

Abstract
Intratumor heterogeneity remains a major obstacle to effective cancer therapy and personalized medicine. Current understanding points to differential therapeutic response among subpopulations of tumor cells as a key challenge to successful treatment. To advance our understanding of how this heterogeneity is reflected in cell-to-cell variations in chemosensitivity and expression of drug-resistance proteins, we optimize and apply a new targeted proteomics modality, single-cell western blotting (scWestern), to a human glioblastoma cell line. To acquire both phenotypic and proteomic data on the same, single glioblastoma cells, we integrate high-content imaging prior to the scWestern assays. The scWestern technique supports thousands of concurrent single-cell western blots, with each assay comprised of chemical lysis of single cells seated in microwells, protein electrophoresis from those microwells into a supporting polyacrylamide (PA) gel layer, and in-gel antibody probing. We systematically optimize chemical lysis and subsequent polyacrylamide gel electrophoresis (PAGE) of the single-cell lysate. The scWestern slides are stored for months then reprobed, thus allowing archiving and later analysis as relevant to sparingly limited, longitudinal cell specimens. Imaging and scWestern analysis of single glioblastoma cells dosed with the chemotherapeutic daunomycin showed both apoptotic (cleaved caspase 8- and annexin V-positive) and living cells. Intriguingly, living glioblastoma subpopulations show up-regulation of a multidrug resistant protein, P-glycoprotein (P-gp), suggesting an active drug efflux pump as a potential mechanism of drug resistance. Accordingly, linking of phenotype with targeted protein analysis with single-cell resolution may advance our understanding of drug response in inherently heterogeneous cell populations, such as those anticipated in tumors.
Individual cells within a tumor vary widely with respect to their chemotherapeutic sensitivity. This cell-to-cell heterogeneity is widely acknowledged to play key roles in chemoresistance and tumor recurrence.1,2 Strikingly, even genetically identical cells in the same local environment can exhibit differences in cellular signaling during drug response.3,4 Potential mechanisms for resistance to chemotherapy are diverse and can include expression of drug efflux pumps (possibly by P-glycoprotein, P-gp5,6) and aberrant activation of NF-kB7 to alterations of apoptosis pathways.8 To tease apart these mechanisms, a wide range of cellular assays are employed, including pooled cell population assays (e.g., slab-gel western blot, cytotoxicity assays). While pooled cell assays are widely used, signal from populations of rare, drug-resistant cells can be lost in cell averaging, thus obscuring critical aspects of cell-to-cell variation in drug response.
Novel single-cell tools are emerging to aid the study of drug response.9,10 For example, single-cell gain-of-function or loss-of-function manipulations with synthetic fluorescent-protein fusion readouts are powerful but potentially perturb protein function.2 Nanostructure initiator mass spectrometry (NIMS) is a label-free, matrix-free tool for studying cellular proliferation in single-cells by analyzing endogenous metabolites in response to drug treatment.11 While NIMS is promising, the technique demands an impressive laboratory infrastructure, which hinders widespread access. Microfluidic approaches that assay both signal (e.g., kinase activity) and response with single-cell resolution are also promising;12 however, the throughput remains low.
Standard workhorse single-cell protein analyses include immunocytochemistry (ICC) via fluorescence microscopy and flow cytometry. While these single-cell immunoassays are ubiquitous, they are unfortunately highly dependent on antibody quality and availability. When antibodies to a target do exist, false positive signals arising from antibody cross-reactivity are a risk.13,14 To mitigate antibody probe cross-reactivity, western blotting has become a workhorse tool, a multistep assay that relies on antibody binding for detection, typically after an upstream protein sizing step that affords enhanced assay specificity. Yet, conventional western blotting does not offer single-cell resolution.
To bring the selectivity of western blotting to single-cell analyses, we have recently introduced single-cell western blotting (scWestern) for simultaneously assaying thousands of cells, with single-cell resolution, in a 4-h workflow.16 The scWestern comprises an array of thousands of microwells stippled into a thin layer of photoactive polyacrylamide (PA) gel. Cells are settled into the microwells and chemically lysed in situ, and then each single-cell lysate is subjected to PA gel electrophoresis (PAGE) and UV-initiated blotting (immobilization via benzophenone methacrylamide monomer cross-linked into the gel). The lysates are then probed with fluorescently labeled antibodies and scanned on a conventional microarray scanner. scWestern supports multiplexed detection of protein targets, including intracellular proteins (i.e., 9 serial stripping and reprobing cycles using 11 antibody sets16). Here, we illustrate the robustness of archival scWestern slides to long-term storage and subsequent reprobing, thus providing the opportunity for study of new protein targets (in the original cell sample) as biological hypotheses mature. The multiplexing capabilities coupled with the unique archival qualities make scWestern an alternative bench top to flow cytometry as a single-cell resolution protein analysis tool.
Here, we interface the scWestern assay with intact whole-cell imaging, thus facilitating imaging-based phenotypic assessment of live cells (in the microwells). Phenotypic data is then correlated with subsequent protein analyses to characterize cell-to-cell variation in drug response in a culture model of human glioblastoma multiforme (GBM). As is needed to understand drug resistance, we conduct phenotype imaging via phase contrast imaging, DNA staining for cell identification, daunomycin (DNR) imaging to assess drug uptake, and Annexin V signal to assess cell viability. We optimize the scWestern assay, including cell lysis, PAGE, and archiving, and then directly correlate drug uptake and cell viability and protein targets (cleaved caspase 8, P-gP) with single-cell resolution, a capability that may help elucidate the molecular mechanisms of drug treatment failure, thus informing treatment regimens.
Experimental Section
Chemicals
Daunomycin (D8809-1MG), tetramethylethylenediamine (TEMED, T9281), ammonium persulfate (APS, A3678), β-mercaptoethanol (M3148), and 30%T, 2.7%C acrylamide/bis-acrylamide (37.5:1) (A3699) were purchased from Sigma-Aldrich. Triton X-100 (BP-151) was purchased from ThermoFisher Scientific. Premixed 10× Tris/glycine/SDS electrophoresis buffer (25 mM Tris, pH 8.3; 192 mM glycine; 0.1% SDS) was purchased from BioRad. Deionized water (18.2 MΩ) was obtained using an Ultrapure water system from Millipore. N-[3-[(3-Benzoylphenyl)formamido]propyl] methacrylamide (BPMAC) was custom synthesized by PharmAgra Laboratories. Note that the BPMAC used in the previous studies was synthesized in-house17,18 and is a positional isomer (para-form, N-[3-[(4-benzoylphenyl)formamido]propyl] methacrylamide) of the BPMAC used in the present study.
