

Abstract
Immunotherapy is a promising strategy for targeting tumors. One emerging approach is to harness the immune effector functions of natural antibodies to destroy tumor cells. Dinitrophenyl (DNP) and the galactose-α-1,3-galactose (αGal) epitope are two haptens that bind endogenous antibodies. One potential alternative is the deoxysugar L-rhamnose. We compared these candidates using a biosensor assay to evaluate human sera for endogenous antibody concentration, antibody isotype distribution, and longevity of antibody-hapten interactions. Antibodies recognizing α–rhamnose are of equal or greater abundance and affinity as those recognizing αGal. Moreover, both rhamnose and αGal epitopes are more effective than DNP at recruiting the IgG antibody subtype. Exposure of tumor cells to rhamnose-bearing glycolipids and human serum promotes complement-mediated cytotoxicity. These data highlight the utility of α-rhamnose-containing glycoconjugates to direct the immune system to target cells.
Keywords: rhamnose, antibody, immunotherapy, surface plasmon resonance, alpha-galactose
Using the immune system to combat disease is a therapeutic strategy that can be exceptionally specific and efficacious.[1-4] One especially valuable application is in the treatment of cancer, where antibody-based therapeutics are now gaining traction.[5]Antibody therapies function through a variety of methods including alteration of signaling, promotion of apoptosis, sequestration of growth factors, and activation of the immune system.[5]Therapeutic monoclonal antibodies must find their target cells, carry out their functional roles, and be compatible with the host. For antibodies to meet these diverse criteria often requires significant engineering. One alternative strategy designed to side-step these challenges is to use compounds that recruit naturally produced antibodies to tumor cells (Figure 1). Cells targeted in this way can be recognized by the immune system as foreign and marked for destruction.
Figure 1.
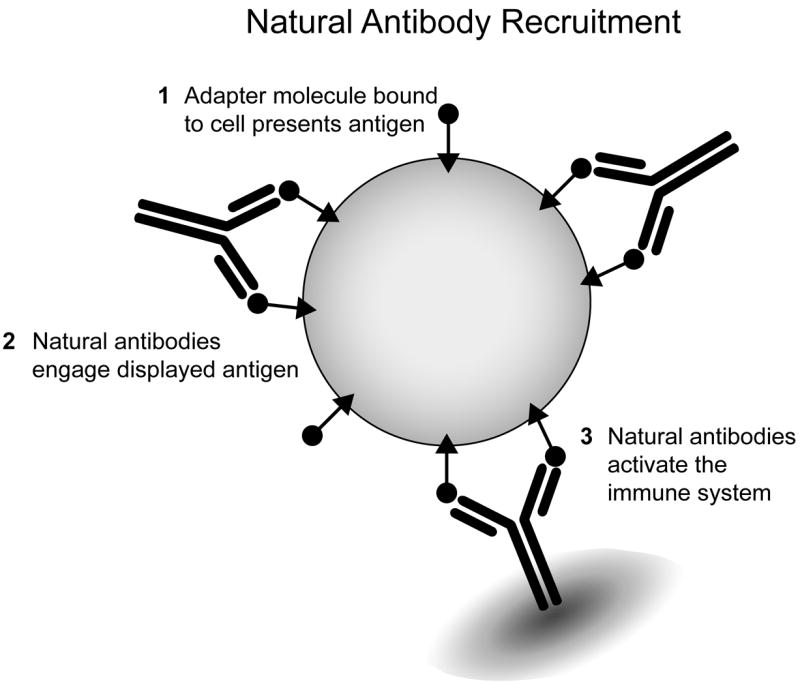
Natural antibody-recruiting molecules applied to the tumor cell surface can mobilize the immune system. Antibody-recruiting molecules function through the following steps: 1. Interaction with the surface of target cells; 2. Recruitment of natural antibodies: 3. Activation of endogenous immune mechanisms.
Recruitment of endogenous antibodies to tumor cells allows for destruction through two antibody-effector mechanisms: complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC). CDC is promoted primarily by antibodies of the IgM isotype. Decoration of target cells with binding epitopes that can recruit antibodies will lead to activation of the classical complement cascade culminating in the formation of membrane attack complex pores in their membranes. This activation of the complement cascade can further amplify immune responses through release of cytokines and inflammatory mediators. These signaling molecules attract immune cells involved in ADCC such as neutrophils, macrophages, and natural killer (NK) cells. Immune effector cells, recognizing surface-bound IgG antibodies, initiate ADCC through activating Fc receptors (FcγRIIIa and FcγRIIa). Both pathways facilitate tumor clearance.[6] Immune system recruitment also has the potential to prime the adaptive immune system to recognize tumor-associated antigens, in essence, to generatean in situ autologous vaccine.[7, 8]
To target tumors using a natural antibody recruitment strategy, a means of adorning cancer cells with antibody-binding groups is needed. Suitable haptens that are recognized by antibodies present in the human population must be identified. Finding such validated haptens is a significant yet poorly addressed challenge. It requires examination of multiple parameters including antibody isotype, affinity, and population distribution. The ideal antigen would be readily accessible or modifiable via chemical synthesis so it could be conjugated to any agent that binds to the tumor cell surface.[9]In principle, any antigen that gives rise to a suitable immune response could be used in conjunction with a vaccination protocol, but antigens that bind endogenous antibodies are advantageous. These antibodies can be present even in individuals that have become partially immunocompromised. To capitalize on both humoral (i.e. CDC) and cellular (i.e. ADCC) immune effector mechanisms, the hapten should bind antibodies of both IgM and IgG isotypes. Several candidate epitopes have been identified.
