

Abstract
In this study the nucleotide diversity in the 5′untranslated region (UTR) of TLR8 gene in riverine as well as swamp buffaloes has been described. Analysis of the 5′UTR of TLR8 gene showed presence of two SNPs in this region, g.-139G>T and g.-128A>G. A PCR–RFLP assay designed for genotyping of g.-139G>T SNP across 667 samples from 2 buffalo populations revealed a striking difference in allele frequency distribution across the swamp and riverine buffaloes. The frequency of T allele was higher in swamp buffalo as compared to riverine buffalo, ranging from 0.71 to 1. The G allele on the other hand exhibited a higher frequency across all the Indian riverine buffalo breeds/populations. The principal component analysis revealed separate clusters for the riverine and swamp buffaloes, as expected; however, the riverine type Assamese buffalo population of eastern India formed a distinct cluster. Since most of the buffalo populations in the eastern region are swamp type, this demarcation may be related to the difference in immune response in riverine and swamp buffaloes. These preliminary results indicate that the genetic variation observed in 5′upstream region of TLR8 gene, which differentiates swamp and riverine buffalo, needs to be further explored for association with disease susceptibility in buffalo, an important dairy and meat animal of Southeast Asia.
Keywords: Buffalo, Toll-like receptor 8, 5′UTR, Polymorphism, Allelic distribution
1. Introduction
Dairy industry of India largely depends on the 105.34 million buffaloes (Livestock Census, 2007, http://dahd.nic.in/dahd/bahs-2012.aspx), which account for 56.7% of the world's buffalo population-. Besides milk, buffaloes are an important source for meat, hide and draft power. As per animal husbandry statistics buffaloes contribute around 52.5% to total milk production in the country. In India there are 13 registered riverine buffalo breeds (Murrah, Niliravi, Mehsana, Jaffarabadi, Banni, Bhadawari, Surti, Nagpuri, Pandharpuri, Marathwada, Toda, Kalahandi and Chilika), alongwith many lesser known populations of significant regional importance. The riverine buffalo (Bubalus bubalis) is found throughout the length and breadth of the country and has 50 chromosomes, while the swamp buffalo (Bubalus bubalis carabanesis) with a chromosome number of 48, is restricted to Northeastern states of the country like Mizoram, Nagaland, Manipur and parts of Assam. The crossbreds of riverine and swamp buffaloes have 49 chromosomes and are distributed mainly in different areas of Assam (Harisah et al., 1989). Riverine buffaloes are mainly used for milk production whereas swamp buffaloes are used for draft and meat purposes (Mishra et al., 2010). Furthermore, there are also reports on comparative study between the two types of buffaloes, showing that swamp buffaloes are immunologically stronger and less susceptible to diseases than riverine buffaloes (Mingala et al., 2009). Therefore, the investigation of genetic diversity in immune genes responsible for the natural immunity in riverine and swamp buffaloes would shed light on the disease susceptibility or resistance in the two types of domestic buffaloes.
Toll-like receptors (TLRs) are evolutionarily conserved membrane-bound proteins that are widely expressed in insects, plants and animals and recognize microbial pathogens to initiate intracellular signaling pathways and trigger the expression of genes responsible for the activation of innate immune response (Takeda and Akira, 2005). Based on ligand–pathogen specificity, ten TLRs have been identified in humans and livestock species, 13 in mice and more than 20 in non-mammalian genomes (Raja et al., 2011; Dubey et al., 2012a). They share a common modular structure that consists of a leucine-rich repeat (LRR) ectodomains, a single transmembrane helix and a cytoplasmic Toll/interleukin-1 receptor (TIR) domain. Among the TLRs reported in mammals, TLR8 recognizes ssRNA derived from viruses as well as small synthetic molecules structurally related to nucleic acids such as imidazoquinolines and immunostimulatory guanosine nucleotides (Zhu et al., 2009). Recently microRNA, important for regulating gene expression has also been reported to induce tumor growth and pro-metastatic inflammatory response through binding to TLR8 in cancerous cells (Fabbri et al., 2012). TLR8 located on the mammalian X chromosome is relatively conserved and lies close to TLR7, both recognize the viral genome/cellular nucleic acids and expressed on intracellular organelles forming an evolutionary cluster with TLR9 (Du et al., 2000; Astakhova et al., 2009). Buffalo TLR8 gene has been characterized and found to consist of a single exon coding for 1041 amino acid long protein similar to other livestock (Dubey et al., 2012b).