Antibodies
Antibodies employed for system characterization include goat anti-GAPDH (1:10, SAB25000450, Sigma, with anti-goat secondary antibody conjugated with Alexa Fluor 555), rabbit anti-β-tubulin (1:10, ab6046, Abcam, with anti-rabbit secondary antibody conjugated with Alexa Fluor 647 or 594), mouse anti-GAPDH (1:10, GTX627408, Genetex, with anti-rabbit secondary antibody conjugated with Alexa Fluor 647), rabbit anti-cleaved caspase 8 (1:10, 9496, Cell Signaling, with anti-rabbit secondary antibody conjugated with Alexa Fluor 647), and rabbit anti-P-glycoprotein (1:10, ab168336, Abcam, with anti-rabbit secondary antibody conjugated with Alexa Fluor 647).
Cell Lines
U373 MG human glioblastoma cells were obtained from the UC Berkeley Tissue Culture Facility via the American Type Culture Collection (ATCC) and maintained in high glucose DMEM (11965, Life Technologies) supplemented with 1 mM sodium pyruvate (11360-070, Life Technologies), 1× MEM nonessential amino acids (11140050, Life Technologies), 1% penicillin/streptomycin (15140122, Invitrogen), and 10% of calf serum (JR Scientific) and maintained in a humidified 37 °C incubator with 5% CO2. For protein diffusion experiments, U373 MG cells stably transduced with GFP by lentiviral infection (multiplicity of infection = 10) were kindly provided by Dr. Ching-Wei Chang in Prof. S. Kumar’s Laboratory. Both U373 MG and U373-GFP cells were maintained in the same media. We note that ATCC U373 MG cells have been discovered to share a common origin with the human glioblastoma line U251 MG, but that the two lines appear to have diverged to exhibit distinct karyotypes and drug sensitivities.19
DNR-Induced Cytotoxicity Assay
DNR cytotoxicity was assessed via a WST-1 cell proliferation assay (5015944, Roche). After seeding in 96-well plates and culturing overnight, the cells were incubated with DNR (with a final concentration ranging from 0.001 to 5 μM, diluted by complete culture media) for 24 h. The cells were incubated with fresh serum-free medium containing 0.5 mg/mL WST-1 for 1 h at 37 °C for the cytotoxicity assay. The absorbance at 450 nm was measured using a microplate reader (uQuant, Biotek). For apoptotic-cell staining, the cells were cultured overnight, treated with DNR (5 μM) for 24 h, harvested, and then resuspended in Annexin V binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2, pH 7.4). The cell suspensions were stained with Annexin V (Alexa Fluor 647 conjugate, A23204, Life Technologies) for 15 min at room temperature, stopped by adding Annexin V binding buffer, and cells were then directly settled on scWestern slides.
SU8 Wafer and scWestern Slide Fabrication
The SU8 wafer and scWestern slide fabrication were performed as detailed previously.16 For the purified protein solution calibration experiments, microwell feature heights and diameters were 60 and 100 μm, respectively. For experiments utilizing cells, microwell feature heights and diameters were 30 and 32 μm, respectively. The PA gel layer on the scWestern slides was chemically polymerized using 0.08% APS and 0.08% TEMED.
scWestern
The scWestern blotting procedure comprises six steps.16 First, 1 million U373 cells are gravity-settled onto the scWestern slide, followed by washing three times by PBS to remove excess cells off the surface. Second, in situ cell lysis is performed by directly pouring the lysis buffer over the slide. Third, lysate is analyzed via PAGE (E = 40 V cm–1). Fourth, protein bands are immobilized by UV activation of the benzophenone (Lightningcure LC5, Hamamatsu). Fifth, the scWestern slides are diffusion-probed by antibodies and finally scanned by a fluorescence microarray scanner (Genepix 4300A, Molecular Devices).
Purified Protein Experiments
Alexa Fluor 488-labeled purified trypsin inhibitor (TI, T23011, 20 kDa) and Alexa Fluor 555-labeled purified bovine serum albumin (BSA, A34786, 65 kDa) were purchased from Life Technologies. The purified proteins were diluted to a final concentration of 1 μM in PBS. We incubated the purified protein with different %T of PA for 30 min and then performed PAGE. After UV immobilization of electrophoresed proteins, the gel slides were washed in TBST for 30 min, air-dried and scanned via the fluorescence microarray scanner.
Fluorescence Imaging
Real-time cell lysis was imaged using a time-lapse acquisition mode controlled by MetaMorph software (Molecular Devices) with 50 ms exposure times, 500 ms time intervals, at 1 × 1 pixel binning through a 10× magnification objective (Olympus UPlanFLN, NA 0.45) on an Olympus IX71 inverted fluorescence microscope equipped with an Andor iXon+ EMCCD camera, ASI motorized stage, and shuttered mercury lamp light source (X-cite, Lumen Dynamics). The first four images were discarded owing to the fluctuation from addition of lysis buffer. Fluorescence signal from a region of interest (ROI) covering the whole microwell was integrated at intervals during lysis. Fluorescence signal from an adjacent, empty microwell was assigned as the background signal. Each integrated intensity value was background-subtracted and normalized to the value at the start of lysis (t = 0). For high-content imaging, the entire microscope slide was imaged using a multidimensional acquisition mode controlled by MetaMorph software and collected through a 4× magnification objective (Olympus UPlanFLN, NA 0.13) on the same microscope. The fluorescence signal was obtained using a standard DAPI filter cube for a DNA stain, Hoechst 33342 (B2261, Sigma), a TRITC filter cube for DNR, and a Cy5 filter cube for Annexin V staining. All images were analyzed by ImageJ 1.46r (NIH). All plots were graphed by OriginPro 8.5.0.
Imaging Processing and Performance Quantification
Quantification of protein PAGE and probing used in-house scripts written in R (http://www.r-project.org).16 Bands widths were characterized by Gaussian curve fitting in MATLAB (R2013b, Curve Fitting Toolbox).
Results and Discussion
In-Gel Probing and High-Performance PAGE
The scWestern architecture comprises an array of microwells stippled into a thin layer (30 μm) of photoactive PA gel, all layered on a glass microscope slide (Figure 1).16 To yield westerns with single-cell resolution, a suspension of cells is first gravity-settled into the microwells. Microwell diameters are selected to favor one cell per microwell occupancy. High-content imaging of intact cells seated in microwells is completed using both phase contrast and fluorescence imaging. After imaging, the cells are chemically lysed in situ. Immediately following lysis, the lysate from each single cell is subjected to western analysis: PAGE, blotting of resolved proteins via in situ UV-initiated immobilization to the benzophenone PA gel, and probing with primary and then fluorescently labeled secondary antibodies.
Figure 1.
