

Conspectus
Decades after its discovery, positron emission tomography (PET) remains the premier tool for imaging neurochemistry in living humans. Technological improvements in radiolabeling methods, camera design, and image analysis have kept PET in the forefront. In addition, the use of PET imaging has expanded because researchers have developed new radiotracers that visualize receptors, transporters, enzymes, and other molecular targets within the human brain.
However, of the thousands of proteins in the central nervous system (CNS), researchers have successfully imaged fewer than 40 human proteins. To address the critical need for new radiotracers, this Account expounds on the decisions, strategies, and pitfalls of CNS radiotracer development based on our current experience in this area.
We discuss the five key components of radiotracer development for human imaging: choosing a biomedical question, selection of a biological target, design of the radiotracer chemical structure, evaluation of candidate radiotracers, and analysis of preclinical imaging. It is particularly important to analyze the market of scientists or companies who might use a new radiotracer and carefully select a relevant biomedical question(s) for that audience. In the selection of a specific biological target, we emphasize how target localization and identity can constrain this process and discuss the optimal target density and affinity ratios needed for binding-based radiotracers. In addition, we discuss various PET test–retest variability requirements for monitoring changes in density, occupancy, or functionality for new radiotracers.
In the synthesis of new radiotracer structures, high-throughput, modular syntheses have proved valuable, and these processes provide compounds with sites for late-stage radioisotope installation. As a result, researchers can manage the time constraints associated with the limited half-lives of isotopes. In order to evaluate brain uptake, a number of methods are available to predict bioavailability, blood–brain barrier (BBB) permeability, and the associated issues of nonspecific binding and metabolic stability. To evaluate the synthesized chemical library, researchers need to consider high-throughput affinity assays, the analysis of specific binding, and the importance of fast binding kinetics. Finally, we describe how we initially assess preclinical radiotracer imaging, using brain uptake, specific binding, and preliminary kinetic analysis to identify promising radiotracers that may be useful for human brain imaging. Although we discuss these five design components separately and linearly in this Account, in practice we develop new PET-based radiotracers using these design components nonlinearly and iteratively to develop new compounds in the most efficient way possible.
Introduction
PET radiotracers are small molecules containing a single positron emitting isotope (e.g., 11C, half-life of 20.38 min, or 18F, half-life of 109.8 min) and are detected in vivo by the measurement of highly tissue-penetrant and coincident γ rays produced upon positron annihilation. Molecular imaging with PET radiotracers can afford a sensitive and relatively noninvasive1,2 quantitation of biochemical parameters within a living human, including within the CNS. These characteristics provide PET imaging with immense potential to fill the void of techniques for assessment of neurophysiological biomarkers of healthy and diseased states in living human subjects and patients.3,4 Applications of CNS PET imaging include establishing proof-of-mechanism and optimal dosing for novel therapeutic agents, allowing for accelerated decision-making in clinical trials. Despite this potential, only 38 central nervous system targets are currently addressed by PET in humans,5 while thousands of brain proteins have yet to be explored.6 This limited availability of CNS PET radiotracers is partly due to the wide range of demanding criteria that must be fulfilled, especially for novel, higher-risk targets, and the empirical nature of radiotracer development.7
In this Account, we present a framework of chemical and biological considerations to optimize radiotracer development, with special attention given to radiotracers for novel CNS targets. Figure 1 showcases the components of the development process, which are represented by individual pools of a fountain. Except for well-studied targets previously vetted during drug discovery, the traditional pipeline approach of lead discovery and optimization may not be strategic for radiotracer development. Instead, the entry point into radiotracer development will vary significantly depending on existing knowledge of the biological target. Data collected from each development component will critically inform decision-making in other components, leading to progressive movement between the different tracer development pools. The streams of water connecting each pool represent one example of the cross-component approach we have found to be maximally efficient for PET radiotracer development.
Figure 1.

Artistic representation of the radiotracer development process. Blue streams highlight one of many potential pathways for initial radiotracer development, which branches into two pathways after chemical design. Purple streams indicate radiotracer development pathways in which previously explored components are revisited for radiotracer optimization. Drawing used with permission of Aaron Keefe.
PET Radiotracer Construction
Preface: Biomedical Question Selection
The end goal for CNS radiotracer development is to address a biomedical question by reporting on the neurochemistry of the living human brain. This biomedical question often arises from an unmet clinical need for which PET imaging can improve treatment paradigms or aid diagnosis in patients. However, the potential of PET imaging for CNS-disease diagnosis, while much touted, is currently low. The other main focus of CNS radiotracer development is clinical brain research. In this arena, basic biomedical research beckons for the development of new PET radiotracers for emerging targets or pathways implicated in human disease, wherein radiotracers are used to discover or validate human neurobiological concepts or as a drug development companion.8−10
Due to the expensive and time-consuming nature of radiotracer development and the low commercial potential for most radiotracers, we must carefully select our biomedical questions. To identify biomedical questions related to unmet medical needs, recent literature can provide a focus, but it is also crucial to complete “market research”. Specifically, insight from physicians who can comment on medical needs in their practice or in clinical research is invaluable. If imaging with a radiotracer for an unmet clinical need would not be “prescribed”, then there may be no need to develop the radiotracer, particularly diagnostic radiotracers. The real power of PET imaging may always lie in the area of basic biomedical research, since only four CNS radiotracers (fludeoxyglucose, florbetapir, flutemetamol, and florbetaben) have been approved by the FDA for diagnostic use and companies vacillate on the value proposition of diagnostic radiotracers. When one considers basic biomedical research, the utility of a radiotracer may be harder to determine a priori but can be gauged by polling colleagues, preclinical scientists, or experts at pharmaceutical companies to determine the human translational potential of a novel radiotracer. Thus, our approach for CNS radiotracer development most often starts with unmet medical needs and uses preclinical imaging to support human translation or to set an expectation in human disease imaging.