The small molecule hapten dinitrophenyl (DNP) was one of the first to be used in generating defined antigens for immunological investigations, and it remains the basis for many antibody-targeting experiments.[3, 7, 10-12] DNP is small, easily manipulated, and immunogenic, but it has some potential liabilities. Although affinity-matured commercial antibodies are available, naturally-occurring anti-DNP antibodies are present in low concentration and have lower affinity than those that have been affinity matured.[3, 10, 13, 14] Additionally, DNP is a small electron-deficient, hydrophobic aromatic compound, and its physical properties complicate its use. Specifically, DNP can bind to hydrophobic biomolecules; it non-specifically interacts with membranes and albumins, limiting the amount of free-antigen available for antibody recruitment.[15, 16]
An alternative natural antigenic epitope that has been extensively exploited for immune recruitment is galactose-α-1,3-galactose (αGal). This epitope is found in most mammals and bacteria, but it is absent in humans, apes, and old world monkeys.[17, 18] Thus, in several primates, including humans, it is recognized as foreign. The pool of antibodies recognizing αGal (termed anti-Gal) is maintained through constant exposure to the epitope, possibly from endogenous gut bacteria.[19] As a result, estimates indicate that anti-Gal comprises up to 2% of circulating IgG and 3-8% of serum IgM.[20, 21] The presence of anti-Gal IgM is one of the major barriers preventing xenotransplantation of porcine organs into primate recipients. It elicits hyperacute rejection.[22]This rejection response resulting from complement activation underscores αGal’s utility for immune recruitment. Still, one drawback to using αGal as bait for endogenous antibodies is its synthetic complexity.[23-26] It is a difficult target for chemical or chemoenzymatic synthesis rendering the creation of conjugates arduous. Current clinical trials that exploit the immunogenicity of αGal rely on biological isolates of αGal species, specifically ceramides obtained from extraction of rabbit erythrocytes.[1] These heterogeneous, animal-derived mixtures are giving rise to intriguing results, yet the active species are not easily amenable to chemical optimization.[1, 8]While DNP and αGal are both currently popular antigens for immune recruitment research, each has distinct disadvantages.
Natural antibodies often recognize carbohydrate determinants, such asαGal or the blood group antigens, which underscores the potential of glycans for antibody recruitment. Although many of these candidates are at least as complexasαGal, recent microarray screens have identified human antibodies that bind the simple monosaccharide L-rhamnose.[27, 28]Rhamnose is a deoxy sugar not observed in humans, but prevalent in microbes and plants.[29-32] Indeed, L-rhamnose differs in configuration from the building blocks of mammalian glycans (except L-fucose), which are carbohydrates of the D configuration. The microarray screens suggest that antibodies recognizing L-rhamnose (anti-Rha) are more abundant than anti-Gal in serum samples, although quantification was difficult.[27, 28]Additionally Anti-Rha was found in a greater percentage of single-donor sera than anti-Gal. These data indicate that anti-Rha may be more prevalent in the human population.
A number of potential natural antibody-recruiting epitopes have been identified, yet a direct comparison is lacking. We therefore sought to directly evaluate the utility of αGal, rhamnose, and DNP as natural antibody targeting agents. To ascertain their utility we compared antibody levels, isotype distribution, and antigen-antibody complex stability in human serum samples.
Our first objective was to determine the relative amount of antibody recognizing each antigen in serum samples. We needed a sensitive method that could also be used to compare affinities. Although antibody titers are often measured by ELISA, a significant amount of carbohydrate-bearing compound is needed – a challenge especially with αGal. In addition, the readout from ELISAs derives from a combination of antibody concentration and avidity. To address this limitation, we envisioned using surface plasmon resonance (SPR) spectroscopy, an information-rich technique requiring only small amounts of material. Moreover, surfaces with immobilized compounds can be regenerated and used multiple times. Although this method is typically associated with determining binding kinetics, it is useful for rank ordering interactions with ligands. It should also provide the means to characterize antibody isotypes, and it is compatible with serum.[33, 34] We therefore developed a biosensor assay to analyze serum antibodies.
We generated hapten conjugates that could be immobilized to a streptavidin-coated surface. The biotin conjugates were synthesized by building upon our published methodology for αGal synthesis, exploiting an efficient rhamnose synthesis developed by the Wang group, and taking advantage of the reactivity of commercially available DNP-lysine.[23, 35] Each epitope was appended to biotin using a spacer of at least twelve ethylene glycol units. Our previous results indicate that the linkers employed should allow each immobilized antigen to engage its target antibodies (Scheme 1).[36] These conjugates were anchored on streptavidin-coated sensor chips and a biotinylated control peptide was used to generate a reference channel. Sera were collected from healthy donors for SPR experiments
Scheme 1.
