

Abstract
Cleft lip and palate is a common human birth defect, and its causes are being dissected through studies of human populations and through the use of animal models. Mouse models in particular have made a substantial contribution to our understanding of the gene pathways involved in palate development and the nature of signaling molecules that act in a tissue-specific manner at critical stages of embryogenesis. Related work has provided further support for investigating the role of common environmental triggers as causal covariates.
Human birth defects arise from many etiologies, including single-gene disorders, chromosome aberrations, exposure to teratogens, and sporadic conditions of unknown cause. Birth defect syndromes include multiple structural abnormalities and/or cognitive delays. However, most human birth defects affect a single organ system, and those disrupting facial structures are found in approximately 1% (or 1 million) of infants born worldwide each year. The most common of these birth defects is cleft lip and/or palate, a complex trait caused by multiple genetic and environmental factors (1).
Genes that play a role in palate development
As shown in Figure 1, the secondary palate develops as an outgrowth of the maxillary prominences at about embryonic day (E) 11.5 in the mouse. The palate shelves initially grow vertically down the side of the tongue (E12.5) and then elevate above the tongue as it drops in the oral cavity (E13.5). With continued growth, the shelves appose in the midline (E14.5) and fuse (E15.5). Growth of the palate shelves depends on the survival and continued proliferation of mesenchymal cells that originate from neural crest and mesodermal cells of the first pharyngeal arch. In this issue of the JCI, using transgenic animal models Rice and coworkers provide details of the interactions between the epithelium and mesenchyme that lead to palate growth and development (2). They demonstrate a signaling process in which Fgf10 is expressed in the mesenchyme, then activates its receptor, FGF receptor 2b (Fgfr2b), which is located in the epithelium. Finally, Fgfr2b mediates expression of sonic hedgehog (Shh) in the epithelium.
Figure 1.
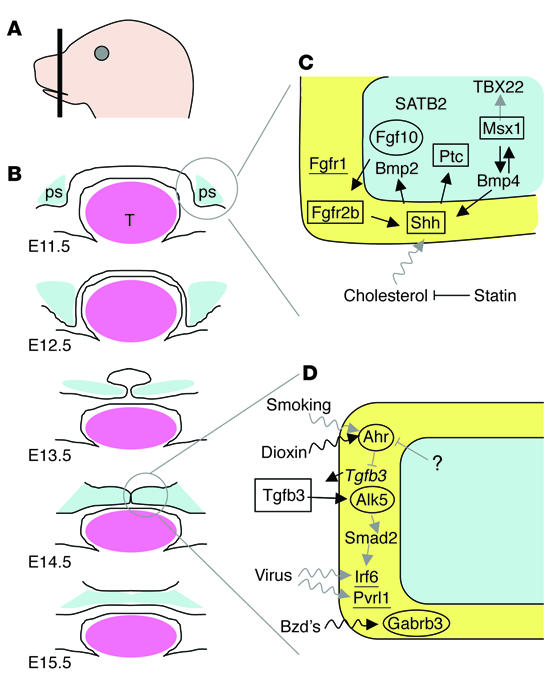
Signaling molecules essential for palate development. (A) Transverse section of embryonic palate. (B) Schematic of sections of normal palate shelf (ps; blue) development at the indicated days after conception. Palate shelves emanate from maxillary prominences (E11.5), grow, and extend vertically past the tongue (E12.5). The tongue (T; pink) drops, allowing the palate shelves to elevate (E13.5), appose (E14.5), and fuse (E15.5). (C and D) Cell-specific expression of signaling molecules during palate growth (C) and fusion (D). Genes involved in palate growth may also be involved in fusion. The γ-aminobutyric acid receptor subunit β3 (Gabrb3) is also involved in palate elevation (not shown). Molecules are expressed in the epithelium (yellow) or mesenchyme (blue). Molecules shown to be essential for palate development are indicated for mouse (oval), human (underlined), and both mouse and human (rectangle). Arrows indicate known (black) or predicted (gray) gene-gene (straight) and gene-environment (wavy) interactions. Ahr, aryl-hydrocarbon receptor; Alk5, activin receptor_like kinase 5; Bmp4, bone morphogenic protein 4; Bzd’s, benzodiazepines; Msx1, msh-like 1 homeo box; Ptc, patched homolog 1; Pvrl1, poliovirus receptor_related 1; SATB2, SATB family member 2; TBX22, T-box 22; Tgfb3, transforming growth factor β3.
Additional genetic factors involved in palate development have been described using mouse transgenic models; in particular, mice lacking the muscle segment–specific homeobox Msx1 (3) or the signaling molecule Tgfb3 (4) exhibit cleft palate. While many other gene knockouts also result in palate or other craniofacial defects, in most cases the gene deletions and/or insertions cause multiple structural or functional defects. Consequently, evaluation of the role of a particular gene in palate formation has not been possible. The function of Msx1 and Tgfb3 in palate development was extended to isolated forms of clefting in humans. Point mutations and/or statistical analyses have indicated a role for these factors in cases of cleft lip and/or palate in which the only other feature was dental abnormalities (1, 5). In parallel with the advances made from the study of animal models, complementary progress has been made to identify additional genes that play a direct role in human palate development.