Biological Target Selection
For any unmet clinical imaging need there may be numerous implicated biological targets and choosing among them is one of the more difficult challenges in PET radiotracer development. Unlike target selection for therapeutic development, a radiotracer target needs only to demonstrate altered expression, occupancy levels, or function in the disease state, making it a secondary, as opposed to primary, marker for the disease. As a result, there is a variety of potential biological targets for PET imaging whose measurement can be applied to broad medical and biological questions, such as differentiating among psychiatric diseases or imaging neurogenesis.8
Two key selection parameters for a biological target are its biochemical function and localization, which alter the strategy for identifying candidate radiotracers and influence the information obtained from the radiotracer–target interaction. Nearly all CNS radiotracers are small-molecules that interact with protein targets (Figure 2). A small subset of radiotracers are substrates for enzymes, including the preeminent CNS PET radiotracer 2-deoxy-2-[18F]fluoroglucose. In addition, radiotracers may bind to and potently inhibit the enzymes they target but without affecting the process being measured due to the small mass being administered.11 Within the intracellular subset of targets exists the smaller group of nuclear targets, which require penetration of the nucleus by the radiotracer. These targets include enzymes involved in epigenetic modulation, such as histone deacetylases (HDACs), which have been a large focus of our lab, as well as the greater imaging community.9,12−15 Protein aggregates that are primarily extracellular have also been targeted for CNS PET imaging, most notably for imaging amyloid and tau deposition.16 These biomolecules are simpler to target due to their extracellular localization; however, the designed radiotracers must penetrate the BBB.
Figure 2.
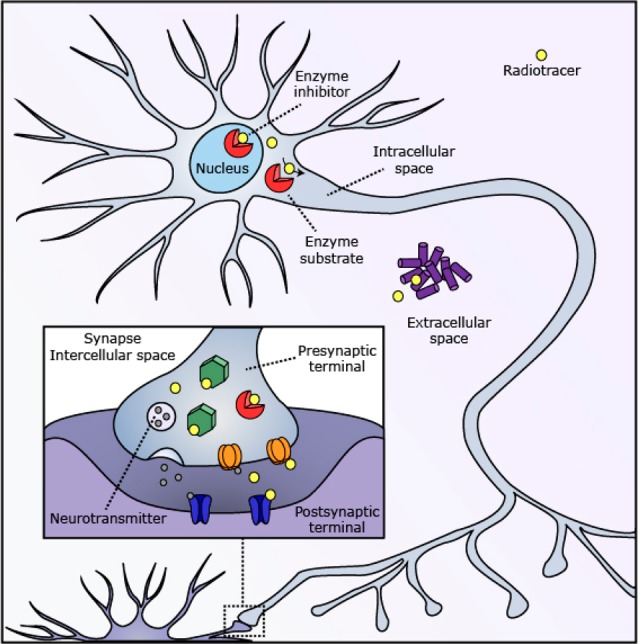
Candidate biological targets for radiotracer development have diverse biochemical function and cellular localization. Established radiotracer targets include enzymes (red), receptors (blue), transporters (orange), and many other intracellular (green) and extracellular (purple) proteins.
Mirroring the development of CNS therapeutic agents, the vast majority of CNS PET radiotracers are targeted toward the intercellular domains of transmembrane proteins, including G-protein coupled receptors (GPCRs), transporters, and ion channels.17 Radiotracers designed to bind a transmembrane protein may compete with a native ligand or be occluded by allosteric regulation. While this interaction may provide useful information on receptor occupancy, variation in the endogenous ligand concentration will confound measurement of receptor density in vivo and set requirements for the radiotracer Kd. In addition, many transmembrane receptors utilize homo- or heteropolymerization and internalization as regulatory mechanisms;18 these altered states are difficult to recapitulate in vitro and may result in drastically different radiotracer binding affinities. New combined MR-PET approaches for relating function to ligand–receptor interactions may elucidate these mechanisms in vivo,19 adding depth to the PET-based interrogation of the rich biology of the synapse.
An essential property that we optimize when developing binding-based radiotracers is the binding potential (BP). The BP provides a measurement of the in vivo radiotracer–target interaction and is comprised of the total biological target density (Bmax) and the binding affinity, represented as the radiotracer dissociation constant (Kd). Collective expertise in the field suggests a BP, or ratio of Bmax to Kd, of at least 5 is suitable for quantitative comparisons with PET imaging, especially in clinical research settings, but there may be scenarios where this “rule” can be violated. Radiotracers used in nonresearch clinical settings typically have a BP greater than 10.20 When the targeted radiotracer is yet to be developed, the dissociation constant will not be known; however, we use the Bmax to estimate the ideal Kd. If the Bmax is unknown, it can be measured using autoradiography or estimated through semiquantitative immunochemical methods.21 For radiotracers that compete with endogenous ligands in vivo, the effective target density available for radiotracer binding (Bavailable) is Bmax scaled to the fraction of targets unoccupied by the native ligand.
The percent change in expression or occupancy of the biological target is also of utmost importance and must exceed the error of the technique. The typical intrasubject test–retest variability for CNS PET radiotracers is 5–15%;22,23 therefore, single-subject, longitudinal changes in target density or receptor occupancy of greater than 15% can be imaged. For population comparisons, there may be high intersubject variability, which will necessarily reduce the chance of detecting these small changes.24 For clinical evaluation of patients, even larger changes in target density or occupancy are ideal as clinicians prefer binary “yes or no” images for ease of diagnosis and treatment monitoring.
The most pragmatic factor in target selection is the existence of high-affinity small-molecule ligands with established structure–affinity relationships, typically resulting from drug development efforts. For highly novel targets without known ligands, compound library screening will be necessary. Due to the time and cost-intensive nature of de novo ligand discovery, this endeavor is best suited for targets that clearly meet the fundamental requirements for a suitable CNS PET target: high Bmax with a large percent change in density or occupancy that correlates strongly with the biomedical question to be addressed.