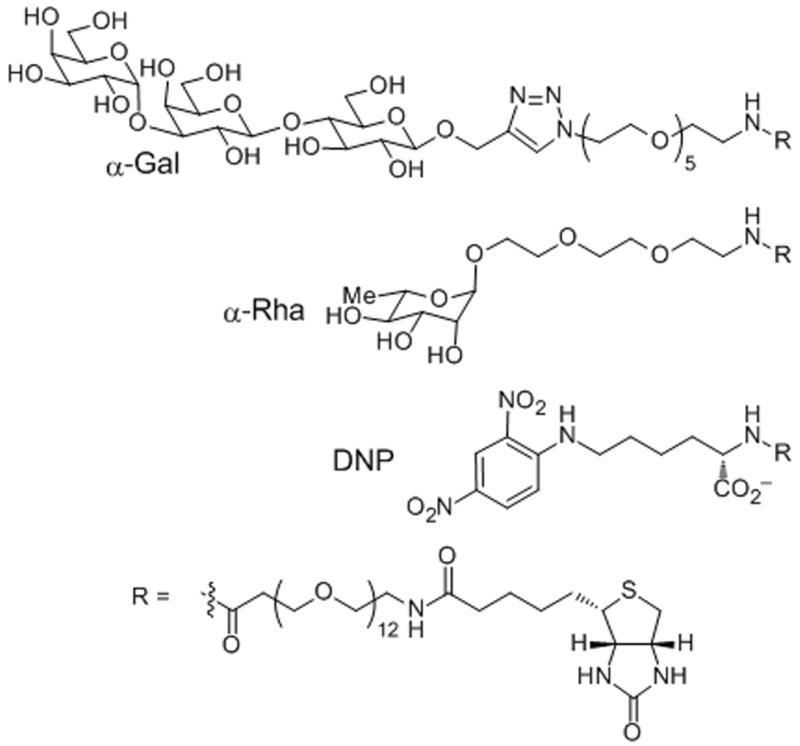
Structural representation of antigens immobilized for SPR: galactose-α-1,3-galactose (αGal), rhamnose (Rha), and dinitrophenyl (DNP).
To evaluate antibody prevalence, we compared corrected response units (RUantigen– RUreference) bound to each flow channel after serum exposure. Higher RUs indicate greater antibody binding and therefore higher titers. For most samples, the flow channel with immobilized rhamnose had the highest signal. Moreover, a clear advantage was observed for rhamnose in that the signals obtained were higher than those from αGal flow channels in all cases (Figures 2A, 2B and S2). These data indicate that antibodies recognizing rhamnose are generally more prevalent than those recognizing αGal. DNP was also recognized by the serum samples, supporting its current usage for immune recruitment in in vitro systems. Still, natural antibodies that bind DNP were less abundant than those binding rhamnose.
Figure 2.
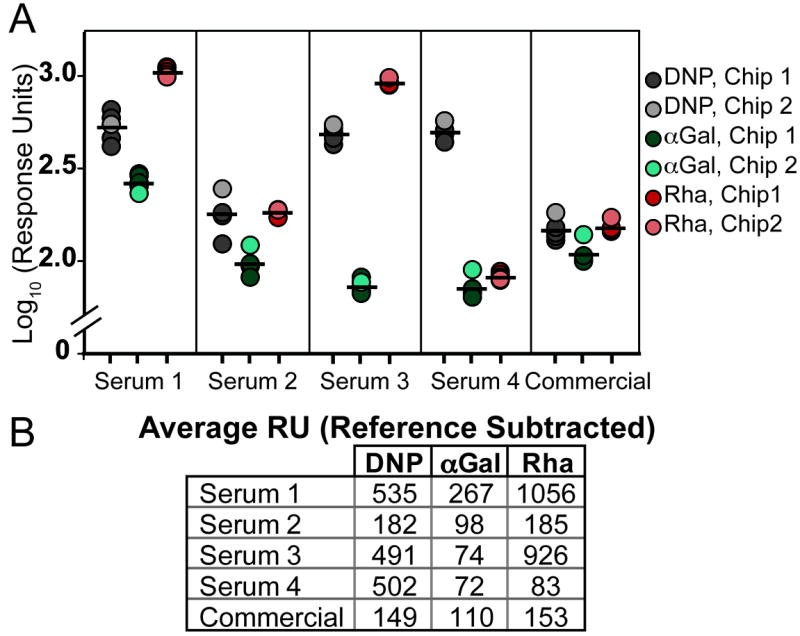
Comparison of the levels of antibodies that recognize rhamnose versus those that bindαGal epitopes from 4 control serum donors (serum 1-4) and from a commercially obtained pool of human serum (commercial).(A) Log plot of individual replicates used for averaging. Grey circles correspond to the DNP flow channel, teal to αGal, and red to rhamnose. Four injections of each serum across one chip (darker circles) and one injection of each serum across a second chip (light circle) were performed to show reproducibility. Black bars indicate average of all five injections. Full response curves are available in supplemental information. (B) Average response units bound to each flow cell after 5 minutes exposure to 10% serum. RU given are from reference subtracted data (RUantigen– RUreference).