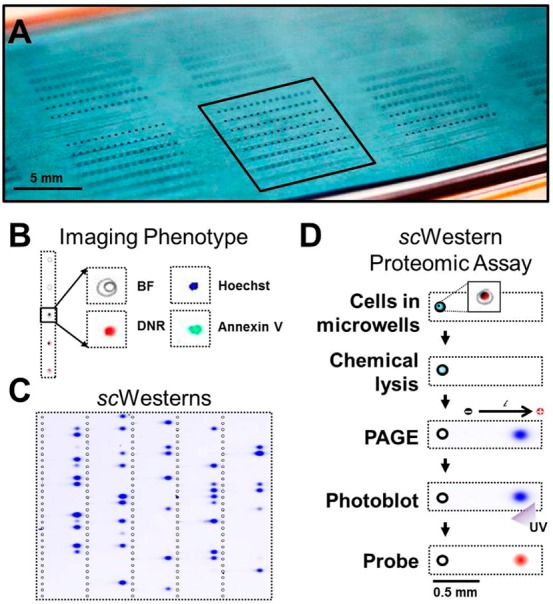
Single-cell phenotype imaging and western blotting workflow. The scWestern array is comprised of a thin layer of photoactive PA gel that (A) houses single-cells in microwells, (B) enables fluorescence and phase contrast imaging of intact cells providing information on cell phenotype, and (C, D) supports subsequent western blotting steps after in situ cell lysis for proteomic analyses.
Importantly, the microwell array is designed such that the pitch between each row of microwells is defined by the length of the PAGE separation axis. This spacing constraint presents a trade-off between PAGE separation resolution, which benefits from long separation distances, and array density, which allows analysis of larger number of single cells. Given a fixed maximum operating power for the high voltage power supplies employed, we sought to design a scWestern with (1) a short, 1 mm long separation axis for each PAGE assay and (2) PAGE performance suitable for analysis of proteins in the 15 to 90 kDa range, thus making the scWestern broadly relevant to cytosolic signaling proteins (Figure S-1 in the Supporting Information).20 To meet these two design specifications, we first investigated the selectivity imparted by the molecular sieving matrix as a function of the PA gel pore size. While gels with smaller pore sizes confer enhanced sizing discrimination and selectivity,21,22 decreasing the gel pore size below a critical value will adversely impact antibody transport into the gel during the probing steps.
Accordingly, to achieve high separation performance we assessed PAGE separation resolution (SR) (Figure 2 and Figure S-2 in the Supporting Information). PA gel densities spanning from 7%T to 10%T (%T, total acrylamide) were assessed using a purified, fluorescently labeled protein ladder (BSA and TI). Full-field, single time-point imaging was employed; thus PAGE was suspended when the highest mobility species (TI) reached the end of the 1 mm separation distance. As such, comparisons are between PAGE assays having the same separation distance (1 mm) but different separation durations.
Figure 2.
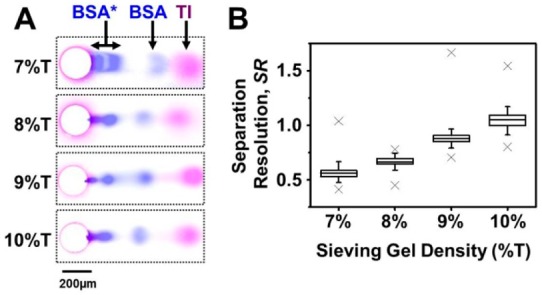
Microscale PAGE performance improves with smaller PA gel pore size. (A) False-color fluorescence micrographs of a 1 mm long PAGE separation distance for a range of PA gel densities. A purified fluorescently labeled two-component protein standard was used: TI (magenta signal) and BSA (blue signal). PAGE was halted when the fastest standard species reached the separation axis terminus (21 s, 7%T; 20 s, 8%T; 23 s, 9%T; 26 s 10%T). Highlighted circular features indicate 30 μm diameter microwells. (B) SR improves as gel pore size is reduced. Box ends indicate 25th and 75th quantiles; line at box middle indicates median value; whiskers extend to 95% confidence limits; and outliers marked with “×”. SR in the 10%T PA gel was significantly higher than that at 9%T PA gel (t test, two tailed, p-value = 2.72 × 10–20). E = 40 V cm–1.
PAGE analysis of BSA revealed both high and low electrophoretic mobility forms, with the lower mobility species likely a covalent dimer of BSA (BSA*; 130 kDa).16,20 As PAGE was designed to analyze the 15–90 kDa molecular mass range relevant to most cytosolic proteins, we observed retention of the large BSA multimer near the edge of the microwell. For the faster mobility species (TI and the highest mobility BSA form), we observed notably more disperse bands in the 7%T PA gel (FWHMTI = 58.9 ± 10.7 μm and FWHMBSA = 35.2 ± 8.1 μm, ± SD, n = 57 separations) as compared to the smaller pore-size 10%T gel (FWHMTI = 44.22 ± 6.91 μm and FWHMBSA = 24.2 ± 4.9 μm, ± SD, n = 103 separations). Accordingly, a marked increase in SR was observed with the smaller pore-size gels (Figure 2B). In particular, while the BSA and TI were not resolved in a 7%T gel (SR = 0.56 ± 0.08, ± SD, n = 57 separations), the species were resolved in the smallest pore-size gel (10%T gel; SR = 1.04 ± 0.09, ± SD, n = 103 separations) with all other conditions comparable. As anticipated for this molecular mass range, higher separation performance was observed with the smallest pore-size gel (10%T). We did not explore lower pore-size gels, as a 150 kDa antibody is ∼30 nm in size23 and gel pore sizes of ∼20 nm are expected in 10.5%T, 5%C PA gels.24 In addition to pore size exclusion limitations, partitioning reduces the local, in-gel antibody concentration compared to the bulk solution.25 A locally lower antibody concentration yields less than optimal probing performance. Taken together, a 10%T PA gel proved suitable for scWestern assays (including PAGE performance and antibody probing) for a broad range of cytosolic signaling proteins.
Microwell in Situ Chemical Lysis of U373 MG Cells
The scWestern workflow dictates that the cell lysis and protein extraction buffer act also as the electrophoresis run buffer. Thus, a single buffer must provide both effective cytoplasmic protein extraction and a low conductivity (to support electrophoresis). To meet these constraints, a modified RIPA buffer16 was employed for cell lysis, protein extraction, and subsequent electrophoresis. The 12 mM Tris/96 mM glycine (pH 8.3) buffer contained 0.5% SDS, 0.1% v/v Triton X-100, and 0.25% sodium deoxycholate. The modified RIPA buffer was poured directly onto the scWestern surface. In comparison, a standard RIPA buffer is 25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 0.1% SDS, 1% NP-40 (nonionic detergent), and 1% sodium deoxycholate. In conventional western blotting, extracted proteins are typically heat denatured (5–10 min) in the presence of reducing reagents prior to protein sizing. In contrast, the multifunctional scWestern lysis and electrophoresis buffer provides denaturing but nonreducing conditions, with a short lysis period (20 s) to minimize protein diffusional losses from the shallow microwells.16,17 The 20 s in situ chemical lysis period is not expected to impact levels of P-glycoprotein (employed in the drug study) owing to an estimated 16 h protein half-life (4 h mRNA half-life).26 While a 20 s lysis period may perturb signaling for species with short signaling time scales, a range of “fast” induced signaling systems are understood to change the protein modification on the order of minutes, not seconds (e.g., phosphorylation of AKT and ERK1/2 after 4 min of EGF stimulation27 or activation of Src by EGF stimulation28).