Radiotracer Chemical Design
Our initial ensemble of candidate radiotracers typically consists of derivatives of a known ligand of the target of interest or the hits from a high-throughput screening campaign. While radiolabeling of known ligands or even an existing therapeutic agent may seem to be the most efficient method for radiotracer development, many examples from our own lab demonstrate that this strategy often results in subpar CNS PET radiotracers due to three main factors: (1) the low mass dose used for radiotracer administration, which requires a high brain/plasma ratio for CNS imaging,15 (2) high nonspecific binding of many therapeutic or inhibitor-adapted radiotracers in brain tissue,14,25−27 and (3) washout kinetics that are too slow for PET imaging.28 Importantly, the last two factors are positive selection traits during therapeutic development because they increase and maintain target-engagement following a single administration, but these properties afford undesirable radiotracers. Efficient prioritization of candidate radiotracers based on synthetic, physiological, and biochemical constraints is required to quickly move forward to validation and preclinical imaging.29
For de novo synthesis of CNS radiotracers, the likelihood of discovering high-affinity, brain-penetrant molecules can be increased by concurrently synthesizing compounds with several different chemical scaffolds, as we did during HDAC radiotracer development. Additionally, scaffolds that offer several sites that can be easily substituted with various functional groups should be targeted to increase the synthetic throughput. This emphasis on high-throughput, modular syntheses is critical: in our group’s experience over 150 compounds were synthesized and over 20 were radiolabeled for non-human primate imaging during the development of a single HDAC CNS radiotracer.15,30 When one develops a compound library, it is also important to synthesize molecules with relatively small changes in chemical structure, since our own studies have shown that addition of a single methyl group can dramatically affect the pharmacokinetics and distribution of a molecule (Figure 3).
Figure 3.
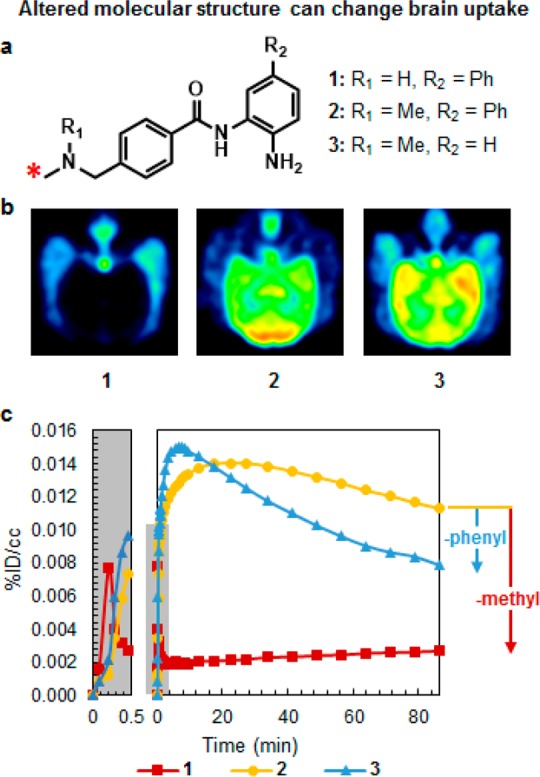
Three structurally related molecules with altered brain uptake and pharmacokinetics. (A) Chemical structure of the three molecules (1–3) that differ in the presence of methyl and phenyl groups; the ∗ indicates the 11C labeling site. (B) Transverse PET images for compounds 1–3 in baboon. (C) Time–activity curves for compounds 1–3. Adapted from ref (15).
While an emphasis on high-throughput synthesis is essential, the chief synthetic constraint is the necessity of a facile, late-stage labeling site that can be applied to numerous derivatives.31 By maintenance the same labeling site for many derivatives, little change in radiolabeling method is necessary between molecules, as demonstrated by the modular, peptide-based strategy for radiotracer development for predominantly peripheral targets.32,33 We often select 11C methylation as the labeling method to increase throughput, due to its facile and straightforward nature and its tolerability for the presence of many functional groups.34 Application of 18F chemistry to a single, well-vetted 11C radiotracer can be achieved during the clinical transition to provide patients access to radiotracers in the absence of an in-house cyclotron.31,35 In some cases, high-throughput 18F fluoralkylations can be applied to a radiotracer library,36,37 but these fluoroalkyl groups are typically not found in CNS radiotracer scaffolds. Thus, there remain chemical limitations for 18F installation, and a continued chemical methodology effort is needed to increase the number of reliable transformations.38−42
Significant physiological constraints are imposed on the chemical structures of candidate CNS radiotracers.29 To maintain radiotracer plasma levels following intravenous administration, the compounds must be bioavailable, often meeting Lipinski’s rule of five.43 The presence of the BBB also generally limits CNS PET radiotracers to molecules that enter the brain via passive diffusion. In addition, candidate radiotracers must exhibit low nonspecific binding or binding that is nonsaturable and for which the molecular details are unclear. One common analogy for considering the impact of nonspecific binding is as follows: although the stars (specific binding) are always in the sky, we can only discern them at night because daylight (nonspecific binding) overwhelms their signal. Several physiochemical properties such as log P, log D, molecular weight, and pKa are somewhat correlated with but not necessarily predictive of the candidate radiotracer’s in vivo behavior. Computational methods to assess the physiochemical properties of potential radiotracers have been developed;17 however, these methods do not show good agreement with a number of radiotracers our lab has developed. In the case of one chemical scaffold, the BBB penetration of the molecules was highly dependent on only two physiochemical properties, presence of a single cation and tPSA.15 Thus, computational tools may prove useful for deciding which compound of a series to radiolabel first, but as compounds in the series are radiolabeled and tested in vivo, valuable trends often develop that can be more predictive of future success.