Genetic polymorphisms in members of the TLR family have been demonstrated to be clinically important. The nucleotide variations in the bovine TLR genes have been associated with inflammatory diseases such as number of somatic cell count, mastitis, keratoconjunctivitis, and tuberculosis (Mucha et al., 2009; Kataria et al., 2011). Polymorphism in human TLR8 is reported to be associated with HIV infection, asthma and tuberculosis (Davila et al., 2008). Gene expression studies have also implicated TLR8 role in the pathogenesis of Johne's disease (JD) in infected sheep tissues (Taylor et al., 2008). Previous studies have reported only three polymorphic sites in the coding and one in the 3′UTR of TLR8 gene of riverine and swamp buffaloes (Dubey et al., 2012b), but variation in the 5′UTR of buffalo TLR8 gene is yet to be investigated. Variation within 5′UTR of TLR8 may have a functional role in governing disease resistance/susceptibility traits. The present study was therefore undertaken with the objective to characterize 5′upstream and 5′untranslated regions of TLR8 gene and document variation in allele frequency distribution across the Indian riverine and swamp buffaloes.
2. Materials and methods
2.1. Sample collection and DNA isolation
Blood samples were collected from the distribution areas of different riverine, alongwith hybrids and swamp types from the region bordering China and Myanmar (Fig. 1, Table 1). Genomic DNA was extracted from whole blood using standard SDS-Proteinase-K digestion and phenol/chloroform extraction procedure (Sambrook and Russell, 2001). Quality and quantity of DNA were assessed by spectrophotometer as well as by 0.8% agarose gel electrophoresis. The riverine, swamp and hybrid status of buffaloes from different parts of Assam as well as north east region were confirmed by analysis of their karyotypes.
Fig. 1.

Geographic distribution of Indian riverine, swamp and hybrid buffalo breeds/populations.
Table 1.
Genotypic and allelic frequency of the Tsp509I alleles (g.-139G>T) across the Indian riverine, swamp and hybrid population/breeds.
| Breeds | Sample no. | Genotype frequency |
Allele frequency 139G>T |
||||
|---|---|---|---|---|---|---|---|
| GG | GT | TT | G | T | |||
| Swamp | MZ | 16 | 0.00 | 0.00 | 1.00 | 0.00 | 1.00 |
| MN | 25 | 0.08 | 0.00 | 0.92 | 0.08 | 0.92 | |
| NG | 35 | 0.03 | 0.23 | 0.74 | 0.14 | 0.86 | |
| ASD | 25 | 0.08 | 0.16 | 0.76 | 0.16 | 0.84 | |
| ASS | 28 | 0.25 | 0.07 | 0.68 | 0.29 | 0.71 | |
| Total | 129 | 0.10 | 0.12 | 0.78 | 0.16 | 0.84 | |
| Hybrid | Total | 4 | 0.75 | 0.00 | 0.25 | 0.75 | 0.25 |
| Riverine | AS | 56 | 0.66 | 0.16 | 0.18 | 0.74 | 0.26 |
| CH | 25 | 0.72 | 0.12 | 0.16 | 0.78 | 0.22 | |
| KH | 10 | 0.90 | 0.10 | 0.00 | 0.95 | 0.05 | |
| DR | 27 | 0.59 | 0.30 | 0.11 | 0.74 | 0.26 | |
| BD | 29 | 0.83 | 0.17 | 0.00 | 0.91 | 0.09 | |
| TD | 46 | 0.78 | 0.13 | 0.09 | 0.85 | 0.15 | |
| SK | 48 | 0.79 | 0.21 | 0.00 | 0.90 | 0.10 | |
| PN | 18 | 0.94 | 0.06 | 0.00 | 0.97 | 0.03 | |
| SR | 9 | 1.0 | 0.0 | 0.0 | 1.0 | 0.0 | |
| MT | 36 | 0.81 | 0.14 | 0.06 | 0.88 | 0.13 | |
| JF | 35 | 0.97 | 0.03 | 0.00 | 0.99 | 0.01 | |