To enhance cell lysis and protein extraction efficiency without the addition of high conductivity species, we explored cell lysis at elevated temperatures (50 °C using the modified RIPA buffer, Figure 3). While 95 °C is typically used for in-tube cell lysate preparation, minimizing lysate diffusion out of the 30-μm deep microwells during lysis is a design constraint of the scWestern. Elevated temperature enhances diffusion. Most proteins are denatured at 50 °C.29−31
Figure 3.
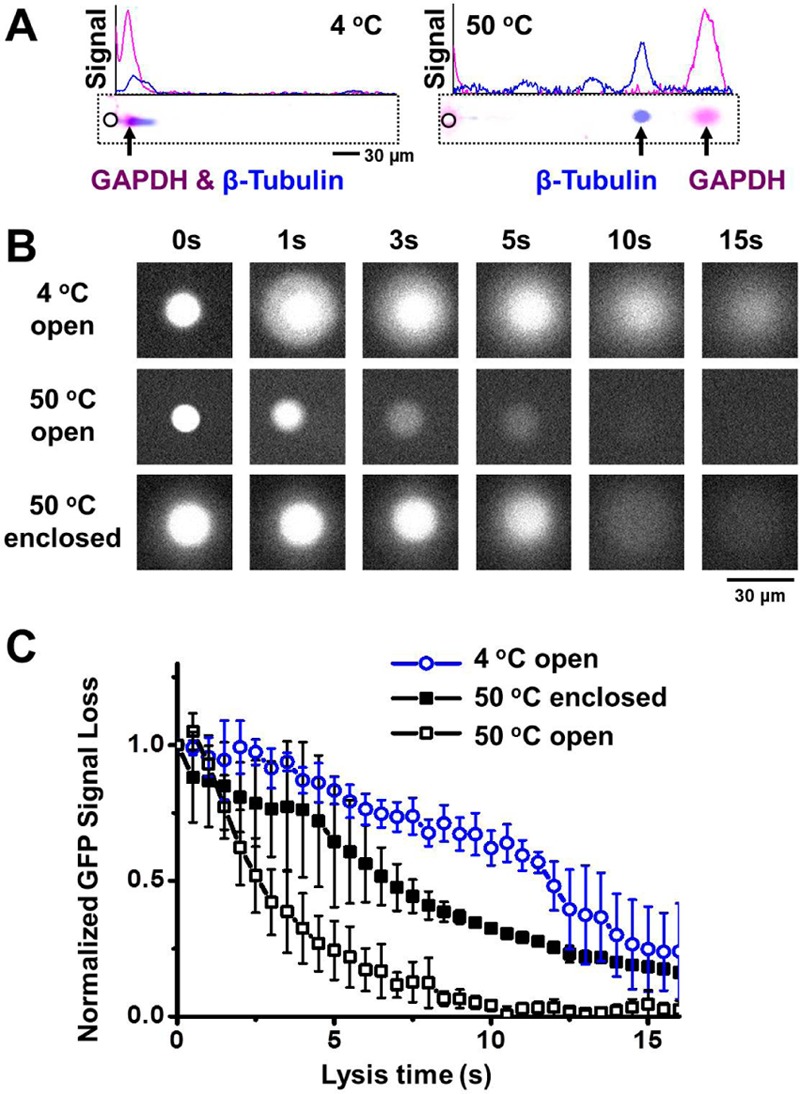
In situ chemical lysis of cells in microwells at elevated temperatures and with enclosed microwells notably improves scWestern performance. (A) False-color fluorescence micrographs and intensity profiles show scWesterns after cool (left, 4 °C) and hot (right, 50 °C) lysis buffer conditions, suggesting a fully resolved protein pair, GAPDH (magenta signal) and β-tubulin (blue signal) under the 50 °C lysis conditions. (B) Fluorescence micrographs during in-microwell lysis of U373-GFP cells under 4 and 50 °C lysis conditions in systems with (enclosed) and without (open) a lid covering the microwell, suggest an enclosed microwell architecture can mitigate protein losses during cell lysis. (C) Time course of the total integrated GFP fluorescence signal from each microwell in part B. Error bars from 3 to 5 independent experiments indicate standard deviation. E = 40 V cm–1, lysis time = 20 s, electrophoresis time = 40 s, 10%T PA gel. (GAPDH, Alexa Fluor 555-labeled secondary antibody; β-tubulin, Alexa Fluor 647-labeled secondary antibody).
To understand the impact of lysis buffer temperature on scWestern performance, we scrutinized (i) downstream PAGE performance and (ii) diffusional losses from the microwell for the different lysis buffer temperature conditions. Glioblastoma (U373 MG) cells were gravity-settled into microwells on a scWestern slide, subjected to either a 4 or 50 °C modified RIPA lysis/extraction/electrophoresis buffer for 20 s and extracted proteins then analyzed by protein PAGE for 40 s (1 mm long separation distance, 10%T PA gel). Lysis buffer temperature was measured with a thermometer immediately before buffer was poured on the scWestern surface. We assayed two widely used internal control proteins, β-tubulin (55 kDa) and GAPDH (35 kDa) in the U373 MG (Figure 3A and Figure S-3 in the Supporting Information).
The scWestern yielded a fully resolved peak pair with SR = 1.36 ± 0.73 (± SD, n = 70 cells) under the 50 °C lysis conditions. In contrast, the 4 °C lysis condition yielded unresolvable peaks for β-tubulin and GAPDH. In the 4 °C lysis condition, the apparent electrophoretic mobility of β-tubulin was 0.44 ± 0.05 × 10–5 cm2 V–1 s–1 (± SD, n = 11 cells) and that of GAPDH was 0.26 ± 0.06 × 10–5 cm2 V–1 s–1 (± SD, n = 11 cells). Under the 50 °C cell lysis condition, the apparent electrophoretic mobilities were 6.60 ± 0.08 × 10–5 cm2 V–1 s–1 (± SD, n = 11 cells) and 8.88 ± 0.14 × 10–5 cm2 V–1 s–1 (± SD, n = 11 cells) for β-tubulin and GAPDH, respectively. Intriguingly, the 50 °C lysis conditions notably improved the electrophoretic injection of both protein standards into the 10%T PA gel. Further, when comparing the 4 and 50 °C conditions, the apparent electrophoretic mobility of the GAPDH “over-speeds” that of the β-tubulin. The exceedingly low mobility of GAPDH in the 4 °C conditions may be attributable to non fully dissociated and non fully denatured native tetramer forms of GAPDH.32 Accordingly, the 50 °C lysis condition resulted in notably less dispersion during PAGE (Figure S-4 in the Supporting Information). As high melting temperature proteins (e.g., proteins with disulfide bonds) may not be fully denatured at 50 °C,33 the addition of DTT, β-mercaptoenthanol, or increased concentrations of SDS to the lysis buffer could be considered if inadequate electrophoretic separation performance is observed.