Although the trend of detecting more anti-rhamnose than anti-Gal is consistent among sera tested, the individual values varied. Serum antibody levels to these antigens reflect immunogenic exposures of individuals to various benign and pathogenic microorganisms that express these carbohydrates. Interestingly, the amount of anti-Rha present in the serum samples has much greater donor-to-donor variability than that of anti-Gal. It is thought that human recognition of αGal primarily stems from exposure to the antigen on gut flora and, to a lesser extent, other infections; a situation perhaps leading to similar exposure levels across a population.[19] Rhamnose, although present on commensal organisms, is also a key component of streptococcal cell walls and capsular polysaccharides.[37]These structures can activate immune cells and induce an antibody response.[38-41] It is interesting to speculate that the higher anti-Rhatiters of donors 1 and 3 may originate from encounters with streptococcal species. There is evidence that immunization with S. pneumoniae type 32F can elicit an anti-rhamnose response.[41] Although type 32F itself is not a component of pneumonococcal vaccines, both Prevnar 13 (Pfizer) and Pneumovax 23 (Merck) contain other strains that incorporate rhamnose into their capsular polysaccharide core structure. Thus, immunization with these vaccines may lead to the production of anti-Rha,[30, 42, 43] which could be co-opted for tumor targeting.
Antibody isotype distribution is important for tuning immune responses and ensuring engagement of both humoral and cellular immunity. To determine relative contributions from IgG and IgM isotypes for each antigen, we measured the binding of anti-human IgG or IgM to flow cells containing αGal, DNP, and rhamnose after they had first been exposed to serum from the normal donors or a commercially pooled sample (Figure 3). As might be anticipated, we found that most antibodies recognizing DNP are from the IgM pool. Rhamnose and αGal are associated with microbial structures and are likely to be presented in an immunological context capable of promoting isotype switching to produce high-affinity IgG. In contrast, DNP is non-natural and unlikely to be encountered unless intentional immunization has occurred. Nevertheless some cross-reactive IgGs are likely recruited.[44, 45] The modest anti-DNP IgG recruitment may reflect the propensity of DNP to bind to hydrophobic sites or stack with tryptophan residues often found in the antigen binding site of antibodies.[15, 46] Both IgG- and IgM-type antibodies were detected bound in channels containing either rhamnose or αGal. The trends for anti-rhamnose IgG and IgM were also confirmed by an ELISA study (Fig. S4). The higher levels of anti-Rha(verses anti-Gal) increases the probability of forming stable and productive antigen-antibody complexes when anti-Rha is recruited to the target cell surface. Because immune functions depend on multivalency, anti-Rha recruitment will depend not only on the concentration of anti-Rha, but also the affinity of antibody for the target.
Figure 3.
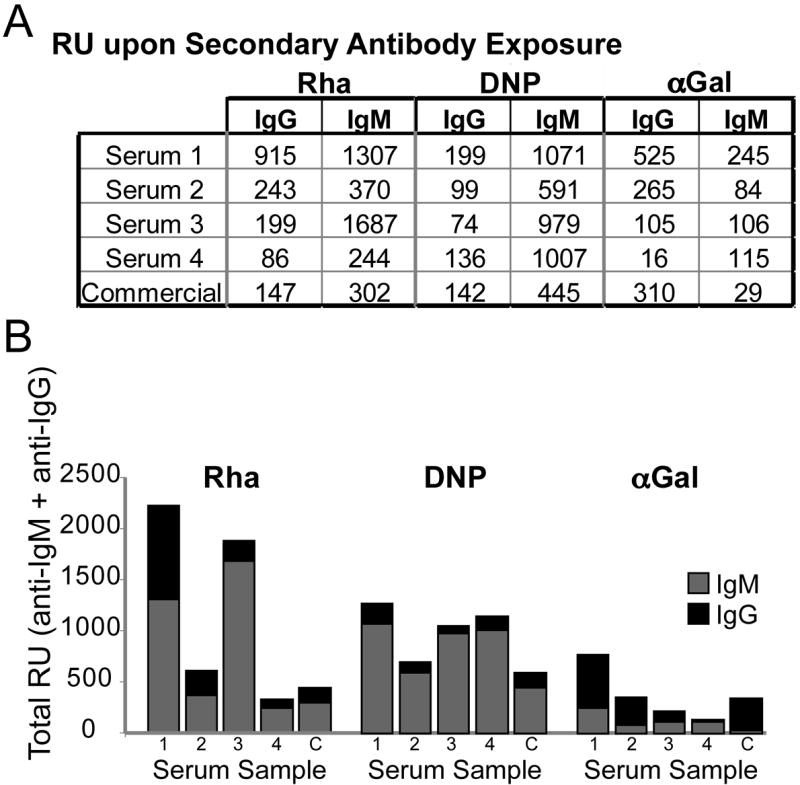
Isotype distribution of antibodies. (A) RU of secondary antibody (anti-IgM or anti-IgG) bound for each condition (reference subtracted). (B) IgG and IgM components of bound antibodies based on response units of secondary antibodies bound. Data from each secondary injection were obtained after a separate serum injection.
