

Abstract
Objective
It was previously reported that docosahexanoic acid (DHA) reduces TNF-α-induced necrosis in L929 cells. However, the mechanisms underlying this reduction have not been investigated. The present study was designed to investigate cellular and biochemical mechanisms underlying the attenuation of TNF-α-induced necroptosis by DHA in L929 cells.
Methods
L929 cells were pre-treated with DHA prior to exposure to TNF-α, zVAD, or Necrostatin-1 (Nec-1). Cell death and survival were assessed by MTT and caspase activity assays, and microscopic visualization. Reactive oxygen species (ROS) were measured by flow cytometry. C16- and C18-ceramide were measured by mass spectrometry. Lysosomal membrane permeabilization (LMP) was evaluated by fluorescence microscopy and flow cytometry using Acridine Orange. Cathepsin L activation was evaluated by immunoblotting and fluorescence microscopy. Autophagy was assessed by immunoblotting of LC3-II and Beclin.
Results
Exposure of L929 cells to TNF-α alone for 24 h induced necroptosis, as evidenced by inhibition of cell death by Nec-1, absence of caspase-3 activity and lamin B cleavage, and morphological analysis. DHA attenuated multiple biochemical events associated with TNF-α-induced necroptosis, including ROS generation, ceramide production, lysosomal dysfunction, cathepsin L activation, and autophagic features. DHA also attenuated zVAD-induced necroptosis but did not attenuate the enhanced apoptosis and necrosis induced by combination of TNF-α with Actinomycin D or zVAD, respectively, suggesting that its protective effects might be limited by the strength of the cell death insult induced by TNF-α.
Conclusions
DHA effectively attenuates TNF-α-induced necroptosis and autophagy, most likely via its ability to inhibit TNF-α-induced sphingolipid metabolism and oxidative stress. These results highlight the role of this Omega-3 fatty acid in antagonizing inflammatory cell death.
Keywords: DHA, L929 cells, lysosomes, necroptosis, oxidative stress, inflammation, TNF-α
Introduction
Tumor necrosis factor alpha (TNF-α) is widely recognized as a key initiator and master regulator of inflammation, playing a key role in promoting immune defense against infectious agents, chronic inflammation, and programmed cell death [1,2]. The inflammatory responses induced by TNF-α are mediated by its interaction with two cell surface receptors, TNFR1 and TNFR2 [2]. Depending on the micro-environmental and cellular context these interactions may not only activate cell death signaling pathways but also survival pathways mediated by transcription factors NF-kB and AP-1 that result in increased cell proliferation, migration, tissue repair, or angiogenesis [3,4]. The molecular mechanisms underlying TNF-α-induced inflammatory cell death have attracted considerable attention recently because of the central role of this cytokine in several pathologies, including neurodegeneration, rheumatic and autoimmune diseases, inflammatory bowel disease, renal failure, and sepsis [2–6].
Due to its heightened sensitivity to TNF-α-induced cytotoxicity, the L929 mouse fibrosarcoma cell line is widely used as the prototype model system to elucidate mechanisms of cytokine-induced inflammatory cell death [7–10]. It is well documented that in this cell line TNF-α preferentially induces a caspase-independent programmed necrotic cell death process termed necroptosis [7,8]. Caspase-dependent apoptosis is typically not observed in L929 cells exposed to TNF-α, unless the cells are also co-treated with transcription or translation inhibitors such as actinomycin D (ActD) or cycloheximide [9,10]. Necroptosis is activated by TNF-α-mediated death receptor signaling leading to the formation of a cytoplasmic protein complex known as the necrosome, which is required for activation of the receptor interacting protein kinases 1 and 3 (RIPK1/3), the key drivers of necroptosis [11–13]. While the mechanisms by which RIPK1 and RIPK3 drive necroptosis in L929 cells are still under intense investigation, it is clear that this mode of cell death is associated with caspase inactivation, generation of reactive oxygen species (ROS), lysosomal and mitochondrial dysfunction, loss of cytoplasmic membrane integrity, and extensive cytoplasmic damage [11–16]. TNF-α-induced necroptosis can be dramatically enhanced in L929 cells under conditions in which caspases are inactive or blocked with broad inhibitors such as zVAD [9,10,14]. TNF-α also activates autophagy, a cellular self-digestion process that may lead to either cell death or survival in L929 cells and other cell types, and available evidence suggests that it contributes to inflammatory cell death [17–19]. Recently, both necroptosis and autophagy have been the subject of intense investigation due to their importance in pathophysiological and inflammatory conditions [20,21].
Omega-3 polyunsaturated fatty acids such as docosahexanoic acid (DHA) and eicosapentaenoic acid (EPA) are potent anti-inflammatory and immunoregulatory agents in a wide variety of diseases associated with chronic inflammation, including metabolic syndrome [22], neurodegeneration [23], inflammatory bowel diseases [24], cardiovascular disease [25], and cancer [26]. DHA- and EPA-induced anti-inflammatory responses are mediated by their respective bioactive mediators such as resolvins and protectins, which act as inhibitors of leukocyte infiltration and signaling activity of inflammatory cytokines, particularly TNF-α [27,28]. DHA also has anti-apoptotic activity, widely documented in neuronal cells, retinal photoreceptor cells, and other cell types [29–31]. Our group reported that DHA-enrichment elicits neuroprotection against lipotoxicity in vitro [32,33], and that DHA pretreatment ameliorates functional deficits and increases tissue sparing in vivo in experimental models of spinal cord injury [34].
Previous reports demonstrated that DHA enrichment reduced TNF-α-induced necrosis in human monocyte U937 cells and in L929 cells, however, the mechanisms underlying this inhibition were not investigated [35,36]. Understanding the inhibitory potential of Omega-3 fatty acids on inflammatory cell death requires uncovering molecular and cellular mechanisms by which these fatty acids antagonize TNF-α function. In this study we demonstrate that DHA antagonizes TNF-α-induced necroptosis in L929 cells via a mechanism that involves attenuation of ROS generation, ceramide production, lysosome dysfunction, and autophagy.
Materials and Methods
Cell culture and reagents
L929 cells were obtained from the American Type Culture Collection (Rockville, MD, USA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 2 mM L-glutamine, 100 units/ml of penicillin, 100 μg/ml of streptomycin (all obtained from Cellgro, Herndon, VA, USA), and 10% fetal bovine serum (Omega Scientific, Tarzana, CA, USA). Acridine Orange (AO), 3-(4,5-dimethylthiazol-2yl-)-2,5-diphenyl tetrazolium bromide (MTT), mouse TNF-α, and necrostatin-1 (Nec-1) were from Sigma-Aldrich (St. Louis, MO, USA). Dimethyl sulfoxide (DMSO) was from Fisher Scientific (Tustin, CA, USA). The broad caspase inhibitor zVAD (benzylocarbonyl-Valine-Alanine-Aspartic acid-fluoromethylketone) was obtained from Enzo Life Sciences (Farmingdale, NY). Cathepsin L specific inhibitor Z-FY-CHO was from Calbiochem (San Diego, CA, USA). The cathepsin L fluorogenic substrate based assay Magic Red MR-(FR)2 and the DNA fluorescent binding dye Hoechst 33342 were purchased from Immunochemistry (Bloomington, MN, USA). The caspase-3/7 substrate Ac-DEVD-AMC, the caspase-8 substrate Ac-IETD-AMC, and Staurosporine were obtained from Enzo Life Sciences and diluted to concentrated stocks in DMSO. Goat polyclonal antibodies to Beclin (BECN1, sc-10086), Lamin B (C-20, sc-6216), and cathepsin L (M-19, sc-6502) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit polyclonal antibody to LC3-II (NB600-1384 lot B1) was obtained from Novus Biologicals (Littleton, CO, USA). Human scleroderma-associated autoantibodies to DNA topoisomerase I (Topo I) were from the serum bank of Dr. Carlos A. Casiano at the Loma Linda University Center for Health Disparities and Molecular Medicine. DHA (Sigma-Aldrich, St. Louis, MI) was dissolved in 1 mM ethanol, and diluted in warm low serum medium with 150 μM fatty acid-free bovine serum albumin (BSA) (EMD Biosciences, La Jolla, CA, USA).