Second, we assessed protein losses from the microwells under the 4 and 50 °C lysis buffer conditions. As a model cell system, we employed a green fluorescent protein (GFP) transfected U373 MG cell line (U373-GFP). GFP (27 kDa) acts as a marker of small molecular mass cytosolic proteins during lysis. Importantly, GFP is expected to emit fluorescence signal under both lysis temperature conditions considered and with SDS treatment (up to 0.5%) at pH 8.5.34−36 The effects of heating and photobleaching on GFP were minimal (Figure S-5 in the Supporting Information).
Comparison of the in-microwell GFP signal after introduction of the lysis buffer shows substantially faster transport of the GFP out of the microwell under the 50 °C lysis conditions, as compared to the 4 °C lysis conditions (Figure 3B). Here, the initial GFP fluorescence signal (prior to cell lysis, t = 0) was normalized to unity. Notably, for the 4 °C lysis condition the GFP fluorescence signal remained above 50% of the initial signal at even 10 s of lysis duration (Figure 3C). However, the GFP fluorescence signal dipped below 50% of the initial signal after just 3 s of lysis with the 50 °C buffer conditions. By 10 s of lysis time, the GFP signal was <10% of the initial signal under the 50 °C lysis buffer conditions.
We next measured GFP fluorescence signal after lysis (20 s at either 4 or 50 °C), protein extraction, and PAGE (Figure S-6 in the Supporting Information). Strong GFP fluorescence was observed after PAGE and immobilization under the 4 °C lysis buffer conditions. In contrast, the GFP fluorescence was almost undetectable after PAGE and UV immobilization under the 50 °C lysis buffer conditions while internal control proteins (GAPDH and β-tubulin) with larger molecular mass than GFP were detectable (Figure 3A). Comparison of the two lysis temperatures across multiple U373-GFP cells revealed that the 4 °C lysis condition (n = 30 cells) yielded a 9-fold higher average GFP signal than that measured under the 50 °C condition (n = 48 cells) (Figure S-6 in the Supporting Information).
We attribute the lower measured signal at elevated temperatures to effective cell lysis with enhanced diffusional losses out of the microwells during the process. We examined the effectiveness of cell lysis and diffusional losses at an intermediate lysis buffer temperature (37 °C, in comparison to a lower temperature 4 °C condition and an elevated temperature 50 °C condition, Figure S-7 in the Supporting Information). As expected, we observed a reduction in diffusional losses of GFP at 37 °C offset by protein extraction and solubilization that yielded a suboptimal subsequent electrophoretic separation of GAPDH and β-tubulin. Consequently, we introduced a lid to enclose the microwell and limit protein mass losses during the lysis process (Figure 3B, bottom panel). The lid-based system is detailed in the Supporting Information (Figure S-8). Use of the lid system improved retention of GFP in the microwell, as compared to the open microwell system.
Integration of High-Content Imaging with scWesterns
We sought to develop scWesterns suitable for the simultaneous study of both drug-induced single-cell phenotypes and targeted proteomics. To accomplish imaging and single-cell protein analysis, we applied imaging (bright-field, fluorescence) and subsequent endpoint scWesterns to study cell-to-cell variation in the response of U373 MG cells to the small-molecule chemotherapeutic daunomycin (a.k.a., daunorubicin, DNR). We assessed the response to DNR treatment using the WST-1 proliferation assay (Figure S-9 in the Supporting Information). U373 MG cells were stimulated with 5 μM DNR treatment for 24 h in a standard culture system, then collected and assessed for apoptosis (via staining, Annexin V conjugated with Alexa Fluor 647). Finally, both attached and buoyant cells (the latter being likely apoptotic) were collected and settled on the scWestern.
We collected bright-field and fluorescence images of control U373 MG cells and DNR-treated U373 MG cells with both positive and negative Annexin V staining (Figure 4A). Immediately after imaging, we performed scWestern blotting and probed for GAPDH, an internal control protein, and cleaved caspase 8, an apoptosis marker37 (Figure 4A and Figure S-10 in the Supporting Information). We observed a positive correlation between the two apoptotic markers, annexin V and cleaved caspase 8 (Pearson correlation coefficient, r = 0.98755, n = 31 cells), confirming a strong association between phenotype and protein expression at the single-cell level (Figure 4B). We also estimated a false positive rate of less than 1.0% (1 single-cell western blot registered as empty during imaging yet reported a positive protein peak upon western blotting, see Figure S-11 in the Supporting Information). False positives may arise from diffusional “cross-talk” between occupied wells and neighboring empty wells. The false negative rate is estimated at ∼30% (6 microwells registered imaging data yet no result upon western blotting). The false negative rate is attributed to diffusional losses of material from occupied wells.
Figure 4.
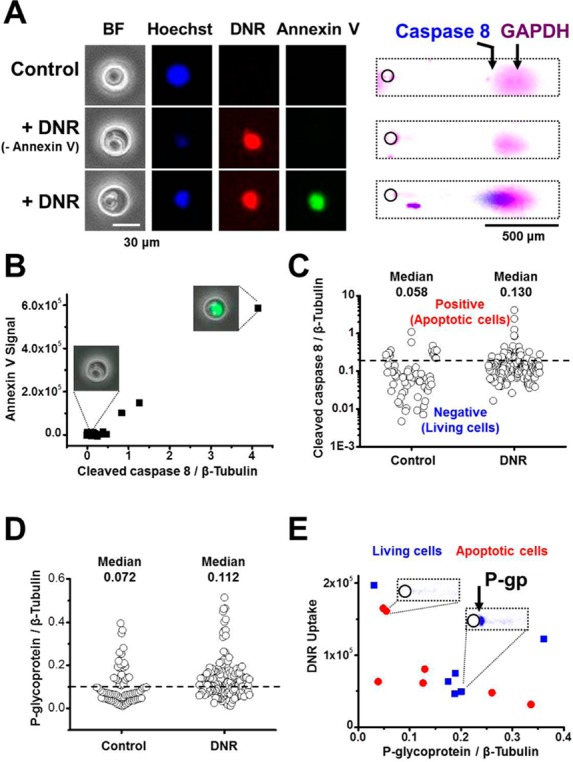
Integration of high-content imaging with scWesterns. (A) Micrograph images and scWesterns of a control cell (upper panel), DNR-treated living cell (middle panel), and DNR-treated apoptotic cell (lower panel). The living cells showed no annexin V staining and cleaved caspase 8 signal, whereas the apoptotic cells showed positive annexin V and cleaved caspase 8 signal. (B) Positive correlation between the apoptotic markers: cleaved caspase 8 and annexin V signal (r = 0.98755). (C) The distribution of cleaved caspase 8-negative and cleaved caspase 8-positive between control and DNR-treated cells defines the positive population of apoptotic cells. Each cleaved caspase 8 signal is normalized to its β-tubulin signal. The dashed line is the 0.2 threshold (control, n = 79 cells; DNR, n = 153 cells). (D) The shifted distribution of P-gp expression in DNR-treated cells compared to control cells suggests the possible existence of adaptive resistance after drug treatment. Each P-gp signal is normalized to its β-tubulin signal. The dashed line is the 0.1 threshold (control, n = 73 cells; DNR, n = 141 cells). (E) Scatter plot of apoptotic (red dots) and living (blue dots) cells as well as P-gp and DNR uptake shows more living cells with moderate to high P-gp expression. E = 40 V cm–1, lysis time = 20 s, electrophoresis time = 30 s, 10%T PA gel.