The abundance of antibodies, their isotype distribution, and affinity are three parameters that determine the effectiveness of immune recruitment and the threshold for activation. More stable, and thus longer-lived, complexes of antibodies with antigens on the cell surface increase the odds of productive immune responses, especially given that both complement and cell-mediated responses depend on multivalent presentation of antibody Fc regions. Because serum samples contain multiple antibody populations at unknown concentrations, specific kinetic parameters cannot be determined from biosensor experiments. Still, the observation of antibody dissociation rates from the surfaces can serve as a proxy for complex longevity. To this end, we monitored antibody dissociation from the surfaces by measuring the fraction of the initial signal remaining after one hour.[47] Generally, the signal loss was greater for antibodies binding αGal- and DNP-modified surfaces than for the corresponding rhamnose one (Figure 4). These findings highlight another potential advantage of anti-rhamnose antibodies over those that bind αGal or DNP.
Figure 4.
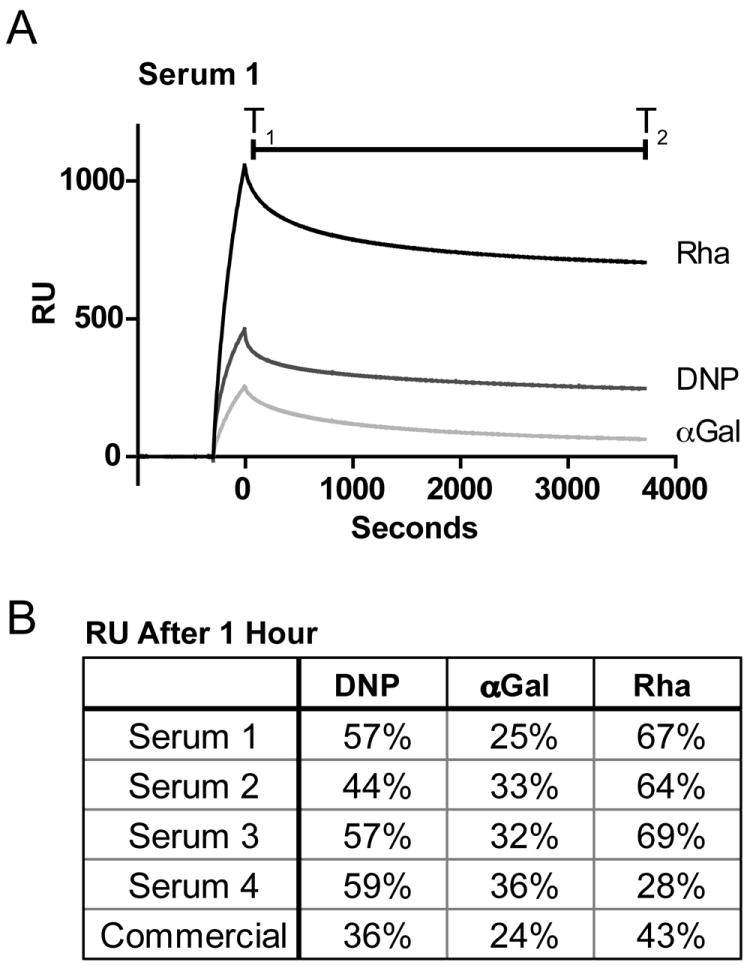
Analysis of the stability of the antigen – antibody complex. A) Sensorgram depicting loss of binding signal over the course of one hour. B) Percentage signal remaining was calculated as RUT2/RuT1. All RU were reference subtracted.
As the first step to exploiting anti-rhamnose antibodies for tumor targeting, we employed a cell culture model. To lure antibodies to the tumor cells, we took advantage of the natural tendency of lipids to insert into membranes[1, 48, 49] by using a synthetic glycolipid displaying rhamnose. The antigen was appended to dipalmitoleoyl phosphoethanolamine (DPoPE) such that two equivalents of rhamnose were present on each phospholipid. Exposure of cells to the conjugate cloaked them ina multivalent display of rhamnose. Specifically, M21 melanoma cells were treated with the lipid-rhamnose conjugate and thenserum. The latter is a source of both anti-rhamnose and human complement. The fraction of viable cells remaining was assessed with a luciferase assay (Figure 5). Tumor cells decorated with rhamnose were subject to killing. This cell death was due to the presence of rhamnose and not the lipid modification. We postulated the cellular toxicity observed resulted from complement-mediated cell killing. Because tumor cells can express membrane proteins that inhibit complement activation, we also tested cell killing in the presence of antibodies blocking the activity of CD55 and CD59, two of the most common complement inhibitory proteins,[50, 51] and these conditions augmented the efficiency of cell killing. Additionally, we demonstrated that complement component C4d deposits on the surface of lipid-rhamnose treated cells (Figure S5). Together, our data indicate that the cytotoxicity observed was complement-mediated.