Cell death and viability assays
For cell viability assays, L929 cells were seeded at 80–90% confluency in 96-well plates, and exposed to 10 ng/ml of mouse TNF-α for up to 24 h, with and without preincubation with DHA (50 μM), followed by addition of the MTT substrate and incubation for 2 h at 37°C in 5% CO2. Supernatants were discarded and the resulting blue precipitates were dissolved by addition of 150 μl of DMSO to each well. Absorbance was measured at 450 nm using a μQuant microplate reader (BioTek Instruments, Winooski, VT, USA). In some experiments, cells were pre-treated with DHA for up to 24 h, or with Nec-1 (30 μM, prepared from concentrated stock dissolved in 1 mM ethanol) for 1 h, prior to addition of TNF-α or zVAD. DHA and Nec-1 were maintained in the cell cultures after addition of TNF-α or zVAD. Morphological analysis of treated cells was performed using an Olympus IX70 inverted microscope equipped with Hoffman Modulation Contrast (Olympus America, Center Valley, PA, USA). Images were acquired using a digital Spot RT3™ camera (Diagnostic Instruments, Sterling Heights, Michigan, USA).
For measurement of caspase-3 activity, L929 cells were seeded in black, clear-bottomed 96-well plates (104 cells per well). At the conclusion of treatment with drugs or inhibitors, cells were incubated with 50 μl of 3X caspase buffer [150mM Hepes pH 7.4, 450 mM NaCl, 150 mM KCl, 30 mM MgCl2, 1.2 mM EGTA, 30% sucrose, 10% CHAPS, and 1.5% NP-40], 30 mM DTT, 3 mM PMSF, and 75 μM of the fluorogenic caspase substrates Ac-DEVD-AMC (caspase 3/7) or Ac-IETD-AMC (caspase-8) for 3 h and 10 h, respectively, at 37°C. Absorbance was then read in a FLX800 Microplate Fluorescent Reader (Bio-Tek Instruments) at excitation of 360 nm and emission of 460 nm. Fold activity was determined by normalizing to one the absorbance values for untreated, control cells. Staurosporine (2 μM) was added to the cells for 8 h as a positive control for caspase activation.
Electrophoresis and immunoblotting
Immunoblotting procedures were carried out as described previously [14]. Briefly, proteins from L929 whole cell lysates were separated by 4–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto polyvinyl difluoride (PVDF) membranes. Protein membranes were blocked with 5% dry milk dissolved in phosphate buffered or Tris-buffered saline-Tween (PBS-T or TBS-T), and incubated with antibodies to cathepsin L, Beclin, LC3-II, or Topo I. Protein bands were detected by chemiluminescence (Perkin Elmer, Boston, MA, USA) after incubation of membranes with appropriate HRP-conjugated secondary antibodies. Antibody to β-actin (Sigma, St Louis, MO, USA) was used as protein loading control.
Flow cytometric analysis of TNF-α-induced ROS
Detection of ROS was performed using the dichlorofluorescein (DCF) flow cytometric method as described previously [33]. Briefly, L929 cells treated with TNF-α for 24 h, with and without pre-incubation with DHA, were incubated with 10 μM of 2′,7′-dichlorodihydrofluorescein deacetate (H2DCFDA, Molecular Probes, Eugene, OR, USA) for 20 min at 37°C. The intensity of DCF fluorescence, proportional to the amount of intracellular H2O2, was measured using a FACSCalibur® flow cytometer at excitation/emission of 488/530 nm respectively. Positive control cells were treated with 300 μm H2O2 for 15 min. Data was collected in log scale and analyzed using Cell Quest Pro® and Flow-Jo® software.
Assessment of TNF-α-induced endogenous ceramide production
Ceramide extraction was performed essentially as described previously [37]. Briefly, L929 cells growing in 6-well plates were treated with TNF-α for 24 h, with and without pre-incubation with DHA. Cells were harvested by removing the medium, washing with PBS and methanol, scraping the cell monolayer, and transferring the cell suspension into a 5.5-ml glass vial with a screw top. Cells were sonicated for 15 min, and 2 ml of chloroform were then added to the suspension followed by centrifugation at 3,000 rpm for 5 min. The lower chloroform layer was transferred to a new glass vial, which was then tightly closed and placed on ice. The upper methanol and middle protein-rich layers were re-extracted by adding 1 ml of methanol and 2 ml of chloroform, followed by vortexing, additional sonication, and centrifugation (3,000 rpm for 5 min). Re-extracted layers were then mixed with the lower chloroform layer and dried under nitrogen at room temperature. Dried samples were reconstituted with 1 ml of acetonitrile, vortexed, and stored at −80°C until analysis by reverse phase high performance liquid chromatography/tanden mass spectrometry (HPLC-MS/MS). C16- and C18-ceramide measurements were normalized for protein content, and protein concentration was measured using the DC Protein Assay kit (Bio-Rad, Hercules, CA, USA).
Determination of lysosomal integrity and detection of intracellular cathepsin L activity
AO was used to analyze lysosomal integrity as described previously [14,33]. Briefly, L929 grown in 6-well plates were treated with TNF-α for 24 h, with and without pre-incubation with DHA. Cells were then exposed to 5 μg/ml AO in complete medium for 15 min at 37°C, rinsed twice with medium, and directly examined under an Olympus BX50 fluorescence microscope using a LUMPlanPl 60X/0.90W water immersion objective. Cathepsin L activity was detected using the fluorogenic substrate-based assay Magic Red MR-(FR)2 [14,33]. Images were acquired using a digital Spot RT3™ camera (Diagnostic Instruments, Sterling Heights, Michigan, USA). LMP was also measured by flow cytometry as described previously [33]. Briefly, L929 cells were stained with AO (5 μg/ml) in DMEM for 15 min at 37°C, washed, and resuspended in PBS. The green (FL1) and red (FL3) fluorescence of 10,000 cells was recorded on a logarithmic scale using a Becton Dickinson FACScan instrument under 488 nm excitation. Decreased FL2 red fluorescence indicated reduced lysosomal integrity.
Statistical analysis
All statistical analyses were done using the Student’s t-test in Microsoft Excel. Standard deviations were calculated based on at least three independent experiments performed in triplicates. Significance was accepted at P < 0.05.
Results
DHA attenuates TNF-α-induced necroptosis in L929 cells
Exposure of L929 cells to TNF-α alone preferentially induces necroptosis, with no apoptosis observed [7,8,10]. As a first step in uncovering mechanisms underlying DHA-mediated attenuation of TNF-α-induced cytotoxicity in L929 cells, we first confirmed that TNF-α induced necroptosis in our cultured cells. Treatment of L929 cells with 10 ng/ml of TNF-α alone for 24 h resulted in significant loss of cell viability as measured by MTT assays (Fig. 1A, P<0.01). Microscopic analysis of TNF-α-treated L929 cells confirmed the results of the cell viability assays, showing necrosis-like morphologic features such as condensed nuclei and swelled cytoplasms, with little or no apoptotic blebbing (Fig. 1B, black arrow). We also observed small round cells in which the condensed nucleus was surrounded by relatively little cytoplasm (Fig. 1B, white arrow). Apoptotic morphology was only observed when TNF-α was applied in combination with ActD (Fig. 1B). Loss of cell viability in the presence of TNF-α alone was significantly inhibited by pre-incubation with Nec-1, an inhibitor of necroptosis that targets RIPK1 [38] (Fig. 1A,B).
Figure 1. Induction of necroptosis by TNF-α in L929 cells.