Another consideration for structural modification is metabolite identity, since all PET radiotracers are extensively metabolized.29 Alterations in radiolabeled metabolite structure can result from changes in radiolabeling site, and this can impact the number of metabolites contributing to the brain signal.29 Likewise, demethylation of 11C-methylated heteroatoms in the periphery can liberate 11C-formaldehyde, 11C-formate, or 11CO2, and defluorination can result in fluoride ion accumulation in the bone of the skull, with the resulting signal "spilling" into the brain.44 The PET detector “sees” the radiotracer and all radiometabolites equally, so the onus is on the investigator to determine the radiochemical species producing the signal. Finally, metabolism is also species-dependent, with compounds typically metabolized more quickly in lower organisms. The differences between species extends to gross anatomy; for example, rats lack a gall bladder. Thus, there is limited validity in ruling out potential radiotracers because they failed in rodent preclinical imaging, and efforts should be made to proceed to non-human primate imaging as rapidly as possible to obtain distribution and kinetic data that is more predictive of radiotracer performance in humans.
Assessment of Radiotracer Library
With molecules in hand, the next challenge in radiotracer development is to narrow the selection of lead compounds to be radiolabeled and tested by preclinical imaging. The initial assessment of candidate radiotracers shares many similarities to the development of any small-molecule probe: affinity, selectivity, and binding kinetics are all major considerations. However, poor in vivo pharmacokinetics and high nonspecific binding are largely responsible for the high attrition rate of candidate radiotracers in the first round of preclinical imaging experiments, and these measures are not readily predictable. Therefore, we strategically assess candidate radiotracers iteratively, returning to in vitro experiments as dictated by preliminary imaging data. A comparison of data obtained from in vitro and in vivo characterization can be found in Table 1 and is discussed in detail in the following sections.
Table 1. Assessment of Key Attributes during Imaging and Nonimaging Components of Radiotracer Development.
| radiotracer attribute | nonimaging assessment | imaging assessment |
|---|---|---|
| binding affinity | potency in in vitro assays | binding potential (Bmax/Kd, smaller Kd = larger BP) |
| binding kinetics | vary preincubation and washing steps in in vitro assays, autoradiography | qualitative TAC analysis |
| quantitative kinetic modeling | ||
| BBB penetration | mass spectrometry of unlabeled tracer | %ID/cc in brain |
| in silico prediction | ||
| specific binding | “no wash” autoradiography | homologous blocking |
| in silico prediction | knockout animals | |
| selective binding | autoradiography | heterologous blocking |
| systematic screening (PDSP) | knockout animals |
In cases where we rank molecules prior to radiolabeling, we measure and compare binding affinities, which often must be subnanomolar to nanomolar, depending on the biological target density.29,45 The nature of this measurement will be highly dependent on the target’s biochemical function and may include displacement of a known ligand, disruption of a protein–protein interaction, formation of a covalent adduct, or inhibition of enzyme catalysis. To maximize efficiency, we optimize binding affinity assays using a known positive control concomitant with the synthesis of the radiotracer library. Once developed, the assay may be modified to measure the association or dissociation rates of the candidate molecules, which need to be relatively fast for radiotracers with short half-lives. For example, we found the kinetics of the hydroxamate class of HDAC inhibitors better suited for PET imaging than the slow-binding benzamides, despite the increased efficacy of benzamides in disease models.21,30 As with all in vitro biochemical assays, test–retest variation may be significant, and the protein preparation may not reflect the in vivo properties of the target. In addition, the precise ranking of the binding affinities of the potential radiotracers is less important than their general clustering.
Candidate radiotracers may also bind to off-target protein(s) that are structurally or functionally similar to the imaging target. This nonselective binding to proteins other than the target of interest is not to be confused with nonspecific binding, discussed previously. The selectivity required for a suitable radiotracer is not fixed and depends on the relative densities of the desired versus off-target proteins, as well as the relative rate of binding of the radiotracer. Furthermore, selectivity may not be problematic if the regional distribution of the off-target proteins has little anatomical overlap with the imaging target. In addition to nonselective binding based on protein homology, potential radiotracers may also have high affinities for other targets that are not easily predictable. These off-targets are best identified by systematic screening, such as the NIMH psychoactive drug screening program.46 While off-target binding should usually be avoided, compounds that exhibit off-target binding can still show in vivo target selectivity when the target protein Bmax is high relative to off-target proteins or when the regional distribution of the target and off-target proteins is nonoverlapping.47
Biochemical analysis of brain tissue bridges the gap between in vitro assays and preclinical imaging. Autoradiography methods allow for facile measurement of tracer association or dissociation rates, and “no-wash” protocols are predictive of in vivo nonspecific binding.48 However, these experiments require radiolabeling and are thus not suitable for prospective screening. If the candidate radiotracers are commercially available, it is advisable to obtain classic pharmacokinetic data prior investing in synthesis and radiolabeling. Newer mass spectrometry methods can provide information about differential distribution of unlabeled compounds throughout the brain for comparison to target protein expression levels and allow for analysis of specific and nonspecific binding across brain regions.49 However, use of unlabeled compounds precludes measurement of the uptake and binding of compound metabolites and a large effort is required to obtain data for full pharmacokinetic analysis, because each animal can only provide information for a single time-point. This contrasts with preclinical PET imaging, wherein metabolite radioactivity can be tracked with blood analysis and full kinetic data is obtained for each injection.
Radiotracer Analysis via Preclinical Imaging
We have found that the most time-efficient assessment of new CNS radiotracers may be to bypass biochemical assessment and move directly to radiolabeling and preclinical imaging, which allows analysis and comparison of radiotracer pharmacokinetics and in vivo target engagement. Direct preclinical imaging also circumvents the disparities often found between in vitro radiotracer assessment and in vivo performance, which can cause researchers to overlook good radiotracers due to a lower binding affinity or lower selectivity between target subtypes.33
When analyzing preclinical imaging data, our first step is verification of radiotracer uptake in the brain. This is accomplished through plotting a time–activity curve (TAC) of the percent injected dose of radiotracer (%ID) per volume (cc) in the total brain or target-rich brain region as a function of time (Figure 3). As a guiding rule in our lab, PET radiotracers with a %ID/cc above 0.1% in rat or 0.01% in non-human primate within 5 min of injection have suitable BBB penetration for CNS PET imaging studies.