We further examined the distribution of cleaved caspase 8 signals between control and DNR-treated U373 MG cells (Figure 4C). On the basis of the distribution of cleaved caspase 8 signal in control U373 MG cells (Figure S-12 in the Supporting Information), we defined a fluorescence signal threshold as 0.2 (the ratio of cleaved caspase 8 to β-tubulin) and further identified 20% apoptotic cells (n = 31/153 cells) in DNR-treated U373 MG cells. Intriguingly, while examining the correlation between DNR uptake and cleaved caspase 8 signal (Figure S-13 in the Supporting Information), we observed that an increase in DNR uptake did not positively correlate with cell apoptosis. For example, the DNR-treated U373 MG cell indicated as cell number 2 had high DNR intensity but was a living cell with no detectable cleaved caspase 8 signal or annexin V signal detectable (Figure S-13 in the Supporting Information).
One possible mechanism for drug resistance in cancer cells is the active efflux of anticancer drugs through the cellular membrane by multidrug resistance proteins, such as the ATP-binding cassette (ABC) transporter family, P-glycoprotein (P-gp).38 Of particular interest, chemotherapy resistance to anthracycline based drugs (e.g., doxorubicin and vincristine) has been attributed to up-regulation of P-gp.39 As such, we employed the scWesterns to assay P-gp expression in DNR-treated U373 MG cells. The size of probed P-gp is 140 kDa, which is excluded from the small pore-size 10%T gel and thus is clearly retained near the edge of the microwells (Figure S-14 in the Supporting Information). On the basis of the scWestern results, we compared P-gp expression between control and DNR-treated U373 MG cells (Figure 4D) and found that the two groups expressed P-gp at significantly different levels (p-value = 0.007, two-tailed t-test; control, n = 73 cells; DNR, n = 141 cells). On the basis of the distribution of P-gp expression in control and DNR-treated U373 MG cells (Figure S-15 in the Supporting Information), we defined a fluorescence signal threshold as 0.1 (the ratio of P-gp to β-tubulin) and further identified 62% cells (n = 87/141 cells) expressing high P-gp levels in DNR-treated U373 MG cells; whereas only 33% cells expressing high P-gp levels (n = 24/73 cells) in control U373 MG cells. The observation suggests the possible existence of adaptive resistance after drug treatment.40,41
As a drug efflux pump, an inverse correlation between drug uptake and P-gp is expected if P-gp is the major drug resistance mechanism. Drug-resistant viable cells are hypothesized to express high levels of P-gp with low DNR uptake, while drug-sensitive apoptotic cells would express the converse (low levels of P-gp expression with high DNR uptake). After DNR treatment, we analyzed the correlation between DNR uptake and P-gp expression in living (cleaved caspase 8-negative) and apoptotic (cleaved caspase 8-positive) cells (Figure 4E). As expected, we observed living cells exhibiting moderate to high P-gp expression and relatively low DNR uptake. The observation suggests the possible existence of a P-gp dependent drug resistant population.
Intriguingly, scWestern analysis also identified a subpopulation of living cells with high DNR uptake and relatively low P-gp expression. Statistical comparison found no correlation between P-gp expression and drug uptake (Pearson correlation coefficient r = −0.4559, p-value = 0.1, two-tailed t-test against the null hypothesis). Our working conclusion finds P-gp is not a major contributor to drug uptake or drug resistance under these conditions. Rare events, such as living cells with high DNR uptake, may be attributable to a non-P-gp associated drug resistance mechanism through, for example, the overexpression of an antiapoptotic protein, Bcl-2,8,38 and is a topic of future inquiry.
Archiving of scWestern Slides
Given the findings of the high-content imaging and scWestern tool regarding non-P-gp drug resistance mechanisms, we sought to facilitate future reanalysis of scWestern slides. Consequently, we evaluated long-term storage for archiving and subsequent reprobing of scWestern slides (Figure 5). To explore archival capability, scWesterns were conducted, probed with GAPDH, rinsed with deionized water, and air-dried before storage. We observed that the thickness of a 10%T PA gel decreases by ∼10% after archival preparation (Figure 5A). We evaluated the GADPH antibody probe fluorescence signal before and after 4 months of archival storage at room temperature (Figure 5B and Figure S-16 in the Supporting Information). The integrated total fluorescence of probed GAPDH after the 4-month storage period was 83.8 ± 8.9% (± SD, n = 28 cells) of the signal before archival storage. While archival storage of probed scWestern slides is feasible, we further attempted to reprobe and strip β-tubulin at a scWestern slide after 7 months of storage (Figure 5C and Figure S-17 in the Supporting Information). For chemical stripping, slides were incubated for 3 h in 55 °C stripping buffer (2.5% SDS and 1% β-mercaptoethanol in 62.5 mM Tris titrated to pH 6.8 with HCl). We observed strong stripping with just 5.3 ± 1.6% (± SD, n = 426 cells) of the initial β-tubulin antibody probe signal retained on the gel (Figure S-17 in the Supporting Information). The apparent probed β-tubulin signal and the subsequent strong stripping suggest the feasibility of reprobing a scWestern slide after long-term archival storage. The robustness of scWestern slides affords the opportunity for probing of new protein targets in the future, as is especially critical in analysis of sparingly available longitudinal clinical biospecimens.
Figure 5.