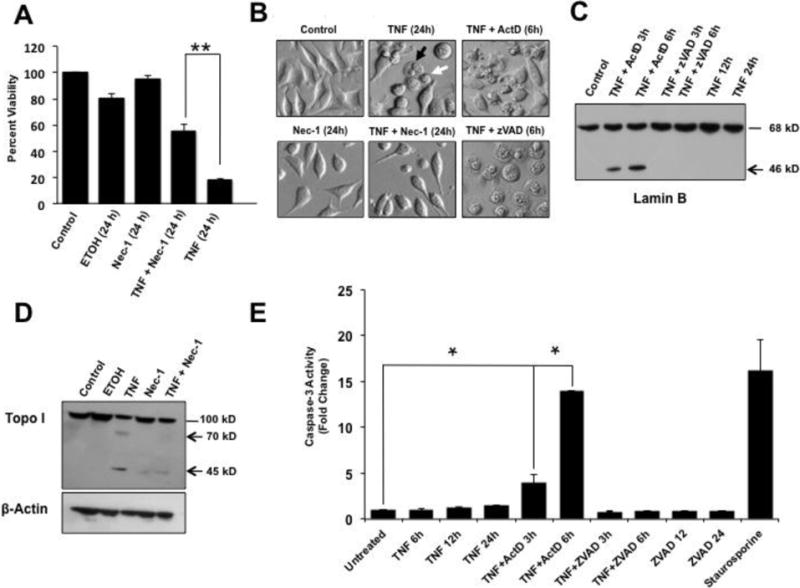
A. MTT viability assays showing that treatment of L929 cells with 30 μM Nec-1 significantly attenuated TNF-α-induced cytotoxicity compared to treatment with TNF-α alone (**P < 0.01). Ethanol (ETOH, 1 mM) was used as vehicle control. B. Microscopic analysis of TNF-α-treated L929 cells showed necrosis-like morphologic features such as condensed nuclei and swelled cytoplasms, with no apoptotic blebbing, and partial restoration of normal cell morphology in the presence of Nec-1. The combination of TNF-α plus Actinomycin D (ActD) induced the classical apoptotic morphology characterized by cellular blebbing and apoptotic bodies, whereas the combination of TNF-α plus zVAD induced extensive necrosis. The black arrow points to a round swelled cell whereas the white arrow points to a small round cell with pyknotic nuclei and little cytoplasm. C. Immunoblots of lysates from L929 cells exposed to TNF-α/ActD showed cleavage of Lamin B into its signature 46 kD apoptotic fragment but no cleavage was observed in cells treated with TNF-α alone or TNF-α/zVAD. D. Immunoblots of lysates from L929 cells exposed to TNF-α alone also showed cleavage of Topo I into its necrotic signature fragments of 70 kD and 45 kD, with Nec-1 inhibiting this cleavage. β-actin was used as loading control. E. Caspase-3 activity, measured with the Ac-DEVD-AMC substrate, was detected in cells treated with TNF-α/ActD but not in cells treated with TNF-α alone or TNF-α/zVAD (*P < 0.05). Staurosporine, a classical inducer of apoptosis and caspase-3, served as positive control for caspase activation. The results shown in A and E are from at least three independent experiments performed in duplicates or triplicates.
To further confirm that TNF-α induced necroptosis in our L929 cultures, we examined the cleavage patterns of Lamin B and Topo I. We and others previously demonstrated that during apoptosis the nuclear envelope and matrix-associated protein Lamin B (68 kD) is cleaved by caspase-3 into a distinct fragment of 46 kD. However, Lamin B is not cleaved during necrosis [39–41]. We also demonstrated previously that during TNF-α/ActD-induced apoptosis, Topo I (100 kD) is cleaved by caspases into a signature 70 kD fragment. However, during necrosis induced by TNF-α and other insults this protein is cleaved into two signature fragments of 70 and 45 kD [14,39,42]. These fragments are readily detected by human autoantibodies to Topo I derived from patients with scleroderma, and can also be detected with commercial antibodies [14]. Immunoblots revealed cleavage of Lamin B into its signature apoptotic 46 kD fragment during TNF-α/ActD-induced apoptosis (Fig. 1C). As expected, no Lamin B cleavage was observed during TNF-α-induced necroptosis, with or without z-VAD (Fig. 1C). Topo I was also cleaved into its signature necrotic cleavage fragments of 70 kD and 45 kD in L929 cells treated with TNF-α-alone, and this cleavage was inhibited by the necroptosis inhibitor Nec-1 (Fig. 1D). Consistent with the preferential induction of necroptosis by TNF-α, treatment of L929 cells with this cytokine alone or in combination with z-VAD failed to inactivate caspase-3, whereas TNF-α/ActD-induced apoptosis significantly activated this protease (Fig. 1E, P<0.05). Similar results were obtained for caspase-8 (data not shown).
Next we sought to reproduce the previous observation by Masuzawa and colleagues [35] that supplementation of L929 cells with BSA-conjugated DHA (50 μM) for up to 24 h reduced TNF-α-induced cytotoxicity. We observed that pre-incubation of L929 cells for 6h and 24 h with 50 μM DHA (diluted in medium containing 150 μM BSA) significantly attenuated TNF-α-induced necroptosis (Fig. 2A, P<0.05). Microscopic analysis of TNF-α-treated L929 cells confirmed the results of the cell viability assays, showing partial restoration of normal cell morphology in the presence of DHA (Fig. 2B). The failure of DHA to protect L929 cells against TNF-α/ActD-induced apoptosis and TNF-α/zVAD-induced enhanced necroptosis (Fig. 2C) suggested that this fatty acid may not be effective in protecting against potent insults that induce extensive and rapid cellular damage and death.
Figure 2. DHA attenuates TNF-α-induced necroptosis in L929 cells.
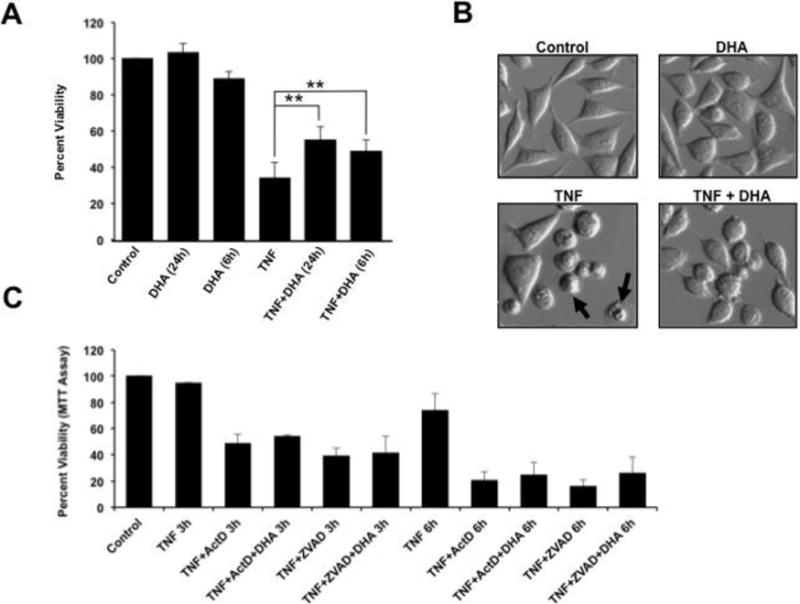
A. MTT viability assays showing that treatment of L929 cells with TNF-α for 24 h resulted in approximately 70% loss of cell viability. Pre-incubation of L929 cells for 24 h with 50 μM DHA significantly attenuated TNF-α-induced necroptosis (**P < 0.01). Pre-incubation of L929 cells with DHA for 6 h also significantly attenuated TNF-α-induced necroptosis. B. Microscopic analysis of TNF-α-treated L929 cells showed necrosis-like morphologic features such as condensed nuclei and swelled cytoplasm, without significant evidence of apoptotic blebbing (black arrows), as well as a partial restoration of normal cell morphology in cultures treated with DHA. C. MTT assays showing that DHA did not protect L929 cells against rapid (< 6 h) apoptosis or necroptosis induced by TNF-α/ActD or TNF-α/zVAD, respectively. The results shown in A and C are from at least three independent experiments performed in duplicates or triplicates.
DHA attenuates the generation of endogenous ROS and ceramide in TNF-α-treated cells
It is well established that enhanced ROS-production by mitochondria is a key mediator of TNF-α-induced necroptosis in L929 cells [15,43]. Our group reported previously that DHA attenuates ROS production during neuronal lipotoxicity [33]. To determine if DHA inhibits TNF-α-induced oxidative stress in L929 cells, we measured ROS generation by flow cytometric analysis using the DCF method. Treatment of L929 cells with TNF-α for 24 h produced a significant 1.7 fold increase in ROS generation (Fig. 3A, P<0.01). DHA by itself did not alter ROS levels relative to the control (cells incubated with DCF), but significantly attenuated TNF-α-induced ROS (Fig. 3A, P<0.05). A representative flow cytometry histogram showing the shifts in DCF fluorescence under the different treatments is provided in Fig. 3B.
Figure 3. DHA attenuates ROS and ceramide generation in TNF-α-treated cells.