| MH | 48 | 0.77 | 0.23 | 0.00 | 0.88 | 0.12 | |
| BN | 28 | 0.83 | 0.17 | 0.00 | 0.91 | 0.09 | |
| NR | 70 | 0.82 | 0.16 | 0.03 | 0.89 | 0.11 | |
| MU | 49 | 0.83 | 0.10 | 0.06 | 0.89 | 0.11 | |
| Total | 534 | 0.82 | 0.14 | 0.05 | 0.89 | 0.12 | |
| Male | Swamp | 26 | 0.27 | 0.00 | 0.73 | 0.27 | 0.73 |
| Assamese | 24 | 0.83 | 0.00 | 0.17 | 0.83 | 0.17 | |
| Riverine | 36 | 0.94 | 0.00 | 0.06 | 0.94 | 0.06 | |
| Total | 86 | 0.68 | 0.0 | 0.32 | 0.68 | 0.32 | |
| Hybrid | 2 | 0.5 | 0.00 | 0.5 | 0.5 | 0.5 | |
2.2. Amplification of 5′upstream region of buffalo TLR8 gene
For the characterization of the 5′upstream region and 5′UTR of buffalo TLR8 gene, three sets of overlapping primers were designed from cattle sequence available in Ensembl browser (ENSBTAG00000031020) for PCR amplification (Supplementary Table 1). Amplification of 5′upstream region TLR8 gene of Bos indicus was also carried out to compare the buffalo data. PCR was performed in a total volume of 20 μl containing 70 ng genomic DNA, 10 × PCR buffer having 15 mM MgCl2, 0.5 μl of 10 mM dNTPs, 10 pmole of each primer and 1 U of Taq DNA Polymerase (Bangalore Genei, India). Following the initial denaturation step (95 °C for 3 min), samples were subjected to 32 cycles of PCR consisting of 94 °C for 30 s, primer specific annealing temperature for 30 s (Supplementary Table 1); and 72 °C for 1 min, followed by a final extension for 5 min at 72 °C. Amplified products were checked on 1.5% agarose gel and sequenced both end after purification. Data was further analyzed using SeqMan and MegAlign program of the Lasergene software (DNASTAR Inc., USA).
2.3. Polymorphism detection and PCR–RFLP assay
The 5′UTR of TLR8 gene was amplified using primer P1 (Supplementary Table 1) on a panel of eight animals of each swamp and riverine buffaloes and purified using PCR purification kit (QIAGEN Inc., CA, USA) and both strand sequencing was performed using forward and reverse primers on ABI 3100 Genetic Analyzer (Applied Biosystems, CA, USA). The sequence of each DNA sample was checked manually in ChromasLite program (http://www.technelysium.com.au/chromas_lite.html) and further subjected to multiple alignments to identify nucleotide variations, using MegAlign program of the Lasergene software. A PCR–RFLP genotyping protocol was developed for the SNP g.-139G>T using the NEB cutter online program (http://tools.neb.com/NEBcutter2/). The PCR products generated using same primer set P1 were digested with the Tsp509I restriction enzyme to differentiate the alleles. The digested products were resolved on 3% agarose gel and the genotypes were recorded manually. This assay was used to screen 667 DNA samples from 20 different riverine and swamp buffalo populations of India (Table 1).
2.4. Statistical analysis
Allele frequency, FST (Weir and Cockerham, 1984) and genetic distance (Nei's standard genetic distances) were calculated using GenAlex6.2 program (Peakall and Smouse, 2006). A principal component analysis (PCA) was carried out to determine breed/population relationships directly based on allele frequencies (Manly, 1986), using GenAlex6.2 program.
