. W.; Labaudiniere R. (2012) Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. U.S.A. 109, 9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine H. III (1999) Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 309, 274–284. [DOI] [PubMed] [Google Scholar]
- Lindgren M.; Sorgjerd K.; Hammarstrom P. (2005) Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys. J. 88, 4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarstrom P.; Jiang X.; Deechongkit S.; Kelly J. W. (2001) Anion shielding of electrostatic repulsions in transthyretin modulates stability and amyloidosis: insight into the chaotrope unfolding dichotomy. Biochemistry 40, 11453–11459. [DOI] [PubMed] [Google Scholar]
- Hurshman Babbes A. R.; Powers E. T.; Kelly J. W. (2008) Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: the relationship between stability and amyloidosis. Biochemistry 47, 6969–6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennion B. J.; Daggett V. (2003) The molecular basis for the chemical denaturation of proteins by urea. Proc. Natl. Acad. Sci. U.S.A. 100, 5142–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagoe C.; Reixach N.; Friske L.; Mustra D.; French D.; Gallo G.; Buxbaum J. N. (2007) In vivo stabilization of mutant human transthyretin in transgenic mice. Amyloid 14, 227–236. [DOI] [PubMed] [Google Scholar]
- Mizuguchi M.; Yokoyama T.; Nabeshima Y.; Kawano K.; Tanaka I.; Niimura N. (2012) Quaternary structure, aggregation and cytotoxicity of transthyretin. Amyloid 19, 5–7. [DOI] [PubMed] [Google Scholar]
- Rappley I.; Monteiro C.; Novais M.; Baranczak A.; Solis G.; Wiseman R. L.; Helmke S.; Maurer M. S.; Coelho T.; Powers E. T.; Kelly J. W. (2014) Quantification of transthyretin kinetic stability in human plasma using subunit exchange. Biochemistry 53, 1993–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S. M.; Wiseman R. L.; Sekijima Y.; Green N. S.; Adamski-Werner S. L.; Kelly J. W. (2005) Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Acc. Chem. Res. 38, 911–921. [DOI] [PubMed] [Google Scholar]
- Colon W.; Kelly J. W. (1992) Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 31, 8654–8660. [DOI] [PubMed] [Google Scholar]
- Lai Z.; Colon W.; Kelly J. W. (1996) The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry 35, 6470–6482. [DOI] [PubMed] [Google Scholar]
- Coelho T.; Carvalho M.; Saraiva M.; Alves I.; Almeida M. R.; Costa P. P. (1993) A strikingly benign evolution of FAP in an individual found to be a compound heterozygote for two TTR mutations: TTR MET 30 and TTR MET 119. J. Rheumatol. 20, 179. [Google Scholar]
- Hammarstrom P.; Schneider F.; Kelly J. W. (2001) Trans-suppression of misfolding in an amyloid disease. Science 293, 2459–2462. [DOI] [PubMed] [Google Scholar]
- Longo A. I.; Hays M. T.; Saraiva M. J. (1997) Comparative stability and clearance of [Met30]transthyretin and [Met119]transthyretin. Eur. J. Biochem. 249, 662–668. [DOI] [PubMed] [Google Scholar]
- Dolado I.; Nieto J.; Saraiva M. J.; Arsequell G.; Valencia G.; Planas A. (2005) Kinetic assay for high-throughput screening of in vitro transthyretin amyloid fibrillogenesis inhibitors. J. Comb. Chem. 7, 246–252. [DOI] [PubMed] [Google Scholar]
- Cardoso I.; Goldsbury C. S.; Muller S. A.; Olivieri V.; Wirtz S.; Damas A. M.; Aebi U.; Saraiva M. J. (2002) Transthyretin fibrillogenesis entails the assembly of monomers: a molecular model for in vitro assembled transthyretin amyloid-like fibrils. J. Mol. Biol. 317, 683–695. [DOI] [PubMed] [Google Scholar]
- Teng M. H.; Yin J. Y.; Vidal R.; Ghiso J.; Kumar A.; Rabenou R.; Shah A.; Jacobson D. R.; Tagoe C.; Gallo G.; Buxbaum J. (2001) Amyloid and nonfibrillar deposits in mice transgenic for wild-type human transthyretin: a possible model for senile systemic amyloidosis. Lab Invest. 81, 385–396. [DOI] [PubMed] [Google Scholar]
- Sousa M. M.; Cardoso I.; Fernandes R.; Guimaraes A.; Saraiva M. J. (2001) Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: evidence for toxicity of nonfibrillar aggregates. Am. J. Pathol. 159, 1993–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Buxbaum J. N.; Kelly J. W. (2001) The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc. Natl. Acad. Sci. U.S.A. 98, 14943–14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhamadsheh M. M.; Connelly S.; Cho A.; Reixach N.; Powers E. T.; Pan D. W.; Wilson I. A.; Kelly J. W.; Graef I. A. (2011) Potent kinetic stabilizers that prevent transthyretin-mediated cardiomyocyte proteotoxicity. Sci. Transl. Med. 3, 97ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman R. L.; Johnson S. M.; Kelker M. S.; Foss T.; Wilson I. A.; Kelly J. W. (2005) Kinetic stabilization of an oligomeric protein by a single ligand binding event. J. Am. Chem. Soc. 127, 5540–5551. [DOI] [PubMed] [Google Scholar]
- Gavrin L. K.; Denny R. A.; Saiah E. (2012) Small molecules that target protein misfolding. J. Med. Chem. 55, 10823–10843. [DOI] [PubMed] [Google Scholar]
- Rosen D. R.; Siddique T.; Patterson D.; Figlewicz D. A.; Sapp P.; Hentati A.; Donaldson D.; Goto J.; O’Regan J. P.; Deng H. X. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. [DOI] [PubMed] [Google Scholar]
- Vassall K. A.; Stubbs H. R.; Primmer H. A.; Tong M. S.; Sullivan S. M.; Sobering R.; Srinivasan S.; Briere L. A.; Dunn S. D.; Colon W.; Meiering E. M. (2011) Decreased stability and increased formation of soluble aggregates by immature superoxide dismutase do not account for disease severity in ALS. Proc. Natl. Acad. Sci. U.S.A. 108, 2210–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.