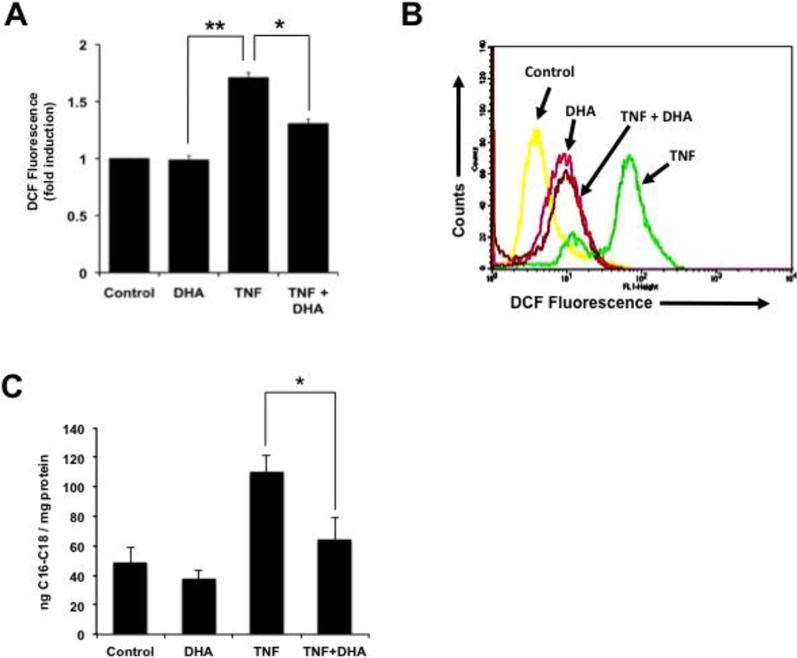
ROS generation was measured with the DCF-flow cytometry assay. A. Treatment with TNF-α for 24 h produced a significant 1.7 fold increase in ROS generation (**P < 0.01). DHA by itself did not alter ROS levels relative to the control (cells incubated with DCF), but significantly attenuated TNF-α-induced ROS (P <0.05). B. Representative flow cytometry histogram showing the shifts in DCF fluorescence under the different treatments. C. DHA attenuated C16–C18 ceramide generation in TNF-α-treated L929 cells (*P < 0.05). Cellular levels of long-chain ceramides-16 and -18 were measured in the absence or presence of TNF-α or DHA using HPLC/MS/MS. The results shown in A and B are from at least three independent experiments performed in duplicates or triplicates.
The attenuation of ROS by DHA suggested that this fatty acid might be inhibiting a key upstream pathway that influences necroptotic cell death. A likely candidate was ceramide, which is a known upstream mediator of TNF-α-induced ROS and necroptosis in L929 cells [44,45]. To determine if DHA interferes with the ability of TNF-α to induce ceramide synthesis in L929 cells, we measured the levels of the abundant ceramides C16 and C18 in these cells, in the absence and presence of TNF-α and/or DHA, using HPLC/MS/MS. Treatment of L929 cells with TNF-α induced elevated levels of C16-C18 ceramides, compared to control cells or cells treated with DHA alone (Fig. 3C). However, pre-incubation with DHA inhibited TNF-α-induced ceramide generation, suggesting that this Omega-3 fatty acid may antagonize the pro-necroptotic signaling of TNF-α by interfering with sphingolipid metabolism.
DHA attenuates lysosomal dysfunction in TNF-α-treated cells
ROS generation during TNF-α-induced cell death may lead to lysosomal damage, as evidenced by our previous observation that treatment of L929 cells with TNF-α triggers enlargement of lysosomes, LMP, and activation and release of cathepsins into the cytoplasm [14]. To determine if DHA exerts a protective effect against TNF-α-induced lysosomal dysfunction in L929 cells, we first analyzed the integrity of lysosomes in cells exposed to AO in the presence and absence of TNF-α and/or DHA. Fluorescence microscopic analysis of lysosomes in control L929 cells, or in cells treated with DHA alone for 24 h, showed intact lysosomal compartments as indicated by small clusters of cytoplasmic red/orange fluorescence separated from green fluorescence (Fig. 4A). Interestingly, most cells exposed to TNF-α alone for 24 h were small and round and displayed enlarged/fused lysosomes with light orange fluorescence (Fig. 4A). This phenomenon was attenuated by 24 h pre-incubation with DHA (Fig. 4A). We did not observe the yellow round cells that are characteristic of the LMP observed during the enhanced necroptosis induced by combined TNF-α/zVAD treatment in L929 cells [14].
Figure 4. DHA attenuates lysosome dysfunction in TNF-α-treated L929 cells.
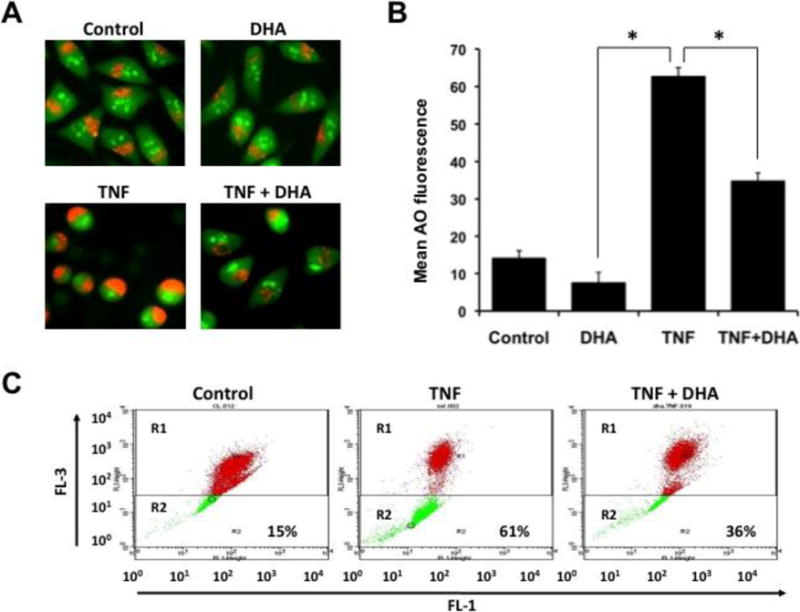
A. Fluorescence microscopic analysis of Acridine Orange (AO)-stained lysosomes in control cells, or cells treated with TNF-α for 24 h in the presence or absence of DHA. B. Quantification of TNF-α induced lysosomal membrane permeabilization (LMP), in the presence and absence of DHA, by AO flow cytometry assay. FL1 green fluorescence and FL3 red fluorescence were measured, and decrease in red fluorescence (R2 window, see panel C) was indicative of reduced lysosomal integrity. Results showed that DHA inhibited TNF-α induced AO fluorescence as indicated by reduction in R2 (*P < 0.05). C. Representative AO flow cytometry dot plot.
To determine if TNF-α induced partial LMP, we evaluated the cells using the AO flow cytometry assay. FL1 green fluorescence and FL3 red fluorescence were measured, and decrease in FL3 was indicative of reduced lysosomal integrity. Figs. 4B and C show that control L929 cells displayed a 15% decrease in FL3 (R2 window), however, a dramatic decrease in FL3 (61%) was observed after 24 h of TNF-α exposure. DHA partially attenuated TNF-α-induced LMP, as evidenced by FL3 decrease of only 36% in TNF-α-treated cells that had been pre-incubated for 24 h with DHA (Fig. 4B,C).
To further confirm that DHA protected L929 cells against TNF-α-induced lysosomal dysfunction, we evaluated the expression and activation of cathepsin L, a lysosomal protease previously implicated by our group in lysosomal dysfunction and Topo I cleavage during TNF-α-induced necrosis and during neuronal lipotoxicity [14,33]. We used the fluorogenic substrate-based assay Magic Red MR-(FR)2, which allows the localization of cathepsin L activity in live cells. As expected, the red fluorescence was localized in untreated cells and in DHA-treated cells in cytoplasmic granules around the nuclei, corresponding to lysosomes, where cathepsin L is normally stored (Fig. 5A). Cells treated with TNF-α for 24 h exhibited very bright red fluorescence, mainly localized to enlarged/fused lysosomes resembling those observed by AO (compare Fig. 5A with Fig. 4A). Cytosolic diffusion of the red fluorescence was observed only in a few cells treated with TNF-α (yellow arrow), suggesting some leakage of activated cathepsin L from the lysosomes (Fig. 5A). Consistent with our other results, preincubation with DHA prior to treatment of cells with TNF-α attenuated the red fluorescence and inhibited the lysosome enlargement/fusion (Fig. 5A). As a control, we used the specific cathepsin L inhibitor Z-FY-CHO, which, like DHA, attenuated TNF-α-induced lysosome enlargement/fusion (Fig. 5A) and cell death, as suggested by morphological analysis of treated cells (Fig. 5B).
Figure 5. DHA attenuates the activation of cathepsin L in TNF-α-treated L929 cells.