If the PET radiotracer exhibits good brain uptake, the TAC can be analyzed further to determine the degree and length of radiotracer retention within in the brain. When brain retention of a radiotracer is low following a high initial brain uptake (Figure 3, compound 1), the radiotracer is potentially being actively effluxed.50 To verify an active efflux mechanism, inhibitors of active efflux proteins, such as cyclosporin A and rifampicin, can be injected prior to the radiotracer to determine whether they increase brain retention.50 Importantly, interspecies differences in active efflux mechanisms have been documented,51 such that a radiotracer that fails in rodents may be suitable for non-human primate or human imaging.
When a candidate radiotracer demonstrates high brain retention, we measure the specificity of binding within the brain via homologous blocking studies, where animals are pretreated with the 12C or 19F (unlabeled) version of the radiotracer prior to radiotracer injection. If the unlabeled compound competes for binding with the radiotracer, the radiotracer signal will be reduced, indicating specific binding. Data analysis for this test requires careful attention, because blockade of binding sites by the unlabeled compound throughout the body may increase the amount of free radiotracer in plasma, resulting in increased total uptake of the radiotracer in the brain relative to untreated control animals (Figure 4a). This effect can be accounted for through normalization of radiotracer uptake to metabolite-corrected plasma radiotracer levels (Figure 4b,c), a quick assessment prior to an investment in more rigorous kinetic modeling quantification.
Figure 4.
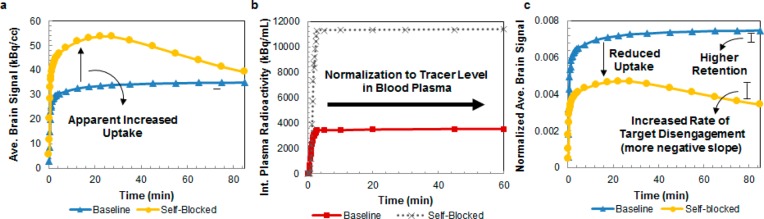
Impact of normalization of brain radiotracer signal to plasma radiotracer level. (A) Non-normalized baseline (blue) and self-blocked (yellow) brain signals for martinostat. (B) Integrated martinostat radioactivity in plasma during baseline (red) and self-blocked (gray) PET scans. (C) Plasma-normalized baseline (blue) and self-blocked (yellow) brain signals for martinostat. Adapted from ref (30).
Following verification of specific binding in the brain, we assess the presence of on-target specific binding, because some radiotracers or their metabolites may specifically bind off-target proteins. On-target specific binding analysis is typically accomplished through heterologous blocking studies in which animals are pretreated with a panel of compounds that are chemically distinct from the radiotracer and that are known to bind the biological target of interest.10 Application of the radiotracer to knockout animal models or autoradiography30 can be used to further verify on-target, specific binding. Autoradiography can additionally be used to correlate the regional distribution of radiotracer binding to the known regional density of the biological target.21
In addition to analyzing the TACs for specific binding, qualitative kinetic analysis can be performed to determine whether the radiotracer is suitable for human imaging. While kinetic properties vary between species, kinetics suitable for robust quantitative analysis can typically be spotted through comparison of TAC slopes at time points after the peak signal. A relatively steeper curve (faster radiotracer washout) following treatment with the nonradioactive radiotracer analog indicates a measurable decrease in BP, even without accounting for changes in plasma radiotracer activity (Figure 4a). A slope near zero may be indicative of a radiotracer with irreversible binding or too-slow kinetics. To interrogate these radiotracers, we complete a bolus or bolus-plus-infusion experiment with injection of a homologous or heterologous blocking agent midscan. A decrease in slope after blocking agent administration indicates a BP decrease, and therefore measurable, reversible binding. The quantitative kinetic analysis of radiotracer binding is the topic of several comprehensive reviews.45,52,53
When moving to preclinical imaging, we have found many instances in which a new radiolabeled compound either did not penetrate the BBB or did not show specific, on-target binding, which required a return to radiotracer design. At times, validation of a new biological target is necessary, for example, when several molecular scaffolds for the initial target have failed to show brain uptake or specific binding. However, with persistence and preclinical assessment of many radiotracers, CNS PET radiotracers with high brain uptake, specific on-target binding, and a suitable kinetic profile can be discovered.
Conclusion and Guiding Principles
Novel radiotracer development is a challenging endeavor, requiring knowledge of disease-related biological targets, chemical synthesis and radiolabeling, in vitro assay development, and image analysis. Biomedical questions should be applicable to human clinical studies and more feasible to answer with PET imaging versus other, possibly less resource-intensive, tools. When the need for a novel radiotracer is established, we make use of the following principles to guide our development program:
1. Know Your Biology
In-house analysis of tissue slices, cell cultures, and protein preparations is paramount to determine the density and regional brain distribution of the target, the change in target density or occupancy, and the binding affinities of lead candidate radiotracers. By doing these assays in the environment of radiotracer discovery, you will gain insight into peculiarities of the biological target and candidate radiotracers that may be valuable during in vivo assessment.
2. Throughput Matters
The structural design of candidate radiotracers must be amenable to late-stage diversification and facile radiolabeling, because many iterations of a chemical series may be needed before a suitable tracer is identified. Biochemical assays of binding affinity can be developed into a high-throughput screen.
3. Aim for Studies in Humans
Low brain penetration, high nonspecific binding, and a poor metabolic profile are the primary factors that eliminate candidate radiotracers in the preclinical imaging stage, but these measures are highly species-dependent. When possible, radiotracers should be assessed in non-human primates, with the goal of moving to human imaging as soon as possible.