3. Results
A total of 1045 bp nucleotide sequence of TLR8 gene was amplified across buffalo along with cattle and compared with reported sequences of other species. The buffalo sequence (GenBank acc no. KC342230) exhibited 99% homology with goat TLR8 gene (GenBank acc no. JQ911705) and only 83% homology with bovine TLR8 sequence (GenBank acc no. KC342231).
3.1. Sequence diversity
In order to identify the genetic polymorphism in the 5′UTR of buffalo TLR8 gene, DNA samples of 8 animals of riverine and swamp buffaloes were amplified by PCR and sequenced. Sequence alignment revealed two polymorphic sites, one G>T transversion 139 nt upstream and another A>G transition 128 nt upstream to the ORF. A PCR–RFLP genotyping assay was developed for the g.-139G>T SNP of buffalo TLR8 gene using Tsp509I restriction enzyme. A single Tsp509I restriction site in the G allele resulted in two fragments of 319 and 123 bp, whereas two restriction sites in the T allele yielded three bands of 253, 123 and 66 bp (Fig. 2). The allele and genotype frequencies of Tsp509I variants in each of the 20 Indian buffalo breeds/populations analyzed have been listed in Table 1. The allele frequency of G and T alleles across all buffalo populations ranged from 0.0 to 0.99 and 0.0 to 1.0 respectively. The allele frequency of T allele was observed to be higher in swamp buffalo as compared to riverine buffalo, ranging from 0.71 to 1. The G allele on the other hand exhibited a higher frequency across all the Indian riverine buffalo breeds/populations (0.89). The frequency of the G allele in the hybrid buffalo population from Assam was observed to be much higher (0.75) than that observed in swamp buffalo but slightly less than that of riverine buffalo. The overall frequency of the GG genotype was observed to be 0.10, 0.75 and 0.82 in swamp, hybrid and riverine buffalo populations respectively. The average frequency of the heterozygous GT genotype was found to be 0.12 in swamp buffalo and this genotype was not detected in the 4 samples of hybrid buffaloes. The TT genotype was not detected in any of the investigated samples of Kalahandi, Bhadawari, South Kanara, Pandharpuri, Surti, Jaffarabadi, Mehsana and Banni riverine buffaloes. However, sixteen swamp buffalo samples from Mizoram exhibited 100% homozygous TT genotype. Heterozygous GT genotype was also absent in the swamp buffalo of Manipur but Nagaland and Assamese buffaloes revealed a higher frequency of the GT genotype. Since TLR8 is located on the X chromosome, the male buffaloes of all the breeds/populations were analyzed separately to assess the diversity among male buffaloes (Table 1). The average frequency of G allele was higher across all male buffaloes (0.94) of riverine compared to swamp type males having frequency of 0.27 for the G allele.
Fig. 2.
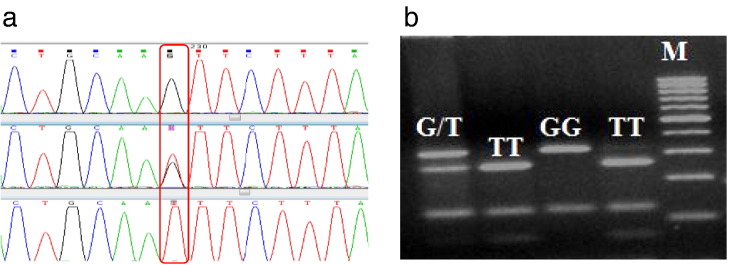
Identification and genotyping of SNP. a. Chromatogram of three different PCR products revealing mutation g.-139G>T in 5′upstream region of buffalo TLR8 gene. b. Restriction digestion of buffalo TLR8 Prom P1 amplified PCR products with Tsp509I enzyme showing different genotypes in 3% agarose gel.