Two recent gene discovery reports are particularly relevant to human palate development. In the first, mutations were identified in the gene that encodes the transcription factor interferon regulatory factor 6 (IRF6), resulting in the autosomal dominant disorder Van der Woude syndrome (VWS) (6). VWS is an especially important model for isolated cleft lip and palate. In the clinic, the only difference between individuals with VWS and those with isolated cleft lip and palate is the presence of pits in the lower lip of most VWS cases. In addition, VWS is caused by mutations in a single gene, whereas the more common isolated cleft is a complex trait caused by multiple gene mutations and/or environmental insults. Very recently it was demonstrated that a common haplotype associated with IRF6 contains a mutation that provides an attributable risk of approximately 12% to all common forms of cleft lip and palate (7). In a second report, nonsense mutations and deletions in the FGFR1 gene were identified in cases of Kallmann syndrome (8), an autosomal dominant disorder typically characterized by infertility and anosmia. However, approximately 5% of Kallmann syndrome cases have clefts of the lip and/or palate and, as with VWS, some individuals may present with clefts as the only component of the phenotype. Other genes that play a role in human palate development were reviewed recently (P63, PVRL1, TGFA, and TBX22; ref 1) or were reported (SATB2) (9).
One remarkable feature of the genes IRF6, MSX1, and FGFR1 is that mutations in any of the three are associated with dental anomalies and “mixed clefting.” Mixed clefting refers to disorders in which cases of isolated cleft palate and cleft lip (with or without cleft palate) occur in the same pedigree. Clefts of the lip or clefts of the lip with the palate arise in the primary palate, whereas clefts of the palate alone occur in the secondary palate. Mixed clefting disorders suggest that identical mechanisms cause these two forms, which previously had been separated based on embryologic and genetic evidence (1). The presence of dental anomalies in some individuals who have mutations in each of these three genes suggests that these same pathways are common to tooth development.
Pathways in palate development
In addition to demonstrating the essential role of the Fgf10/Fgfr2/Shh signaling pathway in palate development, Rice and coworkers (2) integrate this model into the Msx1 pathway. Zhang et al. (10) previously demonstrated that Msx1, bone morphogenetic protein 4 (Bmp4), Shh, and Bmp2 constitute a pathway that is essential for palate development in mice. Figure 1 shows an expansion of the models presented in those papers and incorporates known and speculative interactions between these and other signaling pathways in lip and palate development. First, we show other proteins in pathways, including Shh. These pathways drive the epithelium and mesenchyme interactions that support cell proliferation and palate growth (Figure 1C). In addition, we posit that FGFR1 (11), SATB2 (9), and TBX22 (12) are also involved in palate growth in humans and/or mice, although their exact placement in a known pathway remains to be determined. Second, we show a more speculative pathway that attempts to connect molecules that are involved in palate fusion (Figure 1D). Solid evidence supports a role in palate fusion in mice for Ahr (13), Tgfb3 (4), Alk5 (14), Smad2 (14), Gabrb3 (15), and in humans for IRF6 (6). The involvement of these genes and their hypothesized interactions suggest that a broader view of the major players in palate development is coming into focus and represents additional candidate genes that can be investigated by DNA resequencing and/or statistical analyses.
Pursuit of gene-environment interactions
Although genes play a substantial role in facial embryogenesis, the role that the environment plays in modulating genetic effects is equally critical. At least three major classes of environmental triggers have been studied. One of these is teratogens. Maternal smoking, for example, has been recognized as an important covariate in clefting (16). Other teratogens that increase the risk of cleft lip and palate through maternal ingestion include pharmaceuticals, such as the anticonvulsant phenytoin and benzodiazepines, or pesticides, such as dioxin (17). The effect of a second class of environmental trigger, infection, is less clear. However, we wish to point out that two genes that are essential for palate development, IRF6 (6) and PVRL1 (18), are members of gene families that modulate the immune response to infection. These findings suggest that we need a more critical examination of whether infectious agents increase the risk of clefting after exposure during the first trimester. Finally, both nutrients (e.g., vitamins or trace elements) and cholesterol metabolism also are increasingly seen as being important in influencing embryonic development. Folate in particular is recognized as playing an important role in neural tube formation. The recognition that folic acid supplementation can decrease the risk of neural tube defects represents, along with the treatment of Rh disease and phenylketonuria, one of the great genetic public health successes of the twentieth century (19). Cholesterol is an essential component of Shh signaling. The central role for Shh presented by Rice and colleagues in this issue of the JCI provides further support for the idea that normal variations in cholesterol metabolism and/or disruptions in cholesterol levels through pharmacological intervention might also be risk factors for facial birth defects (Figure 1C). Recently, Edison and Muenke provided preliminary data suggesting that early embryonic exposure to the cholesterol-lowering statin drugs may confer a risk for a wide range of birth defects of the midline, including clefts of the lip and palate (20). The integration of Shh into signaling pathways that include Egf, Fgf, Tgfb or Wnt molecules provides strong justification for critical investigation of the role of cholesterol metabolism in human facial embryogenesis.