Finally, one should remain optimistic. As depicted in Figure 1, the process of PET radiotracer development is often iterative and circuitous, requiring the designer to frequently step back and assess the current best path forward. After the initial biomedical question is posed, the succeeding components (pools) of target selection, chemical design, library assessment, and preclinical imaging may be revisited many times and in variant orders (streams) before a suitable radiotracer (central water burst) is developed that is poised to provide an answer.
Acknowledgments
We acknowledge Aaron Keefe for creating the artwork in Figure 1, which illustrates the authors’ fountain-based view of the radiotracer development process. We thank the members of the Hooker Lab for their insight and advice and Dr. Changning Wang for contribution of data. We acknowledge the National Institutes of Health (Grant NIH R01:5R01DA030321) and the Department of Energy (DOE Training Grant DE-SC0008430) for funding this work.
Glossary
Abbreviations
- PET
positron emission tomography
- CNS
central nervous system
- BBB
blood–brain barrier
- GPCRs
G-protein coupled receptors
- MR-PET
magnetic resonance-positron emission tomography
- BP
binding potential
- TAC
time–activity curve
Biographies
Genevieve Van de Bittner obtained a Ph.D. in Chemistry from the University of California, Berkeley, in 2012. She is currently completing a postdoctoral fellowship at the Martinos Center for Biomedical Imaging at Massachusetts General Hospital, in affiliation with Harvard Medical School. Her current research focus is the development of radiotracers for synapse imaging in humans.
Emily Ricq received her B.Sc. in Chemistry from the University of Arizona in 2010 and is pursuing her Ph.D. in Chemistry & Chemical Biology from Harvard University. Her current research focuses on developing chemical tools to profile epigenetic processes relevant to neurobiology.
Jacob Hooker received his Ph.D. at UC Berkeley in 2007 in Chemistry. After a postdoctoral fellowship at Brookhaven National Laboratory, Prof. Hooker moved to the Martinos Center for Biomedical Imaging at Massachusetts General Hospital in 2009. He is currently an Assistant Professor at Harvard Medical School where he and his team work on new human neuroimaging technologies and their applications to brain disorders.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Lockwood A. H. Invasiveness in Studies of Brain Function by Positron Emission Tomography (PET). J. Cereb. Blood Flow Metab. 1985, 5, 487–489. [DOI] [PubMed] [Google Scholar]
- Eriksson L.; Kanno I. Blood Sampling Devices and Measurements. Med. Prog. Technol. 1991, 17, 249–257. [PubMed] [Google Scholar]
- Zimmer L.; Luxen A. PET Radiotracers for Molecular Imaging in the Brain: Past, Present and Future. NeuroImage 2012, 61, 363–370. [DOI] [PubMed] [Google Scholar]
- Hargreaves R. J.; Rabiner E. A. Translational PET Imaging Research. Neurobiol. Dis. 2014, 61, 32–38. [DOI] [PubMed] [Google Scholar]
- CNS Radiotracer Table http://www.nimh.nih.gov/research-priorities/therapeutics/cns-radiotracer-table.shtml (accessed Jun 17, 2014).
- Martins-de-Souza D.; Carvalho P. C.; Schmitt A.; Junqueira M.; Nogueira F. C. S.; Turck C. W.; Domont G. B. Deciphering the Human Brain Proteome: Characterization of the Anterior Temporal Lobe and Corpus Callosum as Part of the Chromosome 15-Centric Human Proteome Project. J. Proteome Res. 2014, 13, 147–157. [DOI] [PubMed] [Google Scholar]
- Jones T.; Rabiner E. A. PET Research Advisory Company. The Development, Past Achievements, and Future Directions of Brain PET. J. Cereb. Blood Flow Metab. 2012, 32, 1426–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho N. F.; Hooker J. M.; Sahay A.; Holt D. J.; Roffman J. L. In Vivo Imaging of Adult Human Hippocampal Neurogenesis: Progress, Pitfalls and Promise. Mol. Psychiatry 2013, 18, 404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Schroeder F. A.; Hooker J. M. Visualizing Epigenetics: Current Advances and Advantages in HDAC PET Imaging Techniques. Neuroscience 2014, 264, 186–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder F. A.; Wang C.; Van de Bittner G.; Neelamegam R.; Takakura W.; Karunakaran P.; Wey H.-Y.; Reis S.; Gale J. P.; Zhang Y.-L.; Holson E.; Haggarty S.; Hooker J. PET Imaging Demonstrates Histone Deacetylase Target Engagement and Clarifies Brain Penetrance of Known and Novel Small Molecule Inhibitors in Rat. ACS Chem. Neurosci. 2014, 10.1021/cn500162j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegon A.; Kim S. W.; Alexoff D. L.; Jayne M.; Carter P.; Hubbard B.; King P.; Logan J.; Muench L.; Pareto D.; Schlyer D.; Shea C.; Telang F.; Wang G.