3.2. Phylogenetic analyses
The phylogenetic relationship between the investigated buffalo breeds/populations was determined by the DA genetic distance method. Among the swamp buffalo, the maximum genetic distance (0.048) was observed between Assamese Silchar and Mizoram buffaloes, whereas the Assamese Dibrugarh and Manipur buffaloes appeared closely related (Table 2). Among the riverine buffalo highest differentiation was observed between Kalahandi and Surti (0.127). The results revealed that the swamp buffaloes were quite distinct from riverine buffaloes as expected. However, the Assamese buffalo population showed greater identity (0.677) with swamp buffalo than riverine buffalo (genetic identity = 0.429) The Assamese buffalo appeared to be closer to the riverine buffaloes Chilika, Kalahandi and Diara buffaloes of eastern region adjoining to swamp area, as compared to buffalo population of Northern, Western or Southern India. Pairwise FST values between the different buffalo populations also revealed similar results, with the swamp buffalo clearly differentiating from the rest of the buffalo populations (Supplementary Table 2).
Table 2.
Genetic distances DA (Nei et al., 1983) below diagonal and FST above diagonal between different swamp buffalo populations of eastern region of India.
| Buffalo population | Mizoram | Nagaland | Assamese Silchar | Manipuri | Assamese Dibrugarh |
|---|---|---|---|---|---|
| Mizoram | 0.000 | 0.077 | 0.137 | 0.042 | 0.072 |
| Nagaland | 0.014 | 0.000 | 0.016 | 0.010 | 0.000 |
| Assamese Silcher | 0.048 | 0.010 | 0.000 | 0.048 | 0.019 |
| Manipuri | 0.004 | 0.003 | 0.025 | 0.000 | 0.008 |
| Assamese Dibrugarh | 0.012 | 0.000 | 0.012 | 0.002 | 0.000 |
These results were further substantiated by the principal component analysis (PCA) which depicts the spatial distribution of buffalo populations based on genetic distances. The PCA of the three groups of buffalo (swamp, riverine and Assamese-hybrids) revealed a clear differentiation between them (Fig. 3a). When the buffaloes were grouped region wise, the buffalo population from Bihar and Odisha states appeared closer to the Assamese buffalo (Fig. 3b), the remaining riverine buffalo populations clustered together but were distinct from the swamp buffalo. PCA among the swamp buffaloes illustrated a close relationship of the Assamese Dibrugarh and Nagaland buffaloes again (Fig. 3c.). PCA based on allele frequencies among male animals revealed distinct differentiation of riverine, swamp and hybrid buffaloes as expected.
Fig. 3.

Principal component analysis based on allele frequencies. a. Showing placement of riverine, swamp and hybrid buffaloes across quadrants. b. Within swamp types of different regions. c. Individual riverine, swamp and hybrid types, hybrids of Assam placed closer to riverine buffaloes of eastern states of Bihar and Odisha.
4. Discussion
The TLR genes are crucial in triggering an innate immune response against pathogens and have been widely studied in several species (Astakhova et al., 2009; Raja et al., 2011; Dubey et al., 2012a). Nucleotide variation in these genes, associated with disease resistant and susceptibility may help in understanding the factors responsible for an enhanced or reduced defense against pathogenic invasion. Nucleotide changes particularly in the 5′upstream region and 5′UTR of genes may affect their expression by influencing the transcriptional binding factors and stability. Genetic variations in the 5′upstream regions of the TLR2 and TLR4 genes have been associated with somatic cell score and lactation persistency, mastitis etc. in cattle (Sharma et al., 2006; Huang et al., 2011). Maximum homology (99%) of 5′upstream region of buffalo TLR sequence with that of goat was observed to be in concordance with previous reports on TLR8 (Dubey et al., 2012a,b) and DRB*A (Naskar et al., 2012) genes, wherein the water buffalo was observed to be phylogenetically more closer to goat than cattle. High homology of TLR8 region between riverine buffalo and goat suggests that TLR genes have evolved due to challenging by different types of species specific pathogens within populations of livestock species.
4.1. Polymorphism detection and genotyping
Polymorphism within TLR8 gene was associated with high risk to viral infection in Chinese population (Cheng et al., 2007). TLR8 Met1Val and TLR8-129G>C single nucleotide polymorphisms (SNPs rs3764879 and rs3764880) were found in strong association with tuberculosis within male population and associated with coronary artery disease (CAD) in the Chinese population but no significant association within Turkish population was reported (Cheng et al., 2007; Dalgic et al., 2011). Analysis of a stretch of 1045 nucleotides of the 5′-upstream of TLR8 gene led to the identification of two SNPs in this region and three in CDS and one in 3′UTR of buffalo TLR8 gene (Dubey et al., 2012b). This nucleotide diversity was found lower when compared to the polymorphism in the coding region of TLR8 cattle (12), sheep (20), pig (22) and horse (5) polymorphic sites (Cargill and Womack, 2007; Astakhova et al., 2009; Mikula et al., 2010; Uenishi et al., 2011).
The PCR–RFLP assay designed for genotyping the g.-139G>T SNP across 667 buffalo samples of both riverine and swamp types revealed a striking difference in allele frequency distribution of the g.-139G allele across the swamp and riverine buffaloes. The g.-139T allele appeared to be fixed in the Mizoram swamp buffaloes, but it needs to be interpreted with caution as only sixteen samples were analyzed. In general however, all the swamp buffalo populations revealed a higher frequency of the g.-139T allele in contrast to the higher frequency of g.-139G allele in riverine buffaloes.
The significant allele frequency differences between riverine and swamp buffaloes allowed the swamp and riverine buffaloes to be easily distinguished. The genetic distinctness of riverine and swamp buffaloes is well established and this may account for the nucleotide variations observed in this study. However, the near fixation of g-139T allele in swamp buffaloes may also suggest maintenance of this variation by selective pressure during evolution. Previous report on polymorphism in the cytokine promoter region of riverine and swamp buffalo indicates its association with changes in the expression of IFN-γ and TNF-α (Mingala et al., 2009). Therefore, further studies are required to determine the functional relevance of this polymorphism.
4.2. Phylogenetic analyses
The phylogenetic analysis based on g.-139G>T SNP revealed that all the investigated swamp buffalo populations to be closely related (DA ≤ 0.048) however, the greater genetic distance observed between Mizoram and Assamese Silchar buffaloes may be explained by higher frequency of G allele, which was absent in Mizoram buffalo. Earlier studies of allele frequency distribution at another locus g.-2758A>G in the TIR domain of TLR8 gene have also shown similar results in allelic distribution (Dubey et al., 2012b).
The closeness of Nagaland (NG) and Assamese Dibrugarh (ASD) buffalo populations reveals genetic similarity based on polymorphism in 5′upstream of TLR8 gene, which is supported by the geographic propinquity between them. Further, the riverine buffalo breeds of Bihar (Diara) and Odisha (Chilika) appeared phylogenetically closer to the Assamese buffalo. This indicates spread of buffaloes from the northeast region towards the eastern coastal and northwest part of India.
The PCA revealed separate clustering of the riverine and swamp buffaloes, as expected, but the riverine/hybrid buffalo population of Assam region of Eastern India formed a distinct cluster. Earlier studies based on microsatellite markers have also segregated the Assamese buffalo from other buffalo populations (Mishra et al., 2010). In the present study, a general divide between the buffalo population of the eastern region and rest of the country was observed. Since half of the buffalo populations in the eastern region are swamp type, this demarcation may be related to the difference in immune response in riverine and swamp buffaloes. The variation at the identified locus may perhaps suggest its contribution in the disease resistance traits of the swamp and riverine buffaloes, which are known for differential immune response towards pathogens (Mingala et al., 2009).
The information generated will therefore, lead to further investigations involving association of the variations with disease susceptibility or resistance as well as comparative analysis of induction of immune response in buffalo.
Acknowledgments
The authors wish to thank National Agricultural Innovation Project, Indian Council of Agricultural Research, for funding the work under the scheme C2153, reported here. Technical help received from Mr. Naresh Kumar and Anil Kumar is gratefully acknowledged.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary data
Supplementary tables.
References
- Astakhova N.M., Perelygin A.A., Zharkikh A.A., Lear T.L., Coleman S.J., Macleod J.N., Brinton M.A. Characterization of equine and other vertebrate TLR3, TLR7 and TLR8 genes. Immunogenetics. 2009;61:529–539. doi: 10.1007/s00251-009-0381-z. [DOI] [PubMed] [Google Scholar]
- Cargill E.J., Womack J.E. Detection of polymorphisms in bovine Toll-like receptors 3, 7, 8, and 9. Genomics. 2007;89:745–755. doi: 10.1016/j.ygeno.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Cheng P.L., Eng H.L., Chou M.H., You H.L., Lin T.M. Genetic polymorphisms of viral infection-associated Toll-like receptors in Chinese population. Transl. Res. 2007;150:311–318. doi: 10.1016/j.trsl.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Dalgic N., Tekin D., Kayaalti Z., Cakir E., Soylemezoglu T., Sancar M. Relationship between Toll-like receptor 8 gene polymorphisms and pediatric pulmonary tuberculosis. Dis. Markers. 2011;31:33–38. doi: 10.3233/DMA-2011-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila S., Hibberd M.L., Dass H.R., Wong H.E.E., Sahiratmadja E. Genetic association and expression studies indicate a role of Toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet. 2008;4:e1000218. doi: 10.1371/journal.pgen.1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X., Poltorak A., Wei Y., Beutler B. Three novel mammalian Toll-like receptors: gene structure, expression, and evolution. Eur. Cytokine Netw. 2000;11:362–371. [PubMed] [Google Scholar]
- Dubey P.K., Goyal S., Kathiravan P., Mishra B.P., Gahlawat S.K., Kataria R.S. Sequence characterization of river buffalo Toll-like receptor genes 1–10 reveals distinct relationship with cattle and sheep. Int. J. Immunogenet. 2012 doi: 10.1111/j.1744-313X.2012.01135.x. [DOI] [PubMed] [Google Scholar]
- Dubey P.K., Goyal S., Aggarwal J., Gahlawat S.K., Kathiravan P., Mishra B.P., Kataria R.S. Sequence and topological characterization of Toll-like receptor 8 gene of Indian riverine buffalo (Bubalus bubalis) Trop. Anim. Health Prod. 2012 doi: 10.1007/s11250-012-0178-1. [DOI] [PubMed] [Google Scholar]
- Fabbri M., Paone A., Calore F., Galli R., Gaudio E., Santhanam R., Lovat F., Fadda P., Mao C., Nuovo G.J., Zanesi N., Crawford M., Ozer G.H., Wernicke D., Alder H., Caligiuri M.A., Nana-Sinkam P., Perrotti D., Croce C.M. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. U. S. A. 2012;109(31):E2110–E2116. doi: 10.1073/pnas.1209414109. (31) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harisah M., Azmi T.I., Hilmi M., Vidyadaran M.K., Bongso T.A., Nava Z.M., Momongan V., Basrur P.K. Identification of crossbred buffalo genotypes and their chromosome segregation patterns. Genome. 1989;32:999–1002. doi: 10.1139/g89-544. [DOI] [PubMed] [Google Scholar]
- Huang J., Liu L., Wang H., Zhang C., Ju Z., Wang C., Zhong J. Variants and gene expression of the TLR2 gene and susceptibility to mastitis in cattle. Asian J. Anim. Vet. Adv. 2011;6:51–61. [Google Scholar]
- Kataria R.S., Tait R.G., Kumar D., Ortega M.A., Rodiguez J., Reecy M.J. Association of Toll-like receptor 4 single nucleotide polymorphisms with incidence of infectious bovine keratoconjunctivitis (IBK) in cattle. Immunogenetics. 2011;63:115–119. doi: 10.1007/s00251-010-0484-6. [DOI] [PubMed] [Google Scholar]
- Livestock Census Part IV Livestock Census BAHS 2012. 2007 http://dahd.nic.in/dahd/bahs-2012.aspx (Accessed April 2013) [Google Scholar]
- Manly B.F.J. Chapman and Hall; London: 1986. Multivariate Statistical Methods: A Primer. [Google Scholar]
- Mikula I., Mangesh B., Silvia P., Mikula I. Characterization of ovine TLR7 and TLR8 protein coding regions, detection of mutation and Maedi visna virus infection. Vet. Immunol. Immunopathol. 2010;138:51–59. doi: 10.1016/j.vetimm.2010.06.015. [DOI] [PubMed] [Google Scholar]
- Mingala C.N., Konnai S., Cruz L.C., Onuma M., Ohashi K. Comparative moleculo-immunological analysis of swamp- and riverine-type water buffaloes responses. Cytokine. 2009;46:273–282. doi: 10.1016/j.cyto.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Mishra B.P., Prakash B., Kataria R.S., Sadana D.K., Kathiravan P., Das G.C., Goswami R.N., Joshi B.K., Bhasin V., Rasool T.J., Bujarbaruah K.M. Genetic diversity analysis and cytogenetic profiling of Assamese buffaloes from North-East India. Indian J. Anim. Sci. 2010;80:142–147. [Google Scholar]
- Mucha R., Bhide M.R., Chakurkar E.B., Novak M., Mikula I., Sr. Toll-like receptors TLR1, TLR2 and TLR4 gene mutations and natural resistance to Mycobacterium avium subsp. paratuberculosis infection in cattle. Vet. Immunol. Immunopathol. 2009;128:381–388. doi: 10.1016/j.vetimm.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Naskar S., Deb S.M., Niranjan S.K., Kumar S., Sharma D., Sakaram D., Sharma A. Molecular characterization of MHC-DRB cDNA in water buffalo (Bubalus bubalis) Genet. Mol. Biol. 2012;35:95–98. doi: 10.1590/s1415-47572012005000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M., Tajima F., Tateno Y. Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol. 1983;19:153–170. doi: 10.1007/BF02300753. [DOI] [PubMed] [Google Scholar]
- Peakall R., Smouse P.E. GENEALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol. Ecol. Notes. 2006;6:288–295. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja A., Vignesh A.R., Mary B.A., Tirumurugaan K.G., Raj G.D., Kataria R.S., Mishra B.P., Kumanan K. Sequence analysis of Toll-like receptor genes 1–10 of goat (Capra hircus) Vet. Immunol. Immunopathol. 2011;140:252–258. doi: 10.1016/j.vetimm.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Russell D.W. 3rd ed. Cold Spring Harbor Laboratory Press; New York: 2001. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- Sharma B.S., Leyva I., Schenkel F., Karrow N.A. Association of Toll-like receptor 4 polymorphisms with somatic cell score and lactation persistency in Holstein bulls. J. Dairy Sci. 2006;89:3626–3635. doi: 10.3168/jds.S0022-0302(06)72402-X. [DOI] [PubMed] [Google Scholar]
- Takeda K., Akira S. Toll-like receptor in innate immunity. Int. Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- Taylor D.L., Zhong L., Begg D.J., de Silva K., Whittington R.J. Toll-like receptor genes are differentially expressed at the sites of infection during the progression of Johne's disease in outbred sheep. Vet. Immunol. Immunopathol. 2008;124:132–151. doi: 10.1016/j.vetimm.2008.02.021. [DOI] [PubMed] [Google Scholar]
- Uenishi H., Shinkai H., Morozumi T., Muneta Y., Jozaki K., Kojima-Shibata C., Suzuki E. Polymorphisms in pattern recognition receptors and their relationship to infectious disease susceptibility in pigs. BMC Proc. 2011 doi: 10.1186/1753-6561-5-S4-S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir B.S., Cockerham C.C. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Zhu J., Brownlie R., Liu Q., Babiuk L.A., Potter A., Mutwiri G.K. Characterization of bovine Toll-like receptor 8: ligand specificity, signaling essential sites and dimerization. Mol. Immunol. 2009;46:978–990. doi: 10.1016/j.molimm.2008.09.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary tables.