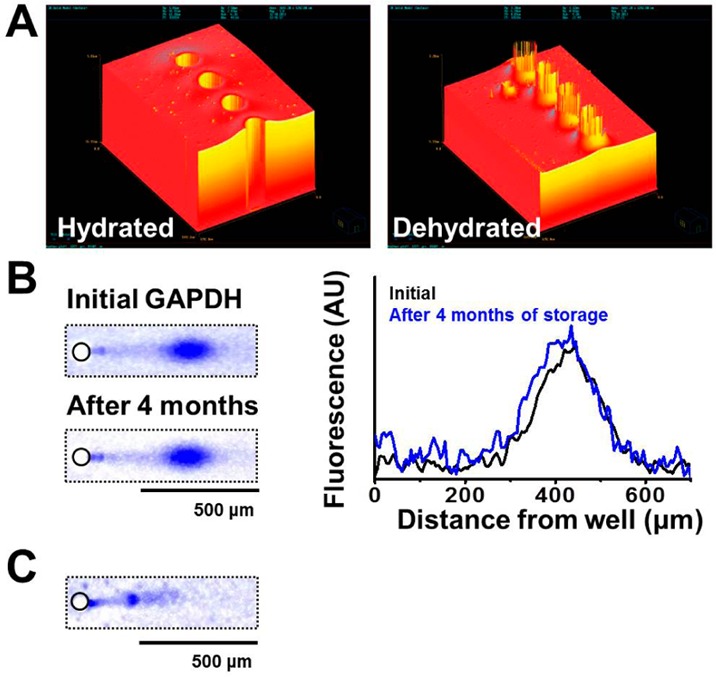
Archival storage of scWestern slides with subsequent successful antibody probing. (A) Optical profilometer images of hydrated and dehydrated scWestern PA gel features. Circular features are microwells, 30 μm in diameter. (B) False-color fluorescence micrographs and intensity traces of the same scWestern device showing antibody probing for GAPDH (blue signal) after fresh preparation and after 4 months of archival storage. (C) Fluorescence micrographs of the same scWestern device reprobed for β-tubulin (blue signal). AU, arbitrary unit. E = 40 V cm–1, lysis time = 25 s, electrophoresis time = 20 s, 12%T PA gel (GAPDH, Alexa Fluor 647-labeled secondary antibody; β-tubulin, Alexa Fluor 594-labeled secondary antibody).
Conclusions
Recent strides to enhance our understanding of cancer have been primarily focused on characterizing genetic aberration to tumor initiation or progression at the genomic or transcriptomic level. With improvements of DNA sequencing technologies, large-scale collaborative initiatives, such as The Cancer Genome Atlas (TCGA), have extensively mapped genotype to phenotype in different cancers.42,43 For example, the integrated information from copy number alterations, exome sequencing, and mRNA assays have helped define breast cancer subtypes and further discover druggable targets.42 However, we know that the proteomic states are dynamic, and crosstalk among proteins occurs frequently in signaling cascades.44 Especially for single time point experiments, findings suggest that fewer than 50% of proteins with measurable changes in abundance are accompanied by changes in the corresponding mRNA.45 Therefore, direct association of proteotype to phenotype is of importance.
Integrated with intact cell imaging, the scWestern platform provides high-specificity protein measurement with single-cell resolution, as it is directly relevant to the study of cell-to-cell variation in drug response. Advances in assay design and development focus first on addressing the coupled requirements of effective molecular sieving by the PA gel and effective subsequent in-gel antibody probing of target proteins in that same gel. A second focus centers on reducing lysate losses from the microwells during the chemical lysis process. A lid-based system is observed to confine lysate to the microwells for a longer duration than possible with open microwells. With enhanced scWestern performance, the scWestern microarray format is integrated with high-content imaging of intact cells in the microwells prior to blotting. Integration of intact single-cell imaging allows direct correlation between phenotypic outcomes and proteomic profiles from the same cell. The multistage, single-cell assay reported here offers a throughput in the electrophoresis stage (5 min handling time) of ∼30 cells/min demonstrated for 153 cells total (Figure 4). A larger or denser microwell array would allow electrophoretic analysis of a larger number of single cells in the same total assay duration. When coupled with the subsequent blotting and probing, a 4 h rate limiting step, that comprise western blotting, the scWestern throughput is ∼0.6 cells/min. This conservative throughput can be easily scaled up by probing multiple slides simultaneously. When integrated with high-content imaging of intact cells prior to electrophoresis, the assay throughput falls considerably to ∼0.1 cells/min, as would be expected without the use of a high-speed live-cell imaging system.
To shed light on a possible mechanism for cancer drug resistance involving the active efflux of anticancer drugs through the cell membrane by P-gp, we employ the scWestern to assay P-gp in DNR-treated glioblastoma cell line (U373 MG). Correlation of intact cell imaging data with single-cell resolution proteomic data may provide a means for identifying small subpopulations of cells that exhibit drug resistance. While single-cell analyses, such as the scWestern, detect rare phenotypes in a population of cancer cells, the importance of those rare and often hard-to-replicate measurements is still being assessed (Figure S-18 in the Supporting Information). Nevertheless, the integration of high-content imaging with high specificity scWesterns offers a new tool for measuring cell-to-cell variation in response to drug treatment.
Acknowledgments
We gratefully acknowledge discussion with Herr Lab members and alumni. We further acknowledge financial support from NIH New Innovator Award (Grant DP2OD007294 to A.E.H.), QB3/Rogers Family Foundation “Bridging-the-Gap” Award (to A.E.H.), NSF Award (Grant CMMI PESO1105539 to S.K.) and IMAT Award (Grant 1R21CA174573 to S.K.). J.M.L. is supported by the UC Berkeley NIH T32 Stem Cell Engineering Training Program. A.E.H. is an Alfred P. Sloan research fellow in chemistry.