-J.; Xu Y.; Fowler J. S. Unique Distribution of Aromatase in the Human Brain: In Vivo Studies with PET and [N-Methyl-11C]vorozole. Synapse 2010, 64, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. W.; Hooker J. M.; Otto N.; Win K.; Muench L.; Shea C.; Carter P.; King P.; Reid A. E.; Volkow N. D.; Fowler J. S. Whole-Body Pharmacokinetics of HDAC Inhibitor Drugs, Butyric Acid, Valproic Acid and 4-Phenylbutyric Acid Measured with Carbon-11 Labeled Analogs by PET. Nucl. Med. Biol. 2013, 40, 912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y. J.; Muench L.; Reid A.; Chen J.; Kang Y.; Hooker J. M.; Volkow N. D.; Fowler J. S.; Kim S. W. Radionuclide Labeling and Evaluation of Candidate Radioligands for PET Imaging of Histone Deacetylase in the Brain. Bioorg. Med. Chem. Lett. 2013, 23, 6700–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Eessalu T. E.; Barth V. N.; Mitch C. H.; Wagner F. F.; Hong Y.; Neelamegam R.; Schroeder F. A.; Holson E. B.; Haggarty S. J.; Hooker J. M. Design, Synthesis, and Evaluation of Hydroxamic Acid-Based Molecular Probes for in Vivo Imaging of Histone Deacetylase (HDAC) in Brain. Am. J. Nucl. Med. Mol. Imaging 2013, 4, 29–38. [PMC free article] [PubMed] [Google Scholar]
- Seo Y. J.; Kang Y.; Muench L.; Reid A.; Caesar S.; Jean L.; Wagner F.; Holson E.; Haggarty S. J.; Weiss P.; King P.; Carter P.; Volkow N. D.; Fowler J. S.; Hooker J. M.; Kim S. W. Image-Guided Synthesis Reveals Potent Blood-Brain Barrier Permeable Histone Deacetylase Inhibitors. ACS Chem. Neurosci. 2014, 5, 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg A.PET Tracers for Beta-Amyloid and Other Proteinopathies. In PET and SPECT of Neurobiological Systems; Dierckx R. A. J. O., Otte A.; de Vries E. F. J.; van Waarde A.; Luiten P. G. M., Eds.; Springer: Berlin Heidelberg, 2014; pp 199–212. [Google Scholar]
- Zhang L.; Villalobos A.; Beck E. M.; Bocan T.; Chappie T. A.; Chen L.; Grimwood S.; Heck S. D.; Helal C. J.; Hou X.; Humphrey J. M.; Lu J.; Skaddan M. B.; McCarthy T. J.; Verhoest P. R.; Wager T. T.; Zasadny K. Design and Selection Parameters to Accelerate the Discovery of Novel Central Nervous System Positron Emission Tomography (PET) Ligands and Their Application in the Development of a Novel Phosphodiesterase 2A PET Ligand. J. Med. Chem. 2013, 56, 4568–4579. [DOI] [PubMed] [Google Scholar]
- Ferré S.; Casadó V.; Devi L. A.; Filizola M.; Jockers R.; Lohse M. J.; Milligan G.; Pin J.-P.; Guitart X. G Protein-Coupled Receptor Oligomerization Revisited: Functional and Pharmacological Perspectives. Pharmacol. Rev. 2014, 66, 413–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander C. Y.; Hooker J. M.; Catana C.; Normandin M. D.; Alpert N. M.; Knudsen G. M.; Vanduffel W.; Rosen B. R.; Mandeville J. B. Neurovascular Coupling to D2/D3 Dopamine Receptor Occupancy Using Simultaneous PET/functional MRI. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 11169–11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S.; Gibson R. In Vivo Site-Directed Radiotracers: A Mini-Review. Nucl. Med. Biol. 2008, 35, 805–815. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang Y.-L.; Hennig K.; Gale J. P.; Hong Y.; Cha A.; Riley M.; Wagner F.; Haggarty S. J.; Holson E.; Hooker J. Class I HDAC Imaging Using [(3)H]CI-994 Autoradiography. Epigenetics 2013, 8, 756–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage H. D.; Voytko M. L.; Ehrenkaufer R. L.; Tobin J. R.; Efange S. M.; Mach R. H. Reproducibility of Repeated Measures of Cholinergic Terminal Density Using [18F](+)-4-Fluorobenzyltrozamicol and PET in the Rhesus Monkey Brain. J. Nucl. Med. 2000, 41, 2069–2076. [PubMed] [Google Scholar]
- Narendran R.; Mason N. S.; May M. A.; Chen C.-M.; Kendro S.; Ridler K.; Rabiner E. A.; Laruelle M.; Mathis C. A.; Frankle W. G. Positron Emission Tomography Imaging of Dopamine D2/3 Receptors in the Human Cortex with [11C]FLB 457: Reproducibility Studies. Synapse 2011, 65, 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl L. M.; Nahmias C. Statistical Power Analysis for PET Studies in Humans. J. Nucl. Med. 1998, 39, 1826–1829. [PubMed] [Google Scholar]
- Stephenson K. A.; van Oosten E. M.; Wilson A. A.; Meyer J. H.; Houle S.; Vasdev N. Synthesis and Preliminary Evaluation of [(18)F]-Fluoro-(2S)-Exaprolol for Imaging Cerebral Beta-Adrenergic Receptors with PET. Neurochem. Int. 2008, 53, 173–179. [DOI] [PubMed] [Google Scholar]
- Wang C.; Wilson C. M.; Moseley C. K.; Carlin S. M.; Hsu S.; Arabasz G.; Schroeder F. A.; Sander C. Y.; Hooker J. M. Evaluation of Potential PET Imaging Probes for the Orexin 2 Receptors. Nucl. Med. Biol. 2013, 40, 1000–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelamegam R.; Hellenbrand T.; Schroeder F. A.; Wang C.; Hooker J. M. Imaging Evaluation of 5HT2C Agonists, [(11)C]WAY-163909 and [(11)C]Vabicaserin, Formed by Pictet-Spengler Cyclization. J. Med. Chem. 2014, 57, 1488–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horti A. G.; Gao Y.; Kuwabara H.; Dannals R. F. Development of Radioligands with Optimized Imaging Properties for Quantification of Nicotinic Acetylcholine Receptors by Positron Emission Tomography. Life Sci. 2010, 86, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike V. W. PET Radiotracers: Crossing the Blood-Brain Barrier and Surviving Metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Schroeder F. A.; Wey H.-Y.; Borra R.; Wagner F.; Reis S.; Kim S. W.; Holson E. B.; Haggarty S. J.; Hooker J. M. In Vivo Imaging of Histone Deacetylases (HDACs) in the Central Nervous System and Major Peripheral Organs. J. Med. Chem. 2014, 10.1021/jm500872p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooker J. M. Modular Strategies for PET Imaging Agents. Curr. Opin. Chem. Biol. 2010, 14, 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe-Goulden J. L.; O’Doherty M. J.; Marsden P. K.; Hart I. R.; Marshall J. F.; Bansal S. S. Rapid Solid Phase Synthesis and Biodistribution of 18F-Labelled Linear Peptides. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 754–759. [DOI] [PubMed] [Google Scholar]