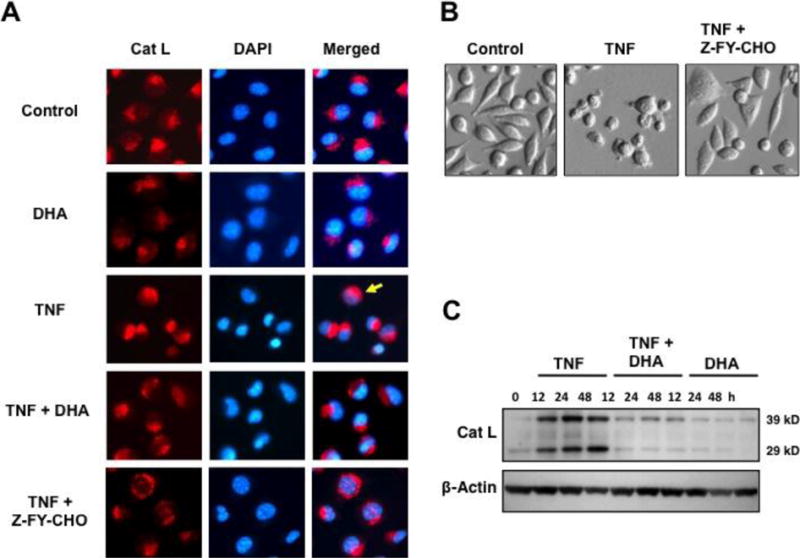
A. Cathepsin L activity assay was performed using the fluorogenic substrate–based assay Magic Red MR-(FR)2, which allows the localization of cathepsin L activity in live cells. The cathepsin L inhibitor Z-FY-CHO was used to inhibit TNF-α-induced cathepsin L activation. Yellow arrow points to a representative cell with diffuse cathepsin L activity. B. Morphological features of cells treated with TNF-α in the presence and absence of Z-FY-CHO. C. Immunoblot showing upregulation and processing of cathepsin L proenzyme (39 kD) into mature form (29 kD) during TNF-α-induced necroptosis, and its inhibition by DHA. β-actin was used as loading control.
Immunoblotting analysis of cathepsin L expression revealed accumulation of both the 39-kd proenzyme and the 29-kD mature form of this protease in L929 cells treated with TNF-α (Fig. 5C). The expression of the 29-kD mature form increased over time in the TNF-α-treated cells, which suggested processing of the proenzyme into the mature form (Fig. 5C). We observed previously a similar increase in cathepsin L expression in L929 cells undergoing enhanced necroptosis after exposure to TNF-α/zVAD [14]. Increased cathepsin L expression and processing was not observed in cells preincubated with DHA in the presence or absence of TNF-α (Fig. 5C), consistent with the observation that DHA attenuated TNF-α-induced lysosomal dysfunction and cathepsin L activation.
DHA attenuates TNF-α-induced autophagic features in L929 cells
The induction of small round cells and lysosomal enlargement and fusion (Figs. 1B, 2B, 4A, 5A, and 5B) by TNF-α suggested that this cytokine induced autophagosome formation and autophagy, which is consistent with reports demonstrating that TNF-α induces autophagy in L929 and other cell types [17–19]. To determine if DHA also attenuated TNF-α-induced autophagic features in L929 cells, we examined the accumulation of the autophagy markers LC3-II and Beclin in TNF-α-treated cells, in the presence and absence of DHA. TNF-α-treated cells displayed a robust time-dependent increase in LC3-II (Fig. 6A), which was attenuated by 24 h pre-incubation with DHA (Fig. 6A). Beclin expression also increased in cells treated with TNF-α for 24 h but subsided by 48 h (Fig. 6B). This increase was also attenuated by DHA.
Figure 6. DHA attenuates the accumulation of autophagy markers in TNF-α-treated L929 cells.
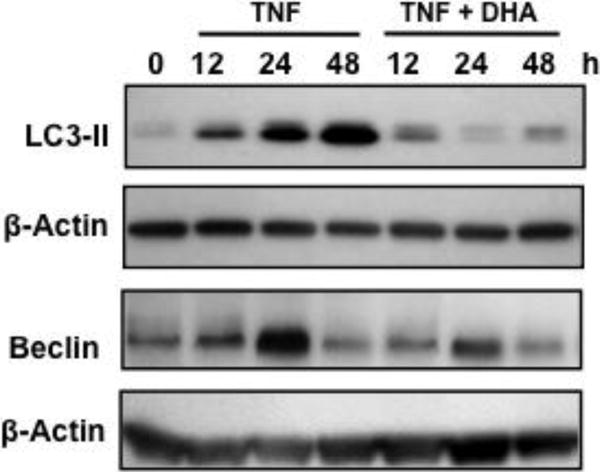
Immunoblots of L929 lysates showing the induction of autophagy markers LC3-II and Beclin by TNF-α, and its partial inhibition by DHA. β-actin was used as loading control.
DHA attenuates zVAD-induced necroptosis and autophagy in L929 cells
Previous reports established that zVAD by itself without exogenous addition of TNF-α, induces necroptosis and autophagy in L929 cells via stimulation of autocrine production of TNF-α [15,46,47]. It was proposed that activation of autophagy in this context could be a cellular attempt to survive zVAD-mediated induction of TNF-α necroptotic signaling. Thus, while zVAD-treated L929 cells display autophagic features, they ultimately die by necroptosis. We sought to determine if DHA also attenuated zVAD-induced necroptosis in these cells. Treatment of L929 cells with zVAD alone for 12 and 24 h significantly reduced cell viability, and consistent with the induction of necroptosis this reduction could be reversed by Nec-1, (Fig. 7A). zVAD-induced necroptosis was significantly attenuated by DHA (Fig. 7A), and morphological analysis of treated cells confirmed these results (Fig. 7B). DHA also inhibited the time-dependent increase of cathepsin L expression (Fig. 7C) and LC3-II conversion (Fig. 7D). These results indicated that DHA is capable of antagonizing both necroptosis and autophagy induced by zVAD alone in L929 cells, consistent with the results obtained with TNF-α alone.
Figure 7. DHA attenuates zVAD-induced necroptosis and autophagy in L929 cells.
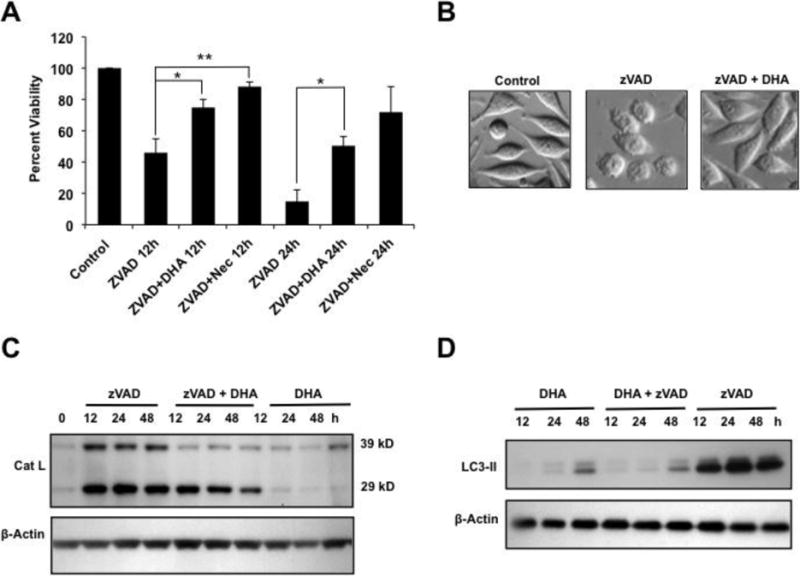
A. MTT viability assays showing that both DHA and Nec-1 significantly attenuated zVAD-induced cytotoxicity compared to treatment with zVAD alone (**P < 0.01). B. Morphological analysis of cells treated with zVAD in the presence and absence of DHA. C. Immunoblots showing induction of cathepsin L expression by zVAD and its partial inhibition by DHA. DHA by itself did not induce cathepsin L. D. Immunoblots of L929 lysates showing the induction of LC3-II by zVAD, and its partial inhibition by DHA. β-actin was used as loading control.
Discussion
Previous reports indicated that DHA enrichment reduced both TNF-α induced necrosis and apoptosis in L929 and U937 cells, respectively, and that among several fatty acids tested, DHA was the most effective in attenuating TNF-α induced cell death [35,36]. Our group also reported that DHA inhibits lipotoxicity-induced apoptosis in neuronal cells via a mechanism that involves protection against ROS and LMP [32,33]. The present study was undertaken to investigate if a similar protective mechanism is also involved in DHA-mediated inhibition of TNF-α-induced necroptosis in L929 cells.
We confirmed that TNF-α induced necroptosis in L929 cells by morphological analysis, inhibition of cell death by Nec-1, absence of Lamin B cleavage and caspase activation, and inhibition of the generation of Topo I necrotic cleavage fragments by Nec-1. We did not observe a complete inhibition of TNF-α-induced necroptosis with Nec-1, a specific inhibitor of RIPK1 that has no effect on RIPK3 [8]. However, we noticed that Nec-1 inhibited more robustly zVAD-induced necroptosis. These results are consistent with the emerging notion that RIPK3 can drive TNF-α-induced necroptosis independently of RIPK1 [8,48,49]. They are also in agreement with the observation that while TNF-α-induced necroptosis in L929 cells is not entirely dependent on RIPK1, zVAD-induced necroptosis is dependent on this kinase [8].