Supporting Information Available
Full blocks of scWestern blots, GFP control experiment, WST-1 proliferation assay, and cleaved caspase 8 histogram. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
C.-C.K., J.-M.L., S.K., and A.E.H. designed the experiments. C.-C.K. and J.-M.L. performed the cell culture and scWesterns. Z.X. designed the software, and C.-C.K., J.-M.L., and Z.X. performed the data analysis. All authors wrote the manuscript. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): C.-C.K., Z.X., and A.E.H. are inventors on pending patents related to scWestern blot methods. A.E.H. holds equity interest in Zephyrus Biosciences.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Niepel M.; Spencer S. L.; Sorger P. K. Curr. Opin Chem. Biol. 2009, 13, 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A. A.; Geva-Zatorsky N.; Eden E.; Frenkel-Morgenstern M.; Issaeva I.; Sigal A.; Milo R.; Cohen-Saidon C.; Liron Y.; Kam Z.; Cohen L.; Danon T.; Perzov N.; Alon U. Science (New York, N.Y) 2008, 322, 1511–1516. [DOI] [PubMed] [Google Scholar]
- Fisher R.; Pusztai L.; Swanton C. Br. J. Cancer 2013, 108, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne K. E.; Taylor S. S. Cancer Cell 2008, 14, 111–122. [DOI] [PubMed] [Google Scholar]
- Vrignaud P.; Londos-Gagliardi D.; Robert J. Oncology 1986, 43, 60–66. [DOI] [PubMed] [Google Scholar]
- Huet S.; Schott B.; Robert J. Br. J. Cancer 1992, 65, 538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotto-Filho A.; Braganhol E.; Schroder R.; de Souza L. H.; Dalmolin R. J.; Pasquali M. A.; Gelain D. P.; Battastini A. M.; Moreira J. C. Biochem. Pharmacol. 2011, 81, 412–424. [DOI] [PubMed] [Google Scholar]
- Eisele G.; Weller M. Cancer Lett. 2013, 332, 335–345. [DOI] [PubMed] [Google Scholar]
- Edwards B. S.; Young S. M.; Saunders M. J.; Bologa C.; Oprea T. I.; Ye R. D.; Prossnitz E. R.; Graves S. W.; Sklar L. A. Exp. Opin. Drug Discovery 2007, 2, 685–696. [DOI] [PubMed] [Google Scholar]
- O’Hara D. M.; Xu Y.; Liang Z.; Reddy M. P.; Wu D. Y.; Litwin V. J. Immunol. Methods 2011, 363, 120–134. [DOI] [PubMed] [Google Scholar]
- O’Brien P. J.; Lee M.; Spilker M. E.; Zhang C. C.; Yan Z.; Nichols T. C.; Li W.; Johnson C. H.; Patti G. J.; Siuzdak G. Cancer Metab. 2013, 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A.; Kolitz S.; Lauffenburger D. A.; Han J. Nat. Commun. 2014, 5, 3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maecker H. T.; Trotter J. Cytometry A 2006, 69, 1037–1042. [DOI] [PubMed] [Google Scholar]
- Stadler C.; Rexhepaj E.; Singan V. R.; Murphy R. F.; Pepperkok R.; Uhlen M.; Simpson J. C.; Lundberg E. Nat. Methods 2013, 10, 315–323. [DOI] [PubMed] [Google Scholar]
- Hughes A. J.; Spelke D. P.; Xu Z.; Kang C. C.; Schaffer D. V.; Herr A. E. Nat. Methods 2014, 11, 749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A. J.; Herr A. E. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 21450–21455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A. J.; Lin R. K.; Peehl D. M.; Herr A. E. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 5972–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanenko A. A.; Kavsan V. M. Gene 2014, 540, 263–265. [DOI] [PubMed] [Google Scholar]
- Shapiro A. L.; Vinuela E.; Maizel J. V. Jr. Biochem. Biophys. Res. Commun. 1967, 28, 815–820. [DOI] [PubMed] [Google Scholar]
- Gallagher S. R. In Current Protocols in Molecular Biology Ausubel F. M.et al. , Eds.; John Wiley & Sons: New York, 2012; Chapter 10, Unit 10 12A. [DOI] [PubMed] [Google Scholar]
- Lo C. T.; Throckmorton D. J.; Singh A. K.; Herr A. E. Lab Chip 2008, 8, 1273–1279. [DOI] [PubMed] [Google Scholar]
- Hansma H. G. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 14678–14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen N. C. Electrophoresis 1998, 19, 1542–1547. [DOI] [PubMed] [Google Scholar]
- Tong J.; Anderson J. L. Biophys. J. 1996, 70, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleman C.; Annereau J. P.; Liang X. J.; Cardarelli C. O.; Taylor B.; Yin J. J.; Aszalos A.; Gottesman M. M. Cancer Res. 2003, 63, 3084–3091. [PubMed] [Google Scholar]
- Henjes F.; Bender C.; von der Heyde S.; Braun L.; Mannsperger H. A.; Schmidt C.; Wiemann S.; Hasmann M.; Aulmann S.; Beissbarth T.; Korf U. Oncogenesis 2012, 1, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na S.; Collin O.; Chowdhury F.; Tay B.; Ouyang M.; Wang Y.; Wang N. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 6626–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaei Tavirani M.; Moghaddamnia S. H.; Ranjbar B.; Amani M.; Marashi S. A. J. Biochem. Mol. Biol. 2006, 39, 530–536. [DOI] [PubMed] [Google Scholar]
- Turi A.; Lu R. C.; Lin P. S. Biochem. Biophys. Res. Commun. 1981, 100, 584–590. [DOI] [PubMed] [Google Scholar]
- Flora K.; Brennan J. D.; Baker G. A.; Doody M. A.; Bright F. V. Biophys. J. 1998, 75, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzola J. L.; Sirover M. A. Biochim. Biophys. Acta 2003, 1622, 50–56. [DOI] [PubMed] [Google Scholar]
- Ku T.; Lu P.; Chan C.; Wang T.; Lai S.; Lyu P.; Hsiao N. Comput. Biol. Chem. 2009, 33, 445–450. [DOI] [PubMed] [Google Scholar]
- Saeed I. A.; Ashraf S. S. Int. J. Biol. Macromol. 2009, 45, 236–241. [DOI] [PubMed] [Google Scholar]
- Alkaabi K. M.; Yafea A.; Ashraf S. S. Appl. Biochem. Biotechnol. 2005, 126, 149–156. [DOI] [PubMed] [Google Scholar]
- Zimmer M. Chem. Rev. 2002, 102, 759–781. [DOI] [PubMed] [Google Scholar]
- Wesselborg S.; Engels I. H.; Rossmann E.; Los M.; Schulze-Osthoff K. Blood 1999, 93, 3053–3063. [PubMed] [Google Scholar]
- Haar C. P.; Hebbar P.; Wallace G. C. t.; Das A.; Vandergrift W. A. 3rd; Smith J. A.; Giglio P.; Patel S. J.; Ray S. K.; Banik N. L. Neurochem. Res. 2012, 37, 1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegl-Kreinecker S.; Buchroithner J.; Elbling L.; Steiner E.; Wurm G.; Bodenteich A.; Fischer J.; Micksche M.; Berger W. J. Neurooncol. 2002, 57, 27–36. [DOI] [PubMed] [Google Scholar]
- Trock B. J.; Leonessa F.; Clarke R. J. Natl. Cancer Inst. 1997, 89, 917–931. [DOI] [PubMed] [Google Scholar]
- Yardley D. A. Int. J. Breast Cancer 2013, 2013, 137414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nature 2012, 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nature 2008, 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boja E. S.; Rodriguez H. Clin. Proteomics 2014, 11, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters K. M.; Liu T.; Quesenberry R. D.; Willse A. R.; Bandyopadhyay S.; Kathmann L. E.; Weber T. J.; Smith R. D.; Wiley H. S.; Thrall B. D. PLoS One 2012, 7, e34515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.