- Gagnon M. K. J.; Hausner S. H.; Marik J.; Abbey C. K.; Marshall J. F.; Sutcliffe J. L. High-Throughput in Vivo Screening of Targeted Molecular Imaging Agents. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 17904–17909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller P. W.; Long N. J.; Vilar R.; Gee A. D. Synthesis of 11C, 18F, 15O, and 13N Radiolabels for Positron Emission Tomography. Angew. Chem., Int. Ed. 2008, 47, 8998–9033. [DOI] [PubMed] [Google Scholar]
- Cole E. L.; Stewart M. N.; Littich R.; Hoareau R.; Scott P. J. H. Radiosyntheses Using Fluorine-18: The Art and Science of Late Stage Fluorination. Curr. Top. Med. Chem. 2014, 14, 875–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.-R.; Suzuki K. [18F]Fluoroalkyl Agents: Synthesis, Reactivity and Application for Development of PET Ligands in Molecular Imaging. Curr. Top. Med. Chem. 2007, 7, 1817–1828. [DOI] [PubMed] [Google Scholar]
- Pretze M.; Pietzsch D.; Mamat C. Recent Trends in Bioorthogonal Click-Radiolabeling Reactions Using Fluorine-18. Molecules 2013, 18, 8618–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E.; Kamlet A. S.; Powers D. C.; Neumann C. N.; Boursalian G. B.; Furuya T.; Choi D. C.; Hooker J. M.; Ritter T. A Fluoride-Derived Electrophilic Late-Stage Fluorination Reagent for PET Imaging. Science 2011, 334, 639–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E.; Hooker J. M.; Ritter T. Nickel-Mediated Oxidative Fluorination for PET with Aqueous [18F] Fluoride. J. Am. Chem. Soc. 2012, 134, 17456–17458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamlet A. S.; Neumann C. N.; Lee E.; Carlin S. M.; Moseley C. K.; Stephenson N.; Hooker J. M.; Ritter T. Application of Palladium-Mediated 18F-Fluorination to PET Radiotracer Development: Overcoming Hurdles to Translation. PLoS One 2013, 8, e59187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Liu W.; Ren H.; Neelamegam R.; Hooker J. M.; Groves J. T. Late Stage Benzylic C–H Fluorination with [18F]Fluoride for PET Imaging. J. Am. Chem. Soc. 2014, 136, 6842–6845. [DOI] [PubMed] [Google Scholar]
- Ren H.; Wey H.-Y.; Strebl M.; Neelamegam R.; Ritter T.; Hooker J. M. Synthesis and Imaging Validation of [18F]MDL100907 Enabled by Ni-Mediated Fluorination. ACS Chem. Neurosci. 2014, 5, 611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Ryu Y. H.; Liow J.-S.; Zoghbi S.; Fujita M.; Collins J.; Tipre D.; Sangare J.; Hong J.; Pike V. W.; Innis R. B. Disulfiram Inhibits Defluorination of (18)F-FCWAY, Reduces Bone Radioactivity, and Enhances Visualization of Radioligand Binding to Serotonin 5-HT1A Receptors in Human Brain. J. Nucl. Med. 2007, 48, 1154–1161. [DOI] [PubMed] [Google Scholar]
- Laruelle M.; Slifstein M.; Huang Y. Positron Emission Tomography: Imaging and Quantification of Neurotransporter Availability. Methods 2002, 27, 287–299. [DOI] [PubMed] [Google Scholar]
- NIMH Psychoactive Drug Screening Program. http://pdsp.med.unc.edu/ (accessed Jun 17, 2014).
- Hooker J. M.; Kim S. W.; Reibel A. T.; Alexoff D.; Xu Y.; Shea C. Evaluation of [11C]Metergoline as a PET Radiotracer for 5HTR in Nonhuman Primates. Bioorg. Med. Chem. 2010, 18, 7739–7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S.; Hamill T.; Hostetler E.; Burns H. D.; Gibson R. E. An in Vitro Assay for Predicting Successful Imaging Radiotracers. Mol. Imaging Biol. 2003, 5, 65–71. [DOI] [PubMed] [Google Scholar]
- Barth V.; Need A. Identifying Novel Radiotracers for PET Imaging of the Brain: Application of LC-MS/MS to Tracer Identification. ACS Chem. Neurosci. 2014, 10.1021/cn500072r. [DOI] [PubMed] [Google Scholar]
- Tournier N.; Saba W.; Cisternino S.; Peyronneau M.-A.; Damont A.; Goutal S.; Dubois A.; Dollé F.; Scherrmann J.-M.; Valette H.; Kuhnast B.; Bottlaender M. Effects of Selected OATP And/or ABC Transporter Inhibitors on the Brain and Whole-Body Distribution of Glyburide. AAPS J. 2013, 15, 1082–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvänen S.; Lindhe Ö.; Palner M.; Kornum B. R.; Rahman O.; Långström B.; Knudsen G. M.; Hammarlund-Udenaes M. Species Differences in Blood-Brain Barrier Transport of Three Positron Emission Tomography Radioligands with Emphasis on P-Glycoprotein Transport. Drug Metab. Dispos. 2009, 37, 635–643. [DOI] [PubMed] [Google Scholar]
- Varnäs K.; Varrone A.; Farde L. Modeling of PET Data in CNS Drug Discovery and Development. J. Pharmacokinet. Pharmacodyn. 2013, 40, 267–279. [DOI] [PubMed] [Google Scholar]
- Kuntner C. Kinetic Modeling in Preclinical Positron Emission Tomography. Z. Med. Phys. 2014, 10.1016/j.zemedi.2014.02.003. [DOI] [PubMed] [Google Scholar]