We showed previously that the generation of 70 and 45 kD cleavage fragments of Topo I is a common feature of TNF-α-induced necroptosis, as well as other modes of primary necrosis, secondary necrosis, and caspase-independent death [39,42]. This would be consistent with the observation that primary necrosis, secondary necrosis, and necroptosis converge into similar cellular and biochemical features [16]. During apoptosis, however, Topo I is cleaved into a detectable 70 kD fragment [14,39,42,50]. It should be emphasized that the apoptotic and necrotic 70 kD fragments of Topo I are different, with the first generated by caspases-3 and -6 by cleavage at aspartic acid residues, and the latter generated by released lysosomal cathepsins, particularly cathepsin L, by cleavage at non-aspartic acid sites possibly located in a protease sensitive region near the caspase-3 motif [14,50]. Thus, the cleavage of Topo I into a 45 kD fragment could be used as a biochemical hallmark of necrotic cell death (primary and secondary, necroptosis, and caspase-independent cell death with necrotic features), similar to the use of signature cleavage patterns of PARP1 to identify particular forms of cell death [51]. In addition, the apoptotic cleavage of Lamin B into a 46 kD, with no cleavage during necrosis, could also be used as a tool to distinguish apoptotic from necrotic cell death.
Our results indicated that DHA attenuated the ability of TNF-α to induce necroptosis in L929 cells via a mechanism that involved inhibition of ceramide and ROS generation, and reduction of LMP. The observed inhibition of cathepsin L activation and LMP by DHA is consistent with our previous observation that this Omega-3 fatty acid inhibits cathepsin L activation and LMP in nerve growth factor differentiated PC12 cells during palmitic acid-induced lipotoxicity, a process that occurs via both caspase-dependent and -independent cell death [33,52]. Since lysosomal dysfunction occurs during both apoptotic and necrotic/necroptotic cell death, these results suggest that DHA acts as an upstream mediator of biochemical events that inhibit signaling pathways activated by ceramide and ROS leading to LMP. This would be consistent with reports indicating that ceramide-induced ROS contributes to TNF-α-induced LMP in both apoptotic and non-apoptotic cell death [44], and that DHA reduces TNF-α and interleukin 1 beta (IL-1β) inflammatory signaling by downregulating the sphingomyelinase pathway [53]. It is also possible that DHA stabilizes lysosomal membranes in L929 cells exposed to TNF-α, or that DHA-mediated changes in plasma membrane properties and microdomains [54] abrogate TNF-α death signaling common to both apoptosis and necrosis.
We observed that exposure of L929 cells to TNF-α also induced autophagic features, as evidenced by the presence of AO-stained fused lysosomes, as well as upregulation of cathepsin L, LC3-II, and Beclin. Consistent with our results, increased cathepsin L activity and expression have been shown to mediate autophagy [55,56]. Our results also confirm the observation that TNF-α induces autophagy in L929 cells, and that this process is associated with upregulation of LC3-II and Beclin 1 [17–19]. Inhibition of TNF-α-induced autophagy with 3-methyladenine (3MA) reduced TNF-α-induced cytotoxicity in L929 cells, suggesting that in this context autophagy contributes to cell death [18]. By contrast, other investigators showed that inhibition of TNF-α-induced autophagy with 3MA exacerbates necroptosis in L929 cells, suggesting that autophagy may negatively regulate TNF-α-induced necroptosis [57]. Autophagy induced by treatment of L929 cells with zVAD, observed in our experiments, has been shown to be a protective mechanism against zVAD-induced necroptosis associated with autocrine production of TNF-α [46,47]. Thus, our observation that DHA attenuated both necroptosis and autophagic features induced by either TNF-α or zVAD is consistent with a role of this fatty acid in antagonizing TNF-α-induced inflammatory cell death.
It should be noted that in our studies DHA did not completely inhibit necroptosis induced by either TNF-α or zVAD, although the inhibitory effect seemed to be more potent against zVAD. These results are in agreement with those of Masuzawa and colleagues [35], who also observed a reduction, rather than complete inhibition, of TNF-α-induced necrosis by DHA in L929 cells. However, these investigators also observed that DHA partially protected L929 cells against TNF-α/ActD–induced apoptosis (which typically occurs in less than 6 h), an observation that we were not able to reproduce in our study. This discrepancy could be attributed to differences in experimental conditions or design, or the reagents used. Our results also demonstrated that DHA did not protect against the enhanced and rapid necroptosis induced by TNF-α/zVAD co-treatment. Taken together, these results suggest that DHA may not protect cells exposed to insults that cause extensive cellular damage with a fast kinetics, whether by apoptosis or necrosis, but may be more effective in partially protecting cells exposed to insults that induce necrotic cell death with slow kinetics, such as TNF-α alone or zVAD alone. It is also possible that DHA selectively interferes with the activity of either RIPK1 or RIPK3 but not with both kinases simultaneously.
Compelling evidence indicates that DHA exerts its anti-inflammatory activity in vitro and in vivo by conversion to lipid mediators such as resolvins that have potent anti-inflammatory and immunoregulatory actions, including inhibiting the production and secretion of TNF-α and other proinflammatory cytokines, and enhancing anti-inflammatory cytokines such as IL-10 [27,28]. An emerging paradox is that while DHA protects neurons, monocytes, fibroblasts and other cell types against certain pro-apoptotic and pro-necrotic insults, it exerts pro-apoptotic effects in vitro in many cancer cell types, including colon, breast, and prostate [52,58–60]. These pro-apoptotic effects are mediated by a plethora of mechanisms that include increasing oxidative stress, decreasing pro-survival signal transduction pathways, increasing the expression and activation of pro-apoptotic proteins while downregulating anti-apoptotic proteins and cell cycle-regulatory proteins, potentiating the effects of anticancer chemotherapeutic drugs, and sensitizing cancer cells to the pro-apoptotic effects of selected TNF family cytokines [52,58–60]. These paradoxical results suggest that the modulatory effects of DHA depend on the cellular context and microenvironment, with certain cancer cell types responding to DHA in vitro by activating apoptosis. It should be noted, however, that the anti-cancer benefit of DHA in humans remains controversial in light of recent epidemiological studies linking high blood levels of Omega-3 with increased risk of prostate cancer [61–63]. These results need to be reconciled with the well-documented role of DHA as an inducer of apoptosis in prostate cancer cells and other tumor cell types in vitro [52,58–60].
In summary, our present study provides evidence that DHA antagonizes TNF-α-mediated necroptosis in L929 cells by attenuating: 1) endogenous levels of ceramide and ROS; 2) lysosomal dysfunction and activation of cathepsin L; and 3) autophagic features. Further evidence to support a role for DHA in antagonizing TNF-α-mediated necroptosis was provided by the observation that DHA also inhibited zVAD induced necroptosis and autophagic features in L929 cells. It would be of interest to determine in future studies if DHA preferentially attenuates RIPK1- or RIPK3-dependent necroptosis. It would be also important to determine if DHA reduces necroptosis induced by other pro-inflammatory cytokines or viruses in various pathophysiological contexts. Administration of DHA in combination with other treatment modalities could serve as a therapeutic strategy to ameliorate necroptosis in pathologies involving inflammatory cell death.
Acknowledgments
We are grateful to Leanna Ursales, Teka-Ann Haynes, Anamika Basu, Tino Sanchez, and other members of the Casiano and De Leon laboratories for their technical and editorial assistance. This work was supported by NIH grants P20MD00988 (MDL and CAC), P20MD001632 (MDL and CAC), and R25GM060507 (MDL)
Contributor Information
Fabio J. Pacheco, Center for Health Disparities and Molecular Medicine and Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350, USA School of Health Sciences, Faculty of Medicine, River Plate Adventist University, Libertador, San Martin-Puiggari, Entre Rios, Argentina.
Frankis G. Almaguel, Center for Health Disparities and Molecular Medicine and Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350, USA
Whitney Evans, Center for Health Disparities and Molecular Medicine and Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350, USA.
Leslimar Rios-Colon, Center for Health Disparities and Molecular Medicine and Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350, USA.
Valery Filippov, Center for Health Disparities and Molecular Medicine and Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350, USA.
Lai S. Leoh, Center for Health Disparities and Molecular Medicine and Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350, USA
Elizabeth Rook-Arena, Center for Health Disparities and Molecular Medicine and Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350, USA; Department of Medicine, Division of Rheumatology, Loma Linda University School of Medicine, Loma Linda, CA, USA.