Figure 5.
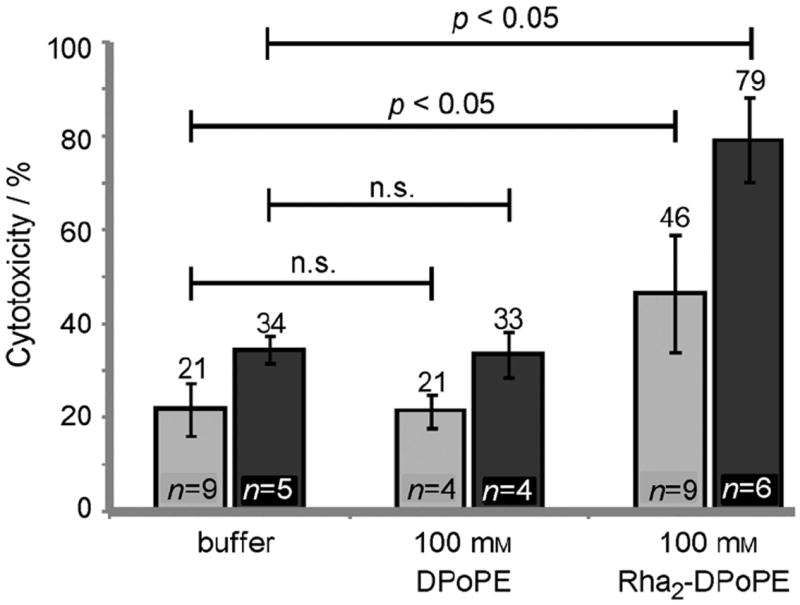
Cytotoxicity of lipid-rhamnose conjugate (see Supporting Information for synthesis). Cells were treated with lipid-rhamnose conjugates and exposed to human serum as a source of both anti-rhamnose and complement. Cytoxicity was assessed by measuring the level of ATP in the sample. Cytotoxicity was also determined in the presence of antibodies that block the complement inhibitors CD55 and CD59 (dark bars).
Our findings provide impetus to employ rhamnosylated conjugates for immune system recruitment. As an antigen for natural antibodies, rhamnose combines some of the best features of both αGal and DNP. Like αGal, rhamnose is a biologically relevant carbohydrate to which a large segment of the population develops immunity. Removing the requirement for prior vaccination is valuable especially when patients are immunocompromised. In addition, L-rhamnose itself is commercially available and can be appended to a variety of tumor targeting agents. Strategies based on anti-Rha targeting would potentially have additional benefits for preclinical in vivo testing. Current mouse models for tumor immunotherapy using αGal conjugates require introduction of tumors in knockout mice lacking the α-1,3-galactosyl transferase and subsequent immunization of these animals against αGal. In contrast, mice do not produce rhamnose-containing glycans. As a result, rhamnose conjugates can be evaluated using standard mouse models. Moreover, immunization of young individuals of normal laboratorystrains affords anti-Rhatiters similar to those of adult humans.[35]
The biosensor assay we have described reveals that antibodies recognizing rhamnose are generally prevalent. In the samples we tested, they also possess higher affinity than those recognizing αGal. The observation that these anti-rhamnose antibodies constitute both IgG and IgM pools suggests they will have the capacity to activate both humoral and cellular immunity. The enhanced stability of rhamnose—anti-rhamnose complexes should increase the levels of productive immune activation. In summary, our results indicate that rhamnose is an excellent candidate for further development in immune recruitment strategies, and is potentially superior to αGal by virtue of its effective antibody recruitment and synthetic tractability. In addition to its therapeutic potential, rhamnose could be integrated into the design of tools to illuminate more specific requirements for successful immune recruitment including antigen presentation methods, antigen presentation density and valency, and antibody profile.
Supplementary Material
Acknowledgments
This work was supported by the Department of Defense (W81XWH-08-1-0648) and the National Institutes of Health (NIAID Grant AI055258). Biacore 2000 SPR data were obtained at the UW–Madison Biophysics Instrumentation Facility, which was established with support from UW–Madison and grants BIR-9512577 (NSF) and S10 RR13790 (NIH). NMR and mass spectra were obtained in UW–Madison Chemistry Department Instrumentation Center supported by the NSF (CHE-1048642, CHE-0342998, CHE-9974839) Flow cytometry data were obtained at the UWCCC Flow Cytometry Laboratory (P30 CA014520) R.T.C.S. was supported by the NIH Chemistry – Biology Interface Training Program (T32 GM008505). We thank Dr. Lingyin Li and Dr. Darrell McCaslin for advice on the SPR experiments. J.H. thanks the Fond québécois de recherche sur la nature et les technologies (FQRNT) for a postdoctoral fellowship.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.20xxxxxxx.