Summary
The elegant work presented by Rice and co-workers (2) affords new opportunities to better understand the pathways involved in human embryogenesis in general and palate development in particular. This work compels the development of additional model systems and also calls for new investigations using human biological and epidemiologic data. The iterative use of human and animal studies will bring the most rapid progress toward enhanced diagnoses, interventions to improve clinical outcomes, and preventative strategies for human birth defects.
Acknowledgments
The ideas presented in this manuscript have benefited from many discussions with our colleagues, in particular Kaare Christensen, Michael Dixon, Peter Jezewski, Andrew Lidral, Mary Marazita, Max Muenke, Paul Romitti, and Alex Vieira. Nancy Davin and Susie McConnell provided critical administrative support. This work was supported by NIH grants to J.C. Murray (DE08559) and B.C. Schutte (DE13513).
Footnotes
See the related article beginning on page 1692.
Nonstandard abbreviations used: embryonic day (E); FGF receptor 2b (Fgfr2b); interferon regulatory factor 6 (IRF6); sonic hedgehog (Shh); Van der Woude syndrome (VWS).
Conflict of interest: The authors serve as unpaid consultants to GeneDx Inc., which offers molecular testing for the IRF6 gene.
References
- 1.Murray JC. Gene/environment causes of cleft lip and/or palate. Clin. Genet. 2002;61:248–256. doi: 10.1034/j.1399-0004.2002.610402.x. [DOI] [PubMed] [Google Scholar]
- 2.Rice R, et al. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. Invest. 2004;113:1692–1700. doi:10.1172/JCI200420384. doi: 10.1172/JCI20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Satokata I, Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat. Genet. 1994;6:348–356. doi: 10.1038/ng0494-348. [DOI] [PubMed] [Google Scholar]
- 4.Proetzel G, et al. Transforming growth factor beta 3 is required for second palate fusion. Nat. Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jezewski P, et al. Complete sequencing demonstrates a role for MSX1 in nonsyndromic cleft lip and palate. J. Med. Genet. 2003;40:399–407. doi: 10.1136/jmg.40.6.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kondo S, et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zucchero, T.M., et al. 2004. Interferon regulatory factor 6 (IRF6) is a modifier for isolated cleft lip and palate. N. Engl. J. Med. In press. [DOI] [PubMed]
- 8.Dode C, et al. Loss of function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- 9.Fitzpatrick DR, et al. Identification of SATB2 as the cleft palate gene on 2q32-q33. Hum. Mol. Genet. 2003;12:2491–2501. doi: 10.1093/hmg/ddg248. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Z, et al. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129:4135–4146. doi: 10.1242/dev.129.17.4135. [DOI] [PubMed] [Google Scholar]
- 11.Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2:reviews3005.1–3005. doi: 10.1186/gb-2001-2-3-reviews3005. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braybrook C, et al. Craniofacial expression of human and murine TBX22 correlates with the cleft palate and ankyloglossia phenotype observed in CPX patients. Hum. Mol. Genet. 2002;11:2793–2804. doi: 10.1093/hmg/11.22.2793. [DOI] [PubMed] [Google Scholar]
- 13.Mimura J, et al. Loss of teratogenic response to 2,3,7,8-tetrachlorodinbenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells. 1997;2:645–654. doi: 10.1046/j.1365-2443.1997.1490345.x. [DOI] [PubMed] [Google Scholar]
- 14.Dudas M, Nagy A, Laping NJ, Moustakeas A, Kaartinen V. Tgf-β3-induced palatal fusion is mediated by Alk-5/Smad pathway. Dev. Biol. 2004;266:96–108. doi: 10.1016/j.ydbio.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 15.Hagiwara N, Katarova Z, Siracusa LD, Brilliant MH. Nonneuronal expression of the GABA(A) beta3 subunit gene is required for normal palate development in mice. Dev. Biol. 2003;254:93–101. doi: 10.1016/s0012-1606(02)00030-1. [DOI] [PubMed] [Google Scholar]
- 16.Little J, Cardy A, Munger RG. Tobacco smoking and oral clefts: a meta-analysis. Bull. World Health Organ. 2004;82:213–218. [PMC free article] [PubMed] [Google Scholar]
- 17.Hayes, C. 2002. Environmental risk factors and oral clefts. In Cleft lip & palate from origin to treatment. D.F. Wyszynski, editor. Oxford University Press. Oxford, United Kingdom. 159–169.
- 18.Suzuki K, et al. Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nat. Genet. 2000;25:427–430. doi: 10.1038/78119. [DOI] [PubMed] [Google Scholar]
- 19.Centers for Disease Control and Prevention (CDC). Spina bifida and anencephaly before and after folic acid mandate--United States, 1995-1996 and 1999-2000. MMWR Morb. Mortal. Wkly. Rep. 2004;53:362–365. [PubMed] [Google Scholar]
- 20.Edison RJ, Muenke M. Central nervous system and limb anomalies in case reports of first-trimester statin exposure. N. Engl. J. Med. 2004;350:1579–1582. doi: 10.1056/NEJM200404083501524. [DOI] [PubMed] [Google Scholar]