Melanie Mediavilla-Varela, Center for Health Disparities and Molecular Medicine and Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350, USA.
Marino De Leon, Center for Health Disparities and Molecular Medicine and Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350, USA.
Carlos A. Casiano, Email: ccasiano@llu.edu, Center for Health Disparities and Molecular Medicine and Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350, USA; Department of Medicine, Division of Rheumatology, Loma Linda University School of Medicine, Loma Linda, CA, USA.
References
- 1.Zelová H, Hošek J. TNF-α signalling and inflammation: interactions between old acquaintances. Inflamm Res. 2013;62:641–51. doi: 10.1007/s00011-013-0633-0. [DOI] [PubMed] [Google Scholar]
- 2.Bradley JR. TNF-mediated inflammatory disease. J Pathol. 2008;214:149–60. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- 3.Chu WM. Tumor necrosis factor. Cancer Lett. 2013;328:222–5. doi: 10.1016/j.canlet.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertazza L, Mocellin S. Tumor necrosis factor (TNF) biology and cell death. Front Biosci. 2008;13:2736–43. doi: 10.2741/2881. [DOI] [PubMed] [Google Scholar]
- 5.Galluzzi L, Kroemer G. Necroptosis turns TNF lethal. Immunity. 2011;35:849–51. doi: 10.1016/j.immuni.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–18. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 7.Remijsen Q, Goossens V, Grootjans S, Van den Haute C, Vanlangenakker N, Dondelinger Y, Roelandt R, Bruggeman I, Goncalves A, Bertrand MJ, Baekelandt V, Takahashi N, Berghe TV, Vandenabeele P. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 2014 doi: 10.1038/cddis.2013.531. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho Y, McQuade T, Zhang H, Zhang J, Chan FK. RIP 1-dependent and independent effects of necrostatin-1 in necrosis and T cell activation. PLoS One. 2011;6:e23209. doi: 10.1371/journal.pone.0023209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humphreys DT, Wilson MR. Modes of L929 cell death induced by TNF-alpha and other cytotoxic agents. Cytokine. 1999;11:773–82. doi: 10.1006/cyto.1998.0492. [DOI] [PubMed] [Google Scholar]
- 10.Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187:1477–85. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christofferson DE, Li Y, Yuan J. Control of life-or-death decisions by RIP1 kinase. Annu Rev Physiol. 2014;76:129–50. doi: 10.1146/annurev-physiol-021113-170259. [DOI] [PubMed] [Google Scholar]
- 12.Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27:1640–9. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011;2:e230. doi: 10.1038/cddis.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pacheco FJ, Servin J, Dang D, Kim J, Molinaro C, Daniels T, Brown-Bryan TA, Imoto-Egami M, Casiano CA. Involvement of lysosomal cathepsins in the cleavage of DNA topoisomerase I during necrotic cell death. Arthritis Rheum. 2005;52:2133–45. doi: 10.1002/art.21147. [DOI] [PubMed] [Google Scholar]
- 15.Ye YC, Wang HJ, Yu L, Tashiro S, Onodera S, Ikejima T. RIP1-mediated mitochondrial dysfunction and ROS production contributed to tumor necrosis factor alpha-induced L929 cell necroptosis and autophagy. Int Immunopharmacol. 2012;14:674–82. doi: 10.1016/j.intimp.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, Guerin CJ, Brunk UT, Declercq W, Vandenabeele P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–30. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- 17.Cheng Y, Qiu F, Tashiro S, Onodera S, Ikejima T. ERK and JNK mediate TNFalpha-induced p53 activation in apoptotic and autophagic L929 cell death. Biochem Biophys Res Commun. 2008;376:483–8. doi: 10.1016/j.bbrc.2008.09.018. [DOI] [PubMed] [Google Scholar]
- 18.Harhaji L, Mijatovic S, Maksimovic-Ivanic D, Popadic D, Isakovic A, Todorovic-Markovic B, Trajkovic V. Aloe emodin inhibits the cytotoxic action of tumor necrosis factor. Eur J Pharmacol. 2007;568:248–59. doi: 10.1016/j.ejphar.2007.04.029. [DOI] [PubMed] [Google Scholar]
- 19.Lin NY, Stefanica A, Distler JH. Autophagy: A key pathway of TNF-induced inflammatory bone loss. Autophagy. 2013;9:1253–5. doi: 10.4161/auto.25467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan FK. Fueling the flames: Mammalian programmed necrosis in inflammatory diseases. Cold Spring Harb Perspect Biol. 2012;4(11) doi: 10.1101/cshperspect.a008805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones SA, Mills KH, Harris J. Autophagy and inflammatory diseases. Immunol Cell Biol. 2013;91:250–8. doi: 10.1038/icb.2012.82. [DOI] [PubMed] [Google Scholar]
- 22.Simopoulos AP. Dietary omega-3 fatty acid deficiency and high fructose intake in the development of metabolic syndrome, brain metabolic abnormalities, and non-alcoholic fatty liver disease. Nutrients. 2013;5:2901–23. doi: 10.3390/nu5082901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crupi R, Marino A, Cuzzocrea S. n-3 fatty acids: role in neurogenesis and neuroplasticity. Curr Med Chem. 2013;20:2953–63. doi: 10.2174/09298673113209990140. [DOI] [PubMed] [Google Scholar]
- 24.Marion-Letellier R, Savoye G, Beck PL, Panaccione R, Ghosh S. Polyunsaturated fatty acids in inflammatory bowel diseases: a reappraisal of effects and therapeutic approaches. Inflamm Bowel Dis. 2013;19:650–61. doi: 10.1097/MIB.0b013e3182810122. [DOI] [PubMed] [Google Scholar]
- 25.Lorente-Cebrián S, Costa AG, Navas-Carretero S, Zabala M, Martínez JA, Moreno-Aliaga MJ. Role of omega-3 fatty acids in obesity, metabolic syndrome, and cardiovascular diseases: a review of the evidence. J Physiol Biochem. 2013;69:633–51. doi: 10.1007/s13105-013-0265-4. [DOI] [PubMed] [Google Scholar]
- 26.Jing K, Wu T, Lim K. Omega-3 polyunsaturated Fatty acids and cancer. Anticancer Agents Med Chem. 2013;13:1162–77. doi: 10.2174/18715206113139990319. [DOI] [PubMed] [Google Scholar]
- 27.Zhang MJ, Spite M. Resolvins: anti-inflammatory and proresolving mediators derived from omega-3 polyunsaturated fatty acids. Annu Rev Nutr. 2012;32:203–27. doi: 10.1146/annurev-nutr-071811-150726. [DOI] [PubMed] [Google Scholar]
- 28.Hong S, Lu Y. Omega-3 fatty acid-derived resolvins and protectins in inflammation resolution and leukocyte functions: targeting novel lipid mediator pathways in mitigation of acute kidney injury. Front Immunol. 2013;4:13. doi: 10.3389/fimmu.2013.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukherjee PK, Chawla A, Loayza MS, Bazan NG. Docosanoids are multifunctional regulators of neural cell integrity and fate: significance in aging and disease. Prostaglandins Leukot Essent Fatty Acids. 2007;77:233–8. doi: 10.1016/j.plefa.2007.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimazawa M, Nakajima Y, Mashima Y, Hara H. Docosahexaenoic acid (DHA) has neuroprotective effects against oxidative stress in retinal ganglion cells. Brain Res. 2009;1251:269–75. doi: 10.1016/j.brainres.2008.11.031. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Y, Calon F, Julien C, Winkler JW, Petasis NA, Lukiw WJ, Bazan NG. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase-and PPARγ-mediated mechanisms in Alzheimer’s disease models. PLoS One. 2011;6:e15816. doi: 10.1371/journal.pone.0015816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Almaguel FG, Liu JW, Pacheco FJ, Casiano CA, De Leon M. Activation and reversal of lipotoxicity in PC 12 and rat cortical cells following exposure to palmitic acid. J Neurosci Res. 2009;87:1207–18. doi: 10.1002/jnr.21918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Almaguel FG, Liu JW, Pacheco FJ, De Leon D, Casiano CA, De Leon M. Lipotoxicity-mediated cell dysfunction and death involve lysosomal membrane permeabilization and cathepsin L activity. Brain Res. 2010;1318:133–43. doi: 10.1016/j.brainres.2009.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Figueroa JD, Cordero K, Llán MS, De Leon M. Dietary omega-3 polyunsaturated fatty acids improve the neurolipidome and restore the DHA status while promoting functional recovery after experimental spinal cord injury. J Neurotrauma. 2013;30:853–68. doi: 10.1089/neu.2012.2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kishida E, Tajiri M, Masuzawa Y. Docosahexaenoic acid enrichment can reduce L929 cell necrosis induced by tumor necrosis factor. Biochim Biophys Acta. 2006;1761:454–62. doi: 10.1016/j.bbalip.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 36.Yano M, Kishida E, Iwasaki M, Kojo S, Masuzawa Y. Docosahexaenoic acid and vitamin E can reduce human monocytic U937 cell apoptosis induced by tumor necrosis factor. J Nutr. 2000;130:1095–101. doi: 10.1093/jn/130.5.1095. [DOI] [PubMed] [Google Scholar]
- 37.Haynes TA, Duerksen-Hughes PJ, Filippova M, Filippov V, Zhang K. C18 ceramide analysis in mammalian cells employing reversed-phase high-performance liquid chromatography tandem mass spectrometry. Anal Biochem. 2008;378:80–6. doi: 10.1016/j.ab.2008.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W, Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casiano CA, Ochs RL, Tan EM. Distinct cleavage products of nuclear proteins in apoptosis and necrosis revealed by autoantibody probes. Cell Death Differ. 1998;5:183–90. doi: 10.1038/sj.cdd.4400336. [DOI] [PubMed] [Google Scholar]
- 40.Carmagnat M, Drénou B, Chahal H, Lord JM, Charron D, Estaquier J, Mooney NA. Dissociation of caspase-mediated events and programmed cell death induced via HLA-DR in follicular lymphoma. Oncogene. 2006;25:1914–21. doi: 10.1038/sj.onc.1209222. [DOI] [PubMed] [Google Scholar]
- 41.Grdović N, Vidaković M, Mihailović M, Dinić S, Uskoković A, Arambasić J, Poznanović G. Proteolytic events in cryonecrotic cell death: Proteolytic activation of endonuclease P23. Cryobiology. 2010 Jun;60(3):271–80. doi: 10.1016/j.cryobiol.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 42.Wu X, Molinaro C, Johnson N, Casiano CA. Secondary necrosis is a source of proteolytically modified forms of specific intracellular autoantigens: implications for systemic autoimmunity. Arthritis Rheum. 2001;44:2642–52. doi: 10.1002/1529-0131(200111)44:11<2642::aid-art444>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 43.Shindo R, Kakehashi H, Okumura K, Kumagai Y, Nakano H. Critical contribution of oxidative stress to TNFα-induced necroptosis downstream of RIPK1 activation. Biochem Biophys Res Commun. 2013;436:212–6. doi: 10.1016/j.bbrc.2013.05.075. [DOI] [PubMed] [Google Scholar]
- 44.Thon L, Möhlig H, Mathieu S, Lange A, Bulanova E, Winoto-Morbach S, Schütze S, Bulfone-Paus S, Adam D. Ceramide mediates caspase-independent programmed cell death. FASEB J. 2005;19:1945–56. doi: 10.1096/fj.05-3726com. [DOI] [PubMed] [Google Scholar]
- 45.Ardestani S, Deskins DL, Young PP. Membrane TNF-alpha-activated programmed necrosis is mediated by Ceramide-induced reactive oxygen species. J Mol Signal. 2013;8:12. doi: 10.1186/1750-2187-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu YT, Tan HL, Huang Q, Kim YS, Pan N, Ong WY, Liu ZG, Ong CN, Shen HM. Autophagy plays a protective role during zVAD-induced necrotic cell death. Autophagy. 2008;4:457–66. doi: 10.4161/auto.5662. [DOI] [PubMed] [Google Scholar]
- 47.Wu YT, Tan HL, Huang Q, Sun XJ, Zhu X, Shen HM. zVAD-induced necroptosis in L929 cells depends on autocrine production of TNFα mediated by the PKC-MAPKs-AP-1 pathway. Cell Death Differ. 2011;18:26–37. doi: 10.1038/cdd.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Linkermann A, Bräsen JH, De Zen F, Weinlich R, Schwendener RA, Green DR, Kunzendorf U, Krautwald S. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-α-induced shock. Mol Med. 2012 May 9;18:577–86. doi: 10.2119/molmed.2011.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014 Mar 21;343(6177):1357–60. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 50.Samejima K, Svingen PA, Basi GS, Kottke T, Mesner PWJ, Stewart L, Durrieu F, Poirier GG, Alnemri ES, Champoux JJ, Kaufmann SH, Earnshaw WC. Caspase-mediated cleavage of DNA topoisomerase I at unconventional sites during apoptosis. J Biol Chem. 1999;274:4335–40. doi: 10.1074/jbc.274.7.4335. [DOI] [PubMed] [Google Scholar]
- 51.Chaitanya GV, Steven AJ, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal. 2010;8:31. doi: 10.1186/1478-811X-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ulloth JE, Casiano CA, De Leon M. Palmitic and stearic fatty acids induce caspase-dependent and -independent cell death in nerve growth factor differentiated PC 12 cells. J Neurochem. 2003;84:655–68. doi: 10.1046/j.1471-4159.2003.01571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Opreanu M, Lydic TA, Reid GE, McSorley KM, Esselman WJ, Busik JV. Inhibition of cytokine signaling in human retinal endothelial cells through downregulation of sphingomyelinases by docosahexaenoic acid. Invest Ophthalmol Vis Sci. 2010;51:3253–63. doi: 10.1167/iovs.09-4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skender B, Vaculova AH, Hofmanova J. Docosahexaenoic fatty acid (DHA) in the regulation of colon cell growth and cell death: a review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2012;156:186–99. doi: 10.5507/bp.2012.093. [DOI] [PubMed] [Google Scholar]
- 55.Kaasik A, Rikk T, Piirsoo A, Zharkovsky T, Zharkovsky A. Up-regulation of lysosomal cathepsin L and autophagy during neuronal death induced by reduced serum and potassium. Eur J Neurosci. 2005;22:1023–31. doi: 10.1111/j.1460-9568.2005.04279.x. [DOI] [PubMed] [Google Scholar]
- 56.Hsu KF, Wu CL, Huang SC, Wu CM, Hsiao JR, Yo YT, Chen YH, Shiau AL, Chou CY. Cathepsin L mediates resveratrol-induced autophagy and apoptotic cell death in cervical cancer cells. Autophagy. 2009;5:451–60. doi: 10.4161/auto.5.4.7666. [DOI] [PubMed] [Google Scholar]
- 57.Ye YC, Wang HJ, Chen L, Liu WW, Tashiro S, Onodera S, Xia MY, Ikejima T. Negatively-regulated necroptosis by autophagy required caspase-6 activation in TNFα-treated murine fibrosarcoma L929 cells. Int Immunopharmacol. 2013;17:548–55. doi: 10.1016/j.intimp.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 58.Hajjaji N, Bougnoux P. Selective sensitization of tumors to chemotherapy by marine-derived lipids: a review. Cancer Treat Rev. 2013;39:473–88. doi: 10.1016/j.ctrv.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 59.Shin S, Jing K, Jeong S, Kim N, Song KS, Heo JY, Park JH, Seo KS, Han J, Park JI, Kweon GR, Park SK, Wu T, Hwang BD, Lim K. The omega-3 polyunsaturated fatty acid DHA induces simultaneous apoptosis and autophagy via mitochondrial ROS-mediated Akt-mTOR signaling in prostate cancer cells expressing mutant p53. Biomed Res Int. 2013;2013:568671. doi: 10.1155/2013/568671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jing K, Wu T, Lim K. Omega-3 polyunsaturated Fatty acids and cancer. Anticancer Agents Med Chem. 2013;13:1162–77. doi: 10.2174/18715206113139990319. [DOI] [PubMed] [Google Scholar]
- 61.Chua ME, Sio MC, Sorongon MC, Morales ML., Jr The relevance of serum levels of long chain omega-3 polyunsaturated fatty acids and prostate cancer risk: A meta-analysis. Can Urol Assoc J. 2013;7:E333–43. doi: 10.5489/cuaj.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sorongon-Legaspi MK, Chua M, Sio MC, Morales M., Jr Blood level omega-3 Fatty acids as risk determinant molecular biomarker for prostate cancer. Prostate Cancer. 2013;2013:875615. doi: 10.1155/2013/875615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brasky TM, Darke AK, Song X, Tangen CM, Goodman PJ, Thompson IM, Meyskens FL, Jr, Goodman GE, Minasian LM, Parnes HL, Klein EA, Kristal AR. Plasma phospholipid fatty acids and prostate cancer risk in the SELECT trial. J Natl Cancer Inst. 2013;105:1132–41. doi: 10.1093/jnci/djt174. [DOI] [PMC free article] [PubMed] [Google Scholar]