

Abstract
Activation of microglial NADPH oxidase (NOX2) plays a critical role in mediating neuroinflammation, which is closely linked with the pathogenesis of a variety of neurodegenerative diseases, including Parkinson’s disease (PD). The inhibition of NOX2-generated superoxide has become an effective strategy for developing disease-modifying therapies for PD. However, the lack of specific and potent NOX2 inhibitors has hampered the progress of this approach. Diphenyleneiodonium (DPI) is a widely used, long-acting NOX2 inhibitor. However, due to its non-specificity for NOX2 and high cytotoxicity at standard doses (μM), DPI has been precluded from human studies. In this study, using ultra-low doses of DPI, we aimed to: 1) investigate whether these problems could be circumvented and 2) determine whether ultra-low doses of DPI were able to preserve its utility as a potent NOX2 inhibitor. We found that DPI at subpicomolar concentrations (10−14 and 10−13 M) displays no toxicity in primary midbrain neuron-glia cultures. More importantly, we observed that subpicomolar DPI inhibited phorbol myristate acetate (PMA)-induced activation of NOX2. The same concentrations of DPI did not inhibit the activities of a series of flavoprotein-containing enzymes. Furthermore, potent neuroprotective efficacy was demonstrated in a post-treatment study. When subpicomolar DPI was added to neuron-glia cultures pretreated with lipopolysaccharide (LPS), 1-methyl-4-phenylpyridinium or rotenone, it potently protected the dopaminergic neurons. In summary, DPI’s unique combination of high specificity towards NOX2, low cytotoxicity and potent neuroprotective efficacy in post-treatment regimens suggests that subpicomolar DPI may be an ideal candidate for further animal studies and potential clinical trials.
Keywords: Microglia, NADPH oxidase, Neuroinflammation, Oxidative stress, Parkinson’s disease
INTRODUCTION
NADPH oxidase (NOX2) is the primary extracellular superoxide-producing enzyme in activated phagocytes, including microglia (Lambeth 2004; Nunes et al. 2013). Studies from our group and others have demonstrated that microglial NOX2 is a key mediator in initiating and maintaining chronic neuroinflammation (Gao et al. 2011; Qin et al. 2013), which subsequently causes progressive dopaminergic neurodegeneration in rodent models of Parkinson’s disease (PD) (Barnum and Tansey 2010; Block and Hong 2005; Block et al. 2007; Gao and Hong 2008; Hirsch and Hunot 2009; Hirsch et al. 2012). Elevated expression and activation of NOX2 have been found in the substantia nigra of PD patients and animal models (Qin et al. 2013; Wu et al. 2003). Moreover, genetic deletion of NOX2 (gp91phox−/−, the catalytic subunit of NOX2) greatly reduces microglia-mediated neuroinflammation and protects dopaminergic neurons against toxin-induced damage (Block et al. 2007). Recently, NOX2 inhibition has become a new strategy for developing effective disease-modifying therapies for PD (Gao et al. 2012; Hernandes and Britto 2012; Sorce et al. 2012; Surace and Block 2012).
In recent decades, a number of peptides and small, molecular compounds have been developed to inhibit NOX2 and treat inflammation-related diseases (Aldieri et al. 2008; Cifuentes-Pagano et al. 2013; Drummond et al. 2011). However, most of these inhibitors lack enzyme specificity and have high toxicity and low efficacy. For these reasons, the progress in developing useful NOX2 inhibitors as potential therapeutic agents has been hampered. Therefore, the need to develop specific, high-potency, non-toxic NOX2 inhibitors is urgent. Diphenyleneiodonium (DPI) is a widely used, long-acting NOX2 inhibitor that covalently binds gp91phox, the catalytic subunit of NOX2 (Doussiere et al. 1999; O’Donnell et al. 1993). Despite its high potency in inhibiting NOX2 activity, DPI also potently inhibits a variety of other electron-transferring flavoprotein enzymes, including inducible nitric oxide synthase (iNOS), xanthine oxidase, NADH-ubiquinone oxidoreductase, cytochrome P450 reductase and thioredoxin reductase at standard micromolar concentrations (Aldieri et al. 2008). Moreover, micromolