

Abstract
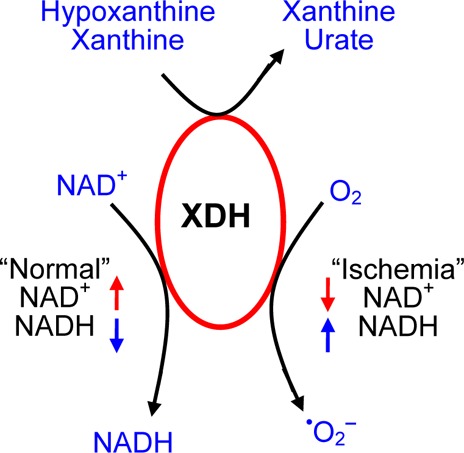
The enzyme xanthine oxidoreductase (XOR) is an important source of oxygen free radicals and related postischemic injury. Xanthine dehydrogenase (XDH), the major form of XOR in tissues, can be converted to xanthine oxidase (XO) by oxidation of sulfhydryl residues or by proteolysis. The conversion of XDH to XO has been assumed to be required for radical generation and tissue injury. It is also possible that XDH could generate significant quantities of superoxide, •O2–, for cellular signaling or injury; however, this possibility and its potential ramifications have not been previously considered. To unambiguously determine if XDH can be a significant source of •O2–, experiments were performed to measure and characterize •O2– generation using XDH from chicken liver that is locked in the dehydrogenase conformation. Electron paramagnetic resonance spin trapping experiments with 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide demonstrated that XDH in the presence of xanthine produces significant amounts of •O2–. NAD+ and NADH inhibited the generation of •O2– from XDH in a dose-dependent manner, with NAD+ exhibiting stronger inhibition than NADH at low physiological concentrations. Decreased amounts of NAD+ and NADH, which occur during and following tissue ischemia, enhanced the generation of •O2– from XDH in the presence of xanthine. It was observed that XDH-mediated oxygen radical generation markedly depressed Ca2+-ATPase activity of isolated sarcoplasmic reticulum vesicles from cardiac muscle, and this was modulated by NAD+ and NADH. Thus, XDH can be an important redox-regulated source of •O2– generation in ischemic tissue, and conversion to XO is not required to activate radical formation and subsequent tissue injury.
Mammalian xanthine dehydrogenase (XDH) is a dimeric protein with a molecular mass of 290 kDa that is composed of two identical independent subunits. Each subunit contains an N-terminal 20 kDa domain with two 2Fe–2S centers, a central 40 kDa flavin adenine dinucleotide (FAD) domain, and a C-terminal 85 kDa molybdopterin binding domain.1,2 Mammalian XDH can be readily converted to xanthine oxidase (XO) by oxidation of sulfhydryl residues or by proteolysis during extraction or purification procedures.3,4 Classically, XDH is considered to be NAD+-dependent, catalyzing the oxidative reaction of xanthine to urate with reduction of NAD+ to NADH, while XO also catalyzes urate formation but in a manner independent of NAD+, using O2 as an electron acceptor from the flavin site (Scheme 1). Therefore, XDH activity is considered primarily dependent on xanthine and NAD+, while XO is dependent on xanthine and O2.3
Scheme 1.

Over the past several decades, XO has been proposed to play a central role in the pathogenesis of oxidative injury in postischemic cells and tissues.5−8 In his seminal report, McCord proposed that ischemia activates free radical generation via a two-step process: (1) the requisite conversion of XDH to XO via a calcium-dependent protease and (2) the formation of the XO substrates hypoxanthine and xanthine secondary to the catabolism of ATP.5 This report triggered myriad biomedical studies in a variety of organ systems, including heart, lung, kidney, liver, and gastrointestinal tract, that have confirmed that XO plays an important role in the injury that occurs upon reperfusion of ischemic tissues.9 In this original report and all of the many biological and biomedical studies that have followed, the conversion of XDH to XO has been considered to be a requisite step for activation of oxygen free radical generation in postischemic tissues.5,10 Most of the xanthine oxidoreductase in mammalian cells is present in the form of XDH.9 Therefore, much attention has focused on the conversion of XDH to XO10,11 because this was considered as the critical first step leading to the formation of oxygen radicals.
In a series of studies applying EPR techniques to directly measure oxygen radical generation in reoxygenated endothelial cells and in cardiac models, it was observed that the process of radical generation was triggered primarily via the formation of the XO substrates hypoxanthine and xanthine due to ischemia-induced degradation of ATP.12−14 Little, if any, increase in the levels of XO occurred, and the magnitude and time course of radical generation precisely followed the cellular levels of XO substrates. Thus, it was reported that the levels of the substrates xanthine and hypoxanthine, not the formation of XO, are the key triggers activating postischemic radical generation.14
In contrast to the hypothesis that conversion of XDH to XO is essential for oxygen radical formation, other researchers reported that XDH itself can reduce O2 with production of •O2– accompanying oxidation of xanthine.15−17 It was also reported that this oxygen-dependent XDH activity is influenced by NADH and NAD+.3,15 In addition, oxidation of NADH by XDH also may produce •O2–.15,16,18−20 Thus, controversy remains regarding whether XDH produces significant amounts of •O2– or whether the enzyme must first be converted to the oxidase form for this to occur.
XDH from chicken liver is locked in the dehydrogenase form and is not converted to XO; however, the enzyme is otherwise very similar to the mammalian form in that it also contains two subunits, including two Fe–S centers, FAD, and molybdopterin.9,21 EPR spectroscopy with the use of spin traps allows measurement and quantitation of •O2– production in enzyme and cellular systems,22−24 and this has been facilitated by the development of the phosphorus-containing nitrone spin trap 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide (DEPMPO) that forms long-lived •O2– adducts. The reaction of •O2– with DEPMPO has been shown to proceed at a rate of ∼90 M–1 s–1 at pH 7 and room temperature with a decay half-life of 890 s.25,26 In another study using both the riboflavin–light and xanthine–xanthine oxidase superoxide-generating systems, the DEPMPO trapping efficiency was shown to be ∼65% with 40-fold more sensitivity than the commonly used cytochrome c reduction method of superoxide detection.27 The cytochrome c reduction method has also been shown to be nonspecific in that cytochrome c can be reduced directly by reactive cysteines and by direct electron transfer from the redox-active centers of XDH and XO.28,29 Cytochrome c may also bind to XDH, slowing the rate of •O2– production.28 Thus, measurement of •O2– by DEPMPO spin trapping has the advantage of greater specificity and a lack of perturbation of XDH or XO. The unique stability and specificity of the DEPMPO •O2– spin trapping method makes it applicable to complex biochemical systems such as XO and XDH.
We performed a series of studies to definitively determine the presence and magnitude of the production of •O2– from purified chicken liver XDH in the presence of xanthine using EPR spin trapping. These measurements of •O2– production are correlated with spectrophotometric measurements of urate production. The concentration-dependent effects of NAD+ and NADH on this process are determined. Finally, measurements are performed to determine if the amount of •O2– production from tissue levels of XDH and xanthine is sufficient to induce oxidative injury of the critical Ca2+-regulating ATPase pump of cardiac muscle, thus contributing to ischemia-reperfusion injury.
Materials and Methods
Materials
XDH was purified as reported previously by homogenization of fresh chicken livers in liquid nitrogen, followed by centrifugation, ammonium sulfate and butanol fractionation, and sequential chromatography on hydroxyapatite, Sephacryl S-300, and a folate affinity column. This procedure avoids the use of acetone extraction, thereby minimizing damage to the Mo center of this enzyme.30,31 XO [grade III, from buttermilk, chromatographically purified, in 2.3 M (NH4)2SO4 and 10 mM sodium phosphate buffer (pH 7.8) containing 1 mM EDTA and 1 mM sodium salicylate] was obtained from Sigma. The salicylate was removed chromatographically with Sephadex G-25 prior to use. Xanthine, NAD+, and NADH were also obtained from Sigma. The spin trap 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide (DEPMPO) was purchased from Oxis.
Spectrophotometric Measurements
UV–visible absorption spectra of either XDH or XO and enzyme assays were performed with a Varian Cary 300 UV–visible spectrophotometer equipped with a temperature-controlled circulator. Xanthine-O2 activity was assayed in air-equilibrated PBS (pH 7.4) solutions at 25 °C after addition of xanthine (360 μM) by measurement of the production of uric acid from the change in absorbance at 295 nm (ε = 9500 M–1 cm–1). Xanthine-NAD+ activity was measured at 25 °C in PBS (pH 7.4) after addition of xanthine (360 μM) and NAD+ by measurement of the production of uric acid.
EPR Measurements
All EPR measurements were performed using Bruker ER 300 or ESP 300E spectrometers operating at X-band with TM110 cavity resonators. The microwave frequency was measured with an EIP model 575 microwave counter (EIP Microwave, Inc., San Jose, CA). To assess •O2– generation, EPR spin trapping studies were performed using the spin trap DEPMPO.22,23 The following instrument settings were used in the spin trapping experiments: modulation amplitude, 0.32 G; time constant, 0.16 s; scan time, 60 s; modulation frequency, 100 kHz; microwave power, 20 mW; microwave frequency, 9.76 GHz. The samples were placed in a quartz EPR flat cell, and spectra were recorded at ambient temperature (25 °C). The collection of EPR data was started 2 min after the addition of xanthine. The component signals observed in these spectra were identified and quantified as reported. The double integrals of DEPMPO-OOH experimental spectra were compared with those of a 1 μM TEMPO sample measured under identical settings to estimate the concentration of the •O2– adduct.22,23
Sarcoplasmic Reticulum Preparation
Cardiac sarcoplasmic reticulum (SR) vesicles were isolated from the left ventricle of healthy, filarial free beagle dogs as follows.32−34 Dogs were anesthetized with sodium pentobarbital (25 mg/kg), and the heart was excised rapidly and placed in an ice-cold saline solution. Muscle (50 g) was minced and homogenized in imidazole buffer (pH 7) at 4 °C in an Excel autohomogenizer (DX-8, Nihon Seiki, Tokyo, Japan). The homogenate was centrifuged at 4000g for 20 min. The supernatant was saved, and the pellet was again homogenized and centrifuged. The combined supernatants were filtered through four layers of cheesecloth and centrifuged at 10000g for 15 min. The supernatant was then filtered through eight layers of cheesecloth and centrifuged at 31000g for 1 h. The pellets were rehomogenized in 1 M KCl, 10 mM imidazole buffer by use of a teflon pestle and then centrifuged at 145000g for 1 h. The final pellet was resusupended in 30% sucrose and 10 mM imidazole (pH 7.0) to a final concentration as determined by the method of Lowry et al. The isolated SR was stored at −80 °C until it was used.
Measurement of Ca2+-ATPase Activity
The Ca2+-ATPase activity of cardiac SR was determined as the rate at which inorganic phosphate (Pi) was liberated during the incubation. The incubation bath (5 mL) was kept at 37 °C and contained 100 mM KCl, 20 mM imidazole buffer (pH 7.0), 10 mM NaN3, 10 mM potassium oxalate, 5 mM Na2ATP, 5 mM MgCl2, and 200 μM CaCl2. The released phosphate in the filtrate was assayed by a colorimetric method.35 The Ca2+-ATPase activity was calculated as the difference in ATPase rate in a bath containing 200 μM Ca2+ compared to one containing 0.02 M EGTA.32−34,36,37
Statistical Analysis
All experiments were performed in triplicate and repeated at least three times. Results are expressed as means ± the standard error of the mean (SEM). Statistical analysis was performed by a Student’s t-test or a one-way analysis of variance. Significance was defined at the p < 0.05 level.
Results
Generation of •O2– from XDH Compared to XO
Initial experiments were performed to measure the magnitude of generation of •O2– from XDH in the presence of xanthine and to compare this to that from similar amounts of XO. Experiments were performed using the spin trap DEPMPO that provides a stable •O2– adduct.22 The DEPMPO was free of any background signal, and in the presence of XO or XDH alone, no signal was seen (Figure 1A,B, traces a and b). However, upon addition of xanthine to XO, a characteristic DEPMPO-OOH adduct spectrum was seen with hyperfine splitting giving rise to 12 resolved peaks (Figure 1A, trace c). In addition to the large signal of DEPMPO-OOH, a small signal of DEPMPO-OH was observed as reported previously.22,23 The •O2–-derived DEPMPO-OOH adduct comprised 92% of the total intensity and the DEPMPO-OH adduct 8%. Upon addition of xanthine to XDH, a similar DEPMPO-OOH signal was observed; however, the intensity of this signal was approximately 40–50% lower than that seen with identical amounts of XO (Figure 1B, trace c). A large signal of DEPMPO-OOH comprising 80% of the total intensity and a small DEPMPO-OH component of 20% was observed. With both XO and XDH, these signals were quenched by SOD (150 units/mL), confirming that they were derived from •O2– (Figure 1A,B, trace d). The time course of •O2– generation was measured, and it was observed that over a period of 30 min both enzymes support a similar sustained process of •O2– generation (Figure 2A). In a series of repeat experiments, a similar process of •O2– generation was seen with the magnitude of generation by XDH being 55% of that of XO (Figure 2B). These data suggest that XDH can generate •O2– in the presence of xanthine in a manner similar to that with XO, although the magnitude is somewhat lower. A simulation of a representative EPR spectrum resulting from the xanthine oxidase–xanthine reaction in the presence of DEPMPO is provided in Figure S1 of the Supporting Information.
Figure 1.
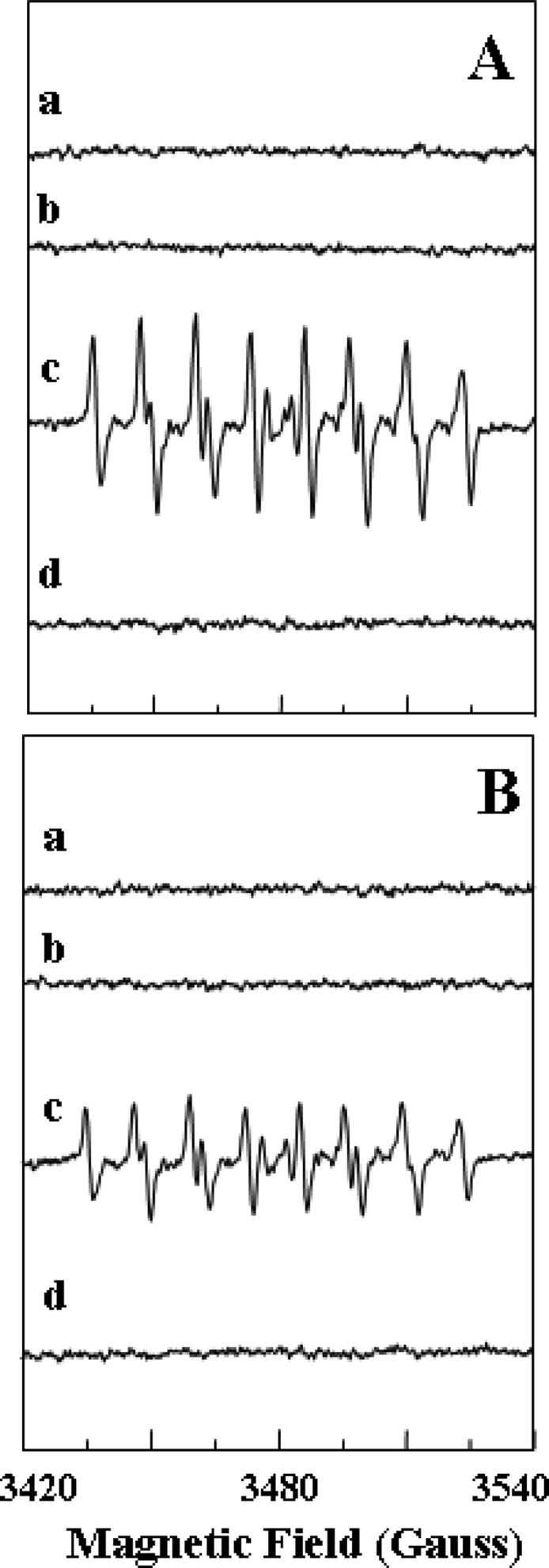
EPR spectra of the DEPMPO-OOH adduct in phosphate-buffered saline. The superoxide radicals were generated from XO or XDH using xanthine. (A) EPR spin trapping measurement of generation of •O2– from XO (0.1 μM) with the addition of xanthine (360 μM) in 0.1 M PBS (pH 7.4): (a) incubation of DEPMPO (10 mM) without XO and xanthine, (b) incubation of DEPMPO (10 mM) and XO without xanthine, (c) incubation of DEPMPO (10 mM) and XO with the addition of xanthine (a prominent spectrum of DEPMPO-OOH with only a small signal of DEPMPO-OH is seen with relative intensities of 92 and 8%, respectively), and (d) incubation of DEPMPO (10 mM), XO, and xanthine in the presence of SOD (150 units/mL) (the intensity of the DEPMPO-OOH signal was greatly diminished). (B) EPR spin trapping measurement of generation of •O2– from XDH (0.1 μM) with the addition of xanthine (360 μM) in 0.1 M PBS (pH 7.4): (a) incubation of DEPMPO (10 mM) without XDH and xanthine, (b) incubation of DEPMPO and XDH without xanthine, (c) incubation of DEPMPO (10 mM) and XDH with the addition of xanthine (a prominent spectrum of DEPMPO-OH is seen with relative intensities of 80 and 20%), and (d) incubation of DEPMPO (10 mM), XDH, and xanthine in the presence of SOD (150 units/mL) (the intensity of the DEPMPO-OOH signal was greatly diminished).
Figure 2.

Time course of generation of •O2– from XO or XDH. (A) Time courses of generation of •O2– from XO (0.1 μM) and XDH (0.1 μM) and pretreatment of XO or XDH with SOD (150 units/mL) measured by EPR spin trapping following the addition of xanthine (360 μM). The reaction mixture was incubated for 2 min for each time course. (B) DEPMPO spin concentration of generation of •O2– from XO (0.1 μM) or XDH (0.1 μM) measured by EPR spin trapping following the addition of xanthine. Data are presented as means ± the standard error of triplicate experiments. *Significant (p < 0.01) difference from the corresponding value of the XO–xanthine system.
Effects of NADH or NAD+ on Generation of •O2– from XDH and XO
Experiments were performed to investigate the effects of NADH and NAD+ on this XDH-mediated •O2– generation. During ischemia, tissue xanthine14 and NADH38 levels are markedly increased compared to those in normally perfused tissues. Oxidation of NADH by XDH also could produce a significant amount of •O2–.15,18−20 We examined the effects of NADH on the generation of •O2– by XDH or XO. When NADH (0.1–1 mM) was added to XDH and this was immediately followed by addition of xanthine, a dose-dependent inhibition of •O2– generation was observed (Figures 3A and 4A). Approximately 50 or 75% inhibition was seen with 0.1 or 0.5 mM NADH, respectively. In experiments when NAD+ (0.1–1 mM) was added to XDH immediately followed by addition of xanthine, a dose-dependent inhibition of •O2– generation was observed (Figures 3B and 4B). Approximately 70 or 90% inhibition was seen with 0.1 or 0.5 mM NAD+, respectively.
Figure 3.
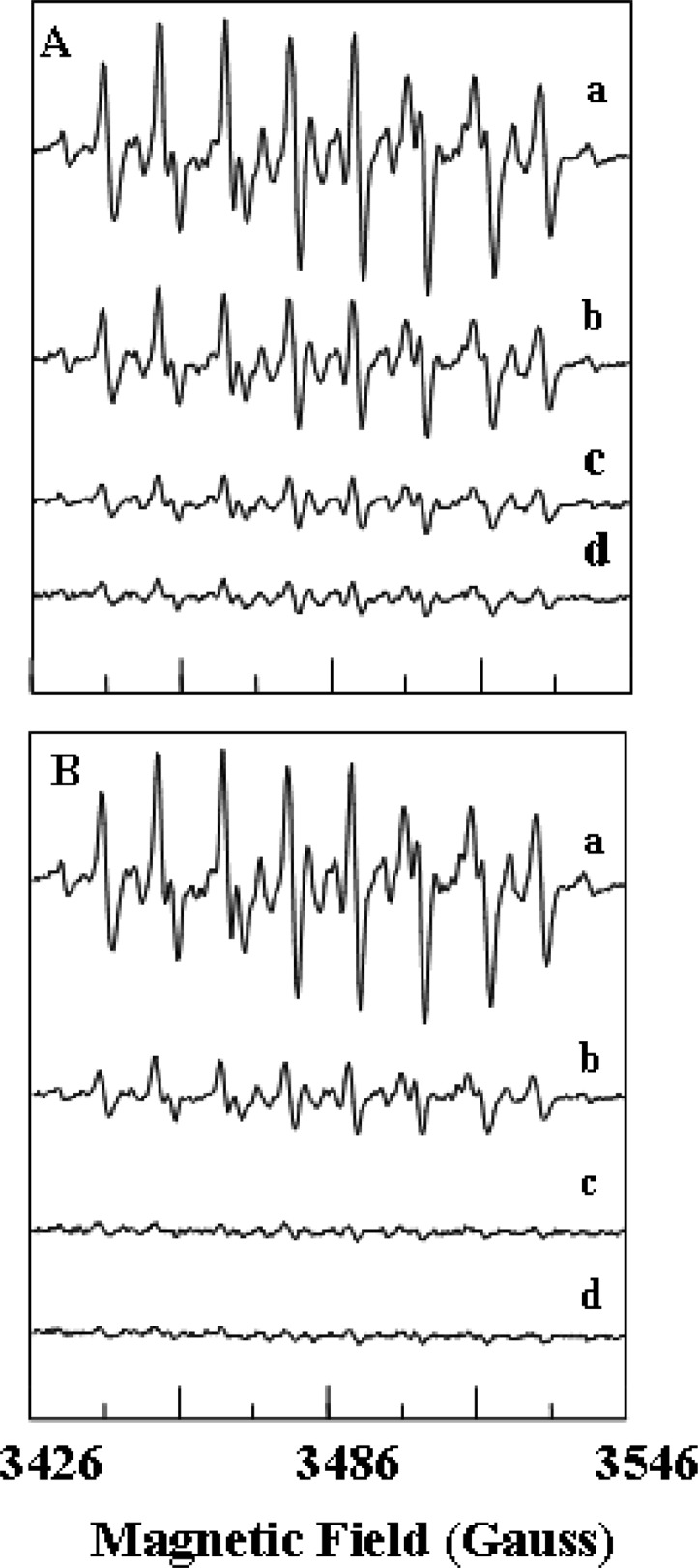
Dose-dependent effects of NADH or NAD+ on the generation of •O2– from XDH. (A) EPR spectra of the DEPMPO-OOH adduct in 0.1 M PBS (pH 7.4). EPR spin trapping measurements of •O2– generation from XDH (0.1 μM) and various concentrations of NADH (0–1 mM) with the addition of xanthine (360 μM) were taken in the presence of DEPMPO (10 mM): (a) 0, (b) 0.1, (c) 0.5, and (d) 1.0 mM NADH. (B) EPR spin trapping measurements of •O2– generation from XDH (0.1 μM) and various concentrations of NAD+ (0–1 mM) with the addition of xanthine (360 μM) were taken in the presence of DEPMPO (10 mM): (a) 0, (b) 0.1, (c) 0.5, and (d) 1.0 mM NAD+.
Figure 4.

Dose-dependent effects of NADH or NAD+ on the generation of •O2– from XDH. (A) XDH (0.1 μM) was pretreated with NADH (0–1 mM) as described in the legend of Figure 3. (B) XDH (0.1 μM) was pretreated with NAD+ (0–1 mM) as described in the legend of Figure 3. Data are presented as means ± the standard error of triplicate experiments.
We also measured the dose-dependent effects of NADH on the generation of •O2– from XO. NADH clearly decreased the rate of •O2– generation in a dose-dependent manner, but much higher levels were required with 50% inhibition seen only above 1 mM NADH (Table 1). The dose-dependent effects of NAD+ on the generation of •O2– from XO were also measured, and it was observed that NAD+ did not inhibit the generation of •O2– from XO (data not shown).
Table 1. Dose-Dependent Effects of NADH on •O2– Generation and Xanthine-O2 Activity of Xanthine Oxidase (XO)a.
| [NADH] (mM) | •O2– generation (% of control) | % of initial xanthine-O2 activity |
|---|---|---|
| 0.1 | 96.09 ± 3.91 | 103.62 ± 3.26 |
| 0.5 | 82.79 ± 2.61b | 90.85 ± 8.89 |
| 1.0 | 66.20 ± 2.04b | 83.45 ± 4.47 |
| 2.0 | 43.92 ± 2.14b | 53.83 ± 3.45b |
| 3.0 | 25.49 ± 2.36b | 33.34 ± 5.92b |
NADH (0–3 mM) was added to XO (0.1 μM) as described in the legend of Figure 3. •O2– generation was measured by EPR spin trapping using DEPMPO, and xanthine-O2 activity was measured via a spectrophotometric assay of urate production monitored at 295 nm. Both were performed with addition of xanthine (360 μM) to the reaction mixture. Data are presented as means ± the standard error of triplicate experiments.
Significantly different (p < 0.05) from the corresponding control value.
Thus, both NADH and NAD+ inhibit the generation of •O2– from XDH in the presence of xanthine, but NAD+ exhibits stronger inhibition at low (0.1–0.5 mM) physiological concentrations. With XO, however, NAD+ did not inhibit •O2– generation even at >2 mM, while 2 mM NADH was required to reach just >50% inhibition.
Effects of NADH on Xanthine-O2 Activity
To determine if NADH inhibits not only •O2– generation but also the conversion of xanthine to urate, we examined the effects of NADH on xanthine-O2 activity of XDH and XO. When NADH (0.1–3 mM) was added to XDH and this was immediately followed by the addition of xanthine, a dose-dependent inhibition of xanthine-O2 activity was observed (Table 2). With NADH concentrations of 2 mM, more than 50% inhibition was seen. The dose-dependent effects of NADH on the xanthine-O2 activity of XO were also measured, and it was seen that NADH decreased xanthine-O2 activity in a dose-dependent manner; however, much like the measurements of •O2– generation, higher NADH levels were required to inhibit the activity of XO versus XDH (Table 1). Thus, these data demonstrate that NADH significantly depressed XDH- or XO-mediated •O2– generation because of the inhibition of xanthine-O2 activity.
Table 2. Dose-Dependent Effects of NADH on the Activity of Xanthine Dehydrogenase (XDH)a.
| [NADH] (mM) | % of initial xanthine-O2 activity | % of initial xanthine-NAD+ activity |
|---|---|---|
| 0.1 | 75.3 ± 1.1b | – |
| 0.5 | 59.7 ± 0.8b | 71.4 ± 3.6c |
| 1.0 | 51.2 ± 0.7b | 51.0 ± 3.4c |
| 2.0 | 44.9 ± 1.1b | 37.6 ± 4.8c |
| 3.0 | 23.8 ± 4.2b | 22.3 ± 3.5c |
NADH (0–3 mM) was added to XDH (0.1 μM) as described in the text. Xanthine-O2 activity was measured from a spectrophotometric assay of urate production monitored at 295 nm. Xanthine-NAD+ activity, in the presence of NAD+ 0.5 mM, was measured by urate formation. Measurements were performed with addition of xanthine (360 μM) to the reaction mixture. Data are presented as means ± the standard error of triplicate experiments.
Significantly different (p < 0.05) from the corresponding control value.
Significantly different (p < 0.05) from the corresponding control value of the treated XDH–xanthine system (0.5 mM NAD+).
Effects of NADH on Xanthine-NAD+ Activity
To further determine the dose-dependent effects of NADH on the conversion of xanthine to urate mediated by XDH in the presence of NAD+, the xanthine-NAD+ activity of XDH was measured. When NADH was added to XDH in the presence of NAD+ and this was immediately followed by the addition of xanthine, a dose-dependent inhibition of urate formation was observed (Table 2). These observations indicate that NADH inhibits XDH-mediated reduction of NAD+.
Effects of NAD+ on Xanthine-NAD+ Activity of XDH and XO
To correlate the effects of NAD+ on •O2– generation with that on enzyme activity measured from the conversion of xanthine to urate, we examined the effects of NAD+ on the activity of XO and XDH. With XO, NAD+ levels of up to 2 mM did not alter XO activity as measured by urate formation. This is consistent with the role of O2 as the requisite electron acceptor. With XDH, however, because NAD+ is the preferred substrate in the oxidative half-reaction, an increased level of uric acid formation was observed in accordance with the NAD+ concentration (Table 3).
Table 3. Dose-Dependent Effects of NAD+ on the Activity of Xanthine Dehydrogenase (XDH)a.
| [NAD+] (mM) | % of the maximal xanthine-NAD+ activity |
|---|---|
| 0.0 | 13.0 ± 0.5 |
| 0.1 | 39.3 ± 2.8b |
| 0.3 | 83.9 ± 6.1b |
| 0.5 | 91.0 ± 3.5b |
| 1.0 | 99.1 ± 5.3b |
| 2.0 | 100 ± 7.0b |
NAD+ (0–2 mM) was added to XDH (0.1 μM) as described in the text. Xanthine-NAD+ activity was measured from a spectrophotometric assay of urate production monitored at 295 nm. Measurements were performed with addition of xanthine (360 μM) to the reaction mixture. Data are presented as means ± the standard error of triplicate experiments.
Significantly different (p < 0.05) from the value in the absence of NAD+.
Effects of Oxygen Radicals Generated by the XDH–Xanthine System on Ca2+-ATPase Activity of Cardiac SR
We and others have shown that the Ca2+-ATPase of SR is highly sensitive to oxidative damage.32−34,36,37 To determine if the process of XDH-mediated radical generation can be physiologically significant and to compare this to that from XO, we examined the effects of oxygen radicals generated from XDH or XO on Ca2+-ATPase activity of isolated SR from canine cardiac muscle. XO and XDH were used at a concentration of 0.1 μM, comparable to that measured in cardiac tissue, with xanthine levels of 360 μM similar to those formed in ischemic myocardium.14,39 Significant inhibition of Ca2+-ATPase activity was seen in both the XDH–xanthine system and the XO–xanthine system (Figure 5). We also measured the effects of NADH or NAD+ on XDH–xanthine-mediated inhibition of Ca2+-ATPase activity. In the presence of either NADH or NAD+, only a modest decrease in the loss of Ca2+-ATPase activity was seen relative to that seen after adding only xanthine with significant inhibition in activity still remaining (Figure 5). These observations clearly suggested that oxygen radicals generated from XDH could cause the disruption of Ca2+-ATPase activity of cardiac SR in a manner similar to that seen with XO.
Figure 5.

Effects of XO or XDH with xanthine, NAD+, and NADH on Ca2+-ATPase activity of cardiac sarcoplasmic reticulum (SR). Cardiac SR (50 μg/mL) was incubated with XO (0.1 μM) or XDH (0.1 μM) alone and with xanthine (X) (360 μM) for 30 min before the reaction was begun with Ca2+, Mg2+, and ATP in the presence or absence of NADH (0.5 mM) or NAD+ (0.5 mM). Ca2+-ATPase activity measured as described in Materials and Methods. Control reaction denoted by C. Data are presented as means ± the standard error of triplicate experiments. *Significantly different (p < 0.01) from the corresponding control value.
Discussion
Xanthine oxidoreductase (XOR) is a critical source of oxygen free radicals in biological cells and tissues and plays an important role in oxygen radical generation and the pathogenesis of injury following postischemic reperfusion.5,40,41 It has been demonstrated that XO is critically important in the mechanism of postischemic radical generation; however, it was previously assumed that all radical generation from (XOR) was solely derived from the oxidase form with none arising from the dehydrogenase, which is typically more than 90% of the total XOR pool.18,42 This extends from the original proposal of McCord that the conversion of XDH to XO was required for oxygen radical generation.5 On this basis, attention over the last two decades has focused largely on conditions that convert XDH to XO10,11,18,20,42 as the critical trigger of radical generation.
We have previously studied and characterized the process and mechanism of postischemic radical generation in isolated hearts.6,43,44 In the isolated rat heart model, we have observed, using EPR spin trapping, that there is a burst of radical generation occurring over the early minutes of reperfusion and that most of this can be inhibited by XOR inhibitors such as allopurinol or oxypurinol.7 The time course of this radical generation paralleled that of XOR substrate levels in the heart that increase during ischemia and then decline upon reflow.14 Only modest conversion of XDH to XO was observed, and the process of radical generation was clearly seen to be linked to that of the XOR substrates xanthine and hypoxanthine. It was further observed that inhibitors of adenosine deaminase that blocked this substrate formation also ameliorated radical generation and injury.45 However, in prior studies in the biomedical literature, it was assumed that XO was absolutely required for this radical generation with no contribution from the predominant dehydrogenase form.
Mammalian XDH can be readily converted to XO by oxidation of cysteine sulfhydryl residues or by proteolysis during extraction or purification procedures.3,9 Therefore, these studies utilized XDH isolated from chicken liver that has the unique property of being locked in the dehydrogenase form while retaining enzymatic properties comparable to those of mammalian XDH.9,21 Detailed in vitro studies of chicken liver XDH have demonstrated that the FAD center is the site of the oxidative half-reaction, with both NAD+ and O2 functioning as electron acceptors.46,47 It was also demonstrated that, as with the oxidase form of the enzyme, •O2– is generated only in the slower steps of the reaction between the FAD semiquinone and O2 at a second-order reaction rate of ∼260 M–1 s–1 (pH 7.8 and 4 °C). For the XDH form of the enzyme, NAD+ is a much more effective oxidant than O2 and outcompetes the O2 for the reducing equivalents in the enzyme. NAD+ reoxidizes reduced chicken liver XDH at a kox of ∼27 s–1 and a Kd of ∼80 μM.47 Very little •O2– is generated because very little O2 is reduced because of the much slower rate of reaction. In addition to NAD+ limiting •O2– generation by reacting with the reduced enzyme faster than O2, binding of NAD+ (and NADH) to the XDH form of the enzyme is expected to block the access of O2 to the FAD center based on the known protein crystal structure.4,48 On the basis of these insights, we performed experiments to directly monitor •O2– generation from both XDH and XO using EPR spin trapping. Our studies provide direct evidence that XDH in the presence of xanthine can produce a large amount of •O2– in a manner similar to that of XO (Figures 1 and 2) and that this •O2– generation can be modulated by the levels of NAD+ and NADH (Figures 3 and 4).
In this study, EPR spin trapping measurements with the use of DEPMPO demonstrated the formation of the DEPMPO-OOH adduct that is specific for •O2–.22 We assessed the quantity and time course of the production of •O2– from XDH by comparing the relative rate of •O2– production from XDH to that from XO, and we observed that the rate of •O2– production from XDH was 55% of that from XO for the xanthine-O2 reaction.
We observed that the production of •O2– from XDH in the presence of xanthine was modulated by both NAD+ and NADH, which exerted prominent concentration-dependent inhibition of •O2– production. NAD+, however, exerted stronger inhibition at lower concentrations than that of NADH, as expected because of its effectiveness as an oxidant for XDH. Approximately 70 or 90% inhibition was seen with 0.1 or 0.5 mM NAD+, respectively, while approximately 50 or 75% inhibition was seen with 0.1 or 0.5 mM NADH, respectively. This is in contrast to the inhibition seen with XO in the presence of xanthine that was affected by only NADH and not by NAD+ (Table 1). Even with 1 mM NADH, only approximately 30% inhibition of XO-derived •O2– generation occurred. With XDH, however, 1 mM NADH or NAD+ induced >90 or >95% inhibition, respectively. Thus, for cellular levels of the NAD+/NADH pool that are typically in the range of 0.1–0.5 mM,49 prominent dose-dependent inhibition of production of •O2– from XDH was greater with NAD+ than NADH, while for XO, only modest inhibition was seen with only NADH and not with NAD+. The turnover numbers for the various reactivities of chicken liver XDH have been determined to be 102 min–1 for the xanthine–NAD+ reductase reaction, 43 min–1 for the xanthine–O2 reductase reaction, and 2 min–1 for the NADH–O2 reductase reaction (pH 7.8 and 4 °C), which suggested that NADH or NAD+ binds near the reduced flavin, blocking access and thereby slowing its reactivity with O2.46
It was also observed that NADH inhibited the activity of XDH measured from urate production in the presence of the substrate xanthine. This was true in the presence or absence of NAD+. This inhibition paralleled the observed decrease in •O2– production. Similarly, higher levels of NADH inhibited the activity of XO. This suggests that NADH associates with the FAD site of XDH or XO and competes with either NAD+ or O2 for binding at this site, thus impeding electron transfer. While the inhibition observed in urate formation was also dose-dependent and largely paralleled that seen for •O2– production, in general this inhibition of activity was somewhat lower than that of •O2– production. Because XO or XDH can reduce O2 to either •O2– or H2O2, this may suggest that NADH induces a shift to the two-electron reduction of O2 to H2O2.50 It should also be noted that both XDH and XO have a NADH–O2 reductase activity in which there is an obligatory two-electron reduction of the FAD center by NADH to give FADH2.
The physiological or pathophysiological relevance of the modulation of •O2– generation and XDH function by NADH or NAD+ can be considered in view of the levels of NADH or NAD+ required to regulate the generation of •O2– from XDH. In normal tissues, the cellular concentrations of the NADH and NAD+ pool have been measured to be in the range of 0.25–0.5 mM.51 In normal oxygenated myocardium, most of the pool would be in the oxidized form as NAD+.49,52,53 However, with the marked hypoxia and reduced state during ischemia, the pool would be almost totally reduced.52 During ischemia, ATP is degraded to form the XO substrates hypoxanthine and xanthine.14 It has also been observed that the levels of the NADH and NAD+ pool are decreased by a factor of more than 2 over the first 40 min after reperfusion, and this has been attributed to the activation of the enzyme poly-ADPribose polymerase.49 Overall, the lower levels of the total pool and NAD+ in particular would enhance the production of •O2– from XDH. During ischemia, this would be limited by the O2 levels in the myocardium, and •O2– generation would then be further stimulated with a prominent increase in the level of O2 upon reperfusion.54
In the heart after ischemia for 30 min, intracellular concentrations of xanthine and hypoxanthine have been observed to increase from preischemic levels of <0.5 to 500–600 μM.14,39 The activity of XO in the ischemic heart has been measured to be ∼10 milliunits/g of protein and that of XDH to be ∼130 milliunits/g of protein.14,39 Thus, most of the XOR is in the reductase form. While after reperfusion we have previously observed a doubling in the levels of XO, still almost 90% of the enzyme remains in the form of XDH. The levels of XOR reported in the heart correspond to concentrations of ∼0.1 μM.
We considered whether the levels of XDH and its substrates in the postischemic heart could result in sufficient radical generation to be functionally significant in the pathogenesis of myocardial injury. A hallmark of postischemic myocardial injury is an alteration in Ca2+ regulation due to oxidant-induced dysfunction of sarcoplasmic reticulum Ca2+-ATPase. We used this phenomenon to determine the effects of tissue levels of XDH in the presence of xanthine levels formed during myocardial ischemia. It was observed that the XDH-mediated oxygen radical generation markedly depressed Ca2+-ATPase activity (Figure 5). Furthermore, while 0.5 mM levels of either NAD+ or NADH lessened the degree of Ca2+-ATPase impairment, depressed activity was still apparent (Figure 5). This suggests that XDH-mediated oxidant injury of cardiac muscle does occur in postischemic tissues and that this process is modulated by the tissue levels of NADH and NAD+.
One can consider the effect of cellular redox state on the production of radicals from XOR. Overall, it was observed that the levels of NADH and NAD+ as well as their relative amount profoundly modulated the generation of •O2– from XDH (Figure 4). The production of •O2– by XDH was found to be redox-regulated with more production under reduced conditions with a higher proportion of the NAD(H) pool in the reduced state. An oxidant shift in the redox state with elevated NAD+ levels would decrease the level of generation of •O2– from XDH and perhaps serve as a feedback mechanism or brake to prevent oxidative injury from XDH. However, major oxidant stress would induce the conversion of the reductase to the oxidase, which is unaffected by NAD+ and only modestly inhibited by higher levels of NADH. This may indicate that XDH, with its lower level and strongly NAD+-regulated •O2– generation, normally serves a signaling role that is tightly redox controlled, while under conditions of major oxidant stress conversion to XO removes this regulation, triggering higher levels of radical generation leading to cellular injury and death. Indeed, in postischemic myocardium, it is anticipated that the initial production of •O2– from XDH, which is enhanced by the reduced state of the myocardium and NAD+ depletion, could then trigger its subsequent partial conversion to XO.
In conclusion, our results indicate that XDH can be an important source of •O2– generation in cells and tissues. Its xanthine-mediated •O2– generation is regulated by the cellular levels of NAD(H) and the cellular redox state. Under normal conditions, XDH-mediated oxygen radical generation is redox-regulated and can serve a homeostatic role in cell signaling; however, under conditions of ischemia and reperfusion, this pathway is greatly enhanced and can lead to oxidative cellular injury with impaired calcium regulation. Because XDH is the predominant form of XOR in normal tissues, XDH-mediated •O2– generation can play an important role in cellular signaling and injury.
Glossary
Abbreviations
- XDH
xanthine dehydrogenase
- Fe–S
iron–sulfur
- FAD
flavin adenine dinucleotide
- XO
xanthine oxidase
- •O2–
superoxide
- EPR
electron paramagnetic resonance
- DEPMPO
5-(diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide
- SR
sarcoplasmic reticulum.
Supporting Information Available
A simulation of a representative EPR spectrum resulting from the xanthine oxidase–xanthine reaction in the presence of DEPMPO (Figure S1) and all the experimental and simulation parameters (figure legend). This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by National Institutes of Health Grants HL63744, HL65608, HL38324 (J.L.Z.), and GM 075036 (R.H.). The part of this work performed at the Research Center of Advanced Technology for Craniomandibular Function of the Kanagawa Dental University was also supported by grants-in-aid for Bioventure Research from the Japanese Ministry of Education, Science, and Culture.
The authors declare no competing financial interest.
Dedication
We acknowledge and dedicate this work to Dr. Vincent Massey who encouraged us to undertake these studies to clarify the role of xanthine dehydrogenase in oxidant production.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Enroth C.; Eger B. T.; Okamoto K.; Nishino T.; Pai E. F. (2000) Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: Structure-based mechanism of conversion. Proc. Natl. Acad. Sci. U.S.A. 97, 10723–10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille R.; Hall J.; Basu P. (2014) The mononuclear molybdenum enzymes. Chem. Rev. 114, 3963–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille R.; Nishino T. (1995) Flavoprotein structure and mechanism. 4. Xanthine oxidase and xanthine dehydrogenase. FASEB J. 9, 995–1003. [PubMed] [Google Scholar]
- Nishino T.; Okamoto K.; Eger B. T.; Pai E. F.; Nishino T. (2008) Mammalian xanthine oxidoreductase: Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 275, 3278–3289. [DOI] [PubMed] [Google Scholar]
- McCord J. M. (1985) Oxygen-derived free radicals in postischemic tissue injury. N. Engl. J. Med. 312, 159–163. [DOI] [PubMed] [Google Scholar]
- Zweier J. L.; Kuppusamy P. (1988) Electron paramagnetic resonance measurements of free radicals in the intact beating heart: A technique for detection and characterization of free radicals in whole biological tissues. Proc. Natl. Acad. Sci. U.S.A. 85, 5703–5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson-Gorman S. L.; Zweier J. L. (1990) Evaluation of the role of xanthine oxidase in myocardial reperfusion injury. J. Biol. Chem. 265, 6656–6663. [PubMed] [Google Scholar]
- De Pascali F.; Hemann C.; Samons K.; Chen C. A.; Zweier J. L. (2014) Hypoxia-Reoxygenation Induces NOS3 Uncoupling in Endothelial Cells Through Tetrahydrobiopterin Depletion and S-glutathionylation. Biochemistry. 53, 3679–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison R. (2002) Structure and function of xanthine oxidoreductase: Where are we now?. Free Radical Biol. Med. 33, 774–797. [DOI] [PubMed] [Google Scholar]
- Kuwabara Y.; Nishino T.; Okamoto K.; Matsumura T.; Eger B. T.; Pai E. F.; Nishino T. (2003) Unique amino acids cluster for switching from the dehydrogenase to oxidase form of xanthine oxidoreductase. Proc. Natl. Acad. Sci. U.S.A. 100, 8170–8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino T.; Okamoto K.; Kawaguchi Y.; Hori H.; Matsumura T.; Eger B. T.; Pai E. F.; Nishino T. (2005) Mechanism of the Conversion of Xanthine Dehydrogenase to Xanthine Oxidase: Identification of the Two Cysteine Disulfide Bonds and Crystal Structure of a Non-convertible Rat Liver Xanthine Dehydrogenase Mutant. J. Biol. Chem. 280, 24888–24894. [DOI] [PubMed] [Google Scholar]
- Abd-Elfattah A. S.; Jessen M. E.; Lekven J.; Doherty N. E.; Brunsting L. A.; Wechsler A. S. (1988) Myocardial reperfusion injury. Role of myocardial hypoxanthine and xanthine in free radical-mediated reperfusion injury. Circulation 78, III224–III235. [PubMed] [Google Scholar]
- Jennings R. B.; Steenbergen C. (1985) Nucleotide metabolism and cellular damage in myocardial ischemia. Annu. Rev. Physiol. 47, 727–749. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Zweier J. L. (1995) Substrate control of free radical generation from xanthine oxidase in the postischemic heart. J. Biol. Chem. 270, 18797–18803. [DOI] [PubMed] [Google Scholar]
- Nishino T.; Nishino T.; Schopfer L. M.; Massey V. (1989) The reactivity of chicken liver xanthine dehydrogenase with molecular oxygen. J. Biol. Chem. 264, 2518–2527. [PubMed] [Google Scholar]
- Maia L.; Duarte R. O.; Ponces-Freire A.; Moura J. J.; Mira L. (2007) NADH oxidase activity of rat and human liver xanthine oxidoreductase: Potential role in superoxide production. JBIC, J. Biol. Inorg. Chem. 12, 777–787. [DOI] [PubMed] [Google Scholar]
- Lindsay S.; Liu T. H.; Xu J. A.; Marshall P. A.; Thompson J. K.; Parks D. A.; Freeman B. A.; Hsu C. Y.; Beckman J. S. (1991) Role of xanthine dehydrogenase and oxidase in focal cerebral ischemic injury to rat. Am. J. Physiol. 261, H2051–H2057. [DOI] [PubMed] [Google Scholar]
- Nishino T.; Nakanishi S.; Okamoto K.; Mizushima J.; Hori H.; Iwasaki T.; Ichimori K.; Nakazawa H. (1997) Conversion of xanthine dehydrogenase into oxidase and its role in reperfusion injury. Biochem. Soc. Trans. 25, 783–786. [DOI] [PubMed] [Google Scholar]
- Sanders S. A.; Eisenthal R.; Harrison R. (1997) NADH oxidase activity of human xanthine oxidoreductase: Generation of superoxide anion. Eur. J. Biochem. 245, 541–548. [DOI] [PubMed] [Google Scholar]
- Nishino T.; Tamura I. (1991) The mechanism of conversion of xanthine dehydrogenase to oxidase and the role of the enzyme in reperfusion injury. Adv. Exp. Med. Biol. 309A, 327–333. [DOI] [PubMed] [Google Scholar]
- Sato A.; Nishino T.; Noda K.; Amaya Y. (1995) The structure of chicken liver xanthine dehydrogenase. cDNA cloning and the domain structure. J. Biol. Chem. 270, 2818–2826. [DOI] [PubMed] [Google Scholar]
- Roubaud V.; Sankarapandi S.; Kuppusamy P.; Tordo P.; Zweier J. L. (1998) Quantitative measurement of superoxide generation and oxygen consumption from leukocytes using electron paramagnetic resonance spectroscopy. Anal. Biochem. 257, 210–217. [DOI] [PubMed] [Google Scholar]
- Lee C. I.; Liu X.; Zweier J. L. (2000) Regulation of xanthine oxidase by nitric oxide and peroxynitrite. J. Biol. Chem. 275, 9369–9376. [DOI] [PubMed] [Google Scholar]
- Roubaud V.; Sankarapandi S.; Kuppusamy P.; Tordo P.; Zweier J. L. (1997) Quantitative measurement of superoxide generation using the spin trap 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide. Anal. Biochem. 247, 404–411. [DOI] [PubMed] [Google Scholar]
- Frejaville C.; Karoui H.; Tuccio B.; Le Moigne F.; Culcasi M.; Pietri S.; Lauricella R.; Tordo P. (1994) 5-Diethoxyphosphoryl-5-methyl-1-pyrroline N-Oxide (DEPMPO): A New Phosphorylated Nitrone for the efficient In Vitro and In Vivo Spin Trapping of Oxygen-centered Radicals. J. Chem. Soc., Chem. Commun. 15, 1793–1794. [DOI] [PubMed] [Google Scholar]
- Frejaville C.; Karoui H.; Tuccio B.; Le Moigne F.; Culcasi M.; Pietri S.; Lauricella R.; Tordo P. (1995) 5-(Diethoxyphosphoryl)-5-methyl-1-pyrroline N-oxide: A new efficient phosphorylated nitrone for the in vitro and in vivo spin trapping of oxygen-centered radicals. J. Med. Chem. 38, 258–265. [DOI] [PubMed] [Google Scholar]
- Roubaud V.; Sankarapandi S.; Kuppusamy P.; Tordo P.; Zweier J. L. (1997) Quantitative measurement of superoxide generation using the spin trap 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide. Anal. Biochem. 247, 404–411. [DOI] [PubMed] [Google Scholar]
- Harris C. M.; Massey V. (1997) The reaction of reduced xanthine dehydrogenase with molecular oxygen. Reaction kinetics and measurement of superoxide radical. J. Biol. Chem. 272, 8370–8379. [DOI] [PubMed] [Google Scholar]
- Komai H.; Massey V.; Palmer G. (1969) The Preparation and Properties of Deflavo Xanthine Oxidase. J. Biol. Chem. 244, 1692–1700. [PubMed] [Google Scholar]
- Ryan M. G.; Ratnam K.; Hille R. (1995) The molybdenum centers of xanthine oxidase and xanthine dehydrogenase. Determination of the spectral change associated with reduction from the Mo(VI) to the Mo(IV) state. J. Biol. Chem. 270, 19209–19212. [DOI] [PubMed] [Google Scholar]
- Ratnam K.; Brody M. S.; Hille R. (1996) Purification of xanthine dehydrogenase and sulfite oxidase from chicken liver. Prep. Biochem. Biotechnol. 26, 143–154. [DOI] [PubMed] [Google Scholar]
- Okabe E.; Hess M. L.; Oyama M.; Ito H. (1983) Characterization of free radical-mediated damage of canine cardiac sarcoplasmic reticulum. Arch. Biochem. Biophys. 225, 164–177. [DOI] [PubMed] [Google Scholar]
- Okabe E.; Fujimaki R.; Murayama M.; Ito H. (1989) Possible mechanism responsible for mechanical dysfunction of ischemic myocardium: A role of oxygen free radicals. Jpn. Circ. J. 53, 1132–1137. [DOI] [PubMed] [Google Scholar]
- Okabe E.; Sugihara M.; Tanaka K.; Sasaki H.; Ito H. (1989) Calmodulin and free oxygen radicals interaction with steady-state calcium accumulation and passive calcium permeability of cardiac sarcoplasmic reticulum. J. Pharmacol. Exp. Ther. 250, 286–292. [PubMed] [Google Scholar]
- Penney C. L. (1976) A simple micro-assay for inorganic phosphate. Anal. Biochem. 75, 201–210. [DOI] [PubMed] [Google Scholar]
- Lee C.; Okabe E. (1995) Hydroxyl radical-mediated reduction of Ca2+-ATPase activity of masseter muscle sarcoplasmic reticulum. Jpn. J. Pharmacol. 67, 21–28. [DOI] [PubMed] [Google Scholar]
- Ishibashi T.; Lee C. I.; Okabe E. (1996) Skeletal sarcoplasmic reticulum dysfunction induced by reactive oxygen intermediates derived from photoactivated rose bengal. J. Pharmacol. Exp. Ther. 277, 350–358. [PubMed] [Google Scholar]
- Williamson J. R. (1966) Glycolytic control mechanisms. II. Kinetics of intermediate changes during the aerobic-anoxic transition in perfused rat heart. J. Biol. Chem. 241, 5026–5036. [PubMed] [Google Scholar]
- Li H.; Samouilov A.; Liu X.; Zweier J. L. (2001) Characterization of the magnitude and kinetics of xanthine oxidase-catalyzed nitrite reduction. Evaluation of its role in nitric oxide generation in anoxic tissues. J. Biol. Chem. 276, 24482–24489. [DOI] [PubMed] [Google Scholar]
- Zweier J. L.; Kuppusamy P.; Lutty G. A. (1988) Measurement of endothelial cell free radical generation: Evidence for a central mechanism of free radical injury in postischemic tissues. Proc. Natl. Acad. Sci. U.S.A. 85, 4046–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier J. L.; Broderick R.; Kuppusamy P.; Thompson-Gorman S.; Lutty G. A. (1994) Determination of the mechanism of free radical generation in human aortic endothelial cells exposed to anoxia and reoxygenation. J. Biol. Chem. 269, 24156–24162. [PubMed] [Google Scholar]
- Nishino T. (1994) The conversion of xanthine dehydrogenase to xanthine oxidase and the role of the enzyme in reperfusion injury. J. Biochem. 116, 1–6. [DOI] [PubMed] [Google Scholar]
- Zweier J. L. (1988) Measurement of superoxide-derived free radicals in the reperfused heart. Evidence for a free radical mechanism of reperfusion injury. J. Biol. Chem. 263, 1353–1357. [PubMed] [Google Scholar]
- Zweier J. L.; Kuppusamy P.; Williams R.; Rayburn B. K.; Smith D.; Weisfeldt M. L.; Flaherty J. T. (1989) Measurement and characterization of postischemic free radical generation in the isolated perfused heart. J. Biol. Chem. 264, 18890–18895. [PubMed] [Google Scholar]
- Xia Y.; Khatchikian G.; Zweier J. L. (1996) Adenosine deaminase inhibition prevents free radical-mediated injury in the postischemic heart. J. Biol. Chem. 271, 10096–10102. [DOI] [PubMed] [Google Scholar]
- Nishino T.; Nishino T.; Schopfer L. M.; Massey V. (1989) The reactivity of chicken liver xanthine dehydrogenase with molecular oxygen. J. Biol. Chem. 264, 2518–2527. [PubMed] [Google Scholar]
- Schopfer L. M.; Massey V.; Nishino T. (1988) Rapid reaction studies on the reduction and oxidation of chicken liver xanthine dehydrogenase by the xanthine/urate and NAD/NADH couples. J. Biol. Chem. 263, 13528–13538. [PubMed] [Google Scholar]
- Nishino T.; Okamoto K.; Kawaguchi Y.; Hori H.; Matsumura T.; Eger B. T.; Pai E. F.; Nishino T. (2005) Mechanism of the conversion of xanthine dehydrogenase to xanthine oxidase: Identification of the two cysteine disulfide bonds and crystal structure of a non-convertible rat liver xanthine dehydrogenase mutant. J. Biol. Chem. 280, 24888–24894. [DOI] [PubMed] [Google Scholar]
- Pieper A. A.; Walles T.; Wei G.; Clements E. E.; Verma A.; Snyder S. H.; Zweier J. L. (2000) Myocardial postischemic injury is reduced by polyADPripose polymerase-1 gene disruption. Mol. Med. 6, 271–282. [PMC free article] [PubMed] [Google Scholar]
- Harris C. M.; Massey V. (1997) The reaction of reduced xanthine dehydrogenase with molecular oxygen. Reaction kinetics and measurement of superoxide radical. J. Biol. Chem. 272, 8370–8379. [DOI] [PubMed] [Google Scholar]
- Tischler M. E.; Friedrichs D.; Coll K.; Williamson J. R. (1977) Pyridine nucleotide distributions and enzyme mass action ratios in hepatocytes from fed and starved rats. Arch. Biochem. Biophys. 184, 222–236. [DOI] [PubMed] [Google Scholar]
- Koretsky A. P.; Katz L. A.; Balaban R. S. (1987) Determination of pyridine nucleotide fluorescence from the perfused heart using an internal standard. Am. J. Physiol. 253, H856–H862. [DOI] [PubMed] [Google Scholar]
- Ceconi C.; Bernocchi P.; Boraso A.; Cargnoni A.; Pepi P.; Curello S.; Ferrari R. (2000) New insights on myocardial pyridine nucleotides and thiol redox state in ischemia and reperfusion damage. Cardiovasc. Res. 47, 586–594. [DOI] [PubMed] [Google Scholar]
- Zhao X.; He G.; Chen Y. R.; Pandian R. P.; Kuppusamy P.; Zweier J. L. (2005) Endothelium-derived nitric oxide regulates postischemic myocardial oxygenation and oxygen consumption by modulation of mitochondrial electron transport. Circulation 111, 2966–2972. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.