References
- 1.Galili U, Wigglesworth K, Abdel-Motal UM. J Immunol. 2007;178:4676–4687. doi: 10.4049/jimmunol.178.7.4676. [DOI] [PubMed] [Google Scholar]
- 2.Carlson CB, Mowery P, Owen RM, Dykhuizen EC, Kiessling LL. ACS Chem Biol. 2007;2:119–127. doi: 10.1021/cb6003788. [DOI] [PubMed] [Google Scholar]
- 3.Murelli RP, Zhang AX, Michel J, Jorgensen WL, Spiegel DA. J Am Chem Soc. 2009;131:17090–17092. doi: 10.1021/ja906844e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Popkov M, Gonzalez B, Sinha SC, Barbas CF. Proc Nat Acad Sci USA. 2009;106:4378–4383. doi: 10.1073/pnas.0900147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sliwkowski MX, Mellman I. Science. 2013;341:1192–1198. doi: 10.1126/science.1241145. [DOI] [PubMed] [Google Scholar]
- 6.Natsume A, Niwa R, Satoh M. Drug Design Devel Ther. 2009;3:7–16. [PMC free article] [PubMed] [Google Scholar]
- 7.Berd D. Vaccine. 2001;19:2565–2570. doi: 10.1016/s0264-410x(00)00490-4. [DOI] [PubMed] [Google Scholar]
- 8.Galili U, Albertini MR, Sondel PM, Wigglesworth K, Sullivan M, Whalen GF. Cancers. 2010;2:773–793. doi: 10.3390/cancers2020773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McEnaney PJ, Parker CG, Zhang AX, Spiegel DA. ACS Chem Biol. 2012:1139–1151. doi: 10.1021/cb300119g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farah FS. Immunology. 1973;25:217–226. [PMC free article] [PubMed] [Google Scholar]
- 11.Ortega E, Kostovetzky M, Larralde C. Mol Immunol. 1984;21:883–888. doi: 10.1016/0161-5890(84)90143-3. [DOI] [PubMed] [Google Scholar]
- 12.Shokat KM, Schultz PG. J Am Chem Soc. 1991;113:1861–1862. [Google Scholar]
- 13.Farah FS, Awdeh ZL. Journal of immunological methods. 1972;1:353–361. doi: 10.1016/0022-1759(72)90028-2. [DOI] [PubMed] [Google Scholar]
- 14.Parker CG, Domaoal RA, Anderson KS, Spiegel DA. J Am Chem Soc. 2009;131:16392–16394. doi: 10.1021/ja9057647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang AX, Murelli RP, Barinka C, Michel J, Cocleaza A, Jorgensen WL, Lubkowski J, Spiegel DA. J Am Chem Soc. 2010;132:12711–12716. doi: 10.1021/ja104591m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung H, Yang T, Lasagna MD, Shi J, Reinhart GD, Cremer PS. Biophys J. 2008;94:3094–3103. doi: 10.1529/biophysj.107.115519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galili U, Clark MR, Shohet SB, Buehler J, Macher BA. Proc Nat Acad Sci USA. 1987;84:1369–1373. doi: 10.1073/pnas.84.5.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galili U, Shohet SB, Kobrin E, Stults CL, Macher BA. J Biol Chem. 1988;263:17755–17762. [PubMed] [Google Scholar]
- 19.Galili U, Mandrell RE, Hamadeh RM, Shohet SB, Griffiss JM. Infect Immun. 1988;56:1730–1737. doi: 10.1128/iai.56.7.1730-1737.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galili U, Rachmilewitz EA, Peleg A, Flechner I. J Exp Med. 1984;160:1519–1531. doi: 10.1084/jem.160.5.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parker W, Bruno D, Holzknecht ZE, Platt JL. J Immunol. 1994;153:3791–3803. [PubMed] [Google Scholar]
- 22.Galili U. Biochimie. 2001;83:557–563. doi: 10.1016/s0300-9084(01)01294-9. [DOI] [PubMed] [Google Scholar]
- 23.Owen RM, Carlson CB, Xu J, Mowery P, Fasella E, Kiessling LL. Chembiochem. 2007;8:68–82. doi: 10.1002/cbic.200600339. [DOI] [PubMed] [Google Scholar]
- 24.Gege C, Kinzy W, Schmidt RR. Carbohydr Res. 2000;328:459–466. doi: 10.1016/s0008-6215(00)00145-2. [DOI] [PubMed] [Google Scholar]
- 25.Naicker KP, Li H, Heredia A, Song H, Wang L-X. Org Biomol Chem. 2004;2:660–664. doi: 10.1039/b313844e. [DOI] [PubMed] [Google Scholar]
- 26.Fang J, Li J, Chen X, Zhang Y, Wang J, Guo Z, Zhang W, Yu L, Brew K, Wang PG. J Am Chem Soc. 1998;120:6635–6638. [Google Scholar]
- 27.Oyelaran O, McShane LM, Dodd L, Gildersleeve JC. J Proteome Res. 2009;8:4301–4310. doi: 10.1021/pr900515y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huflejt ME, Vuskovic M, Vasiliu D, Xu H, Obukhova P, Shilova N, Tuzikov A, Galanina O, Arun B, Lu K, Bovin N. Mol Immunol. 2009;46:3037–3049. doi: 10.1016/j.molimm.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 29.Martinez V, Ingwers M, Smith J, Glushka J, Yang T, Bar-Peled M. J Biol Chem. 2012;287:879–892. doi: 10.1074/jbc.M111.287367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bentley SD, Aanensen DM, Mavroidi A, Saunders D, Rabbinowitsch E, Collins M, Donohoe K, Harris D, Murphy L, Quail MA, Samuel G, Skovsted IC, Kaltoft MS, Barrell B, Reeves PR, Parkhill J, Spratt BG. PLoS Genet. 2006;2:e31. doi: 10.1371/journal.pgen.0020031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kocíncová D, Lam JS. Biochemistry (Mosc) 2011;76:755–760. doi: 10.1134/S0006297911070054. [DOI] [PubMed] [Google Scholar]
- 32.Samuel G, Reeves P. Carbohydr Res. 2003;338:2503–2519. doi: 10.1016/j.carres.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 33.de Boer AR, Hokke CH, Deelder AM, Wuhrer M. Glycoconj J. 2008;25:75–84. doi: 10.1007/s10719-007-9100-x. [DOI] [PubMed] [Google Scholar]
- 34.Bolduc OR, Pelletier JN, Masson J-F. Anal Chem. 2010;82:3699–3706. doi: 10.1021/ac100035s. [DOI] [PubMed] [Google Scholar]
- 35.Chen W, Gu L, Zhang W, Motari E, Cai L, Styslinger TJ, Wang PG. ACS Chem Biol. 2011;6:185–191. doi: 10.1021/cb100318z. [DOI] [PubMed] [Google Scholar]
- 36.Klim JR, Fowler AJ, Courtney AH, Wrighton PJ, Sheridan RTC, Wong ML, Kiessling LL. ACS Chem Biol. 2012;7:518–525. doi: 10.1021/cb2004725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hancock LE, Gilmore MS. Proc Natl Acad Sci USA. 2002;99:1574–1579. doi: 10.1073/pnas.032448299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soell M, Lett E, Holveck F, Scholler M, Wachsmann D, Klein J. J Immunol. 1995;154:851–860. [PubMed] [Google Scholar]
- 39.Panos C, Cohen M. Biochim Biophys Acta. 1966;117:98–106. doi: 10.1016/0304-4165(66)90156-5. [DOI] [PubMed] [Google Scholar]
- 40.Roberts WSL, Stewart FS. Microbiology. 1961;24:253–260. doi: 10.1099/00221287-24-2-253. [DOI] [PubMed] [Google Scholar]
- 41.Pazur JH, Erikson MS, Tay E. Carbohydr Res. 1983;124:253–263. doi: 10.1016/0008-6215(83)88461-4. [DOI] [PubMed] [Google Scholar]
- 42.Gruber WC, Scott DA, Emini EA. Ann N Y Acad Sci. 2012;1263:15–26. doi: 10.1111/j.1749-6632.2012.06673.x. [DOI] [PubMed] [Google Scholar]
- 43.Schütz K, Hughes RG, Parker A, Quinti I, Thon V, Cavaliere M, Würfel M, Herzog W, Gessner JE, Baumann U. J Clin Immunol. 2012 doi: 10.1007/s10875-012-9792-y. [DOI] [PubMed] [Google Scholar]
- 44.Karjalainen K, Mäkelä O. Eur J Immunol. 1976;6:88–93. doi: 10.1002/eji.1830060204. [DOI] [PubMed] [Google Scholar]
- 45.Brandriss MW. Nature. 1969;221:960–962. doi: 10.1038/221960a0. [DOI] [PubMed] [Google Scholar]
- 46.Little JR, Eisen HN. Biochemistry. 1967;6:3119–3125. doi: 10.1021/bi00862a020. [DOI] [PubMed] [Google Scholar]
- 47.Mytych DT, La S, Barger T, Ferbas J, Swanson SJ. J Pharm Biomed Anal. 2009;49:415–426. doi: 10.1016/j.jpba.2008.11.028. [DOI] [PubMed] [Google Scholar]
- 48.Hudak JE, Canham SM, Bertozzi CR. Nat Chem Biol. 2014;10:69–U111. doi: 10.1038/nchembio.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu HP, Kwong B, Irvine DJ. Angew Chem -Int Edit. 2011;50:7052–7055. doi: 10.1002/anie.201101266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mikesch JH, Buerger H, Simon R, Brandt B. Biochim Biophys Acta-Rev Cancer. 2006;1766:42–52. doi: 10.1016/j.bbcan.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Stefanova I, Hilgert I, Kristofova H, Brown R, Low MG, Horejsi V. Mol Immunol. 1989;26:153–161. doi: 10.1016/0161-5890(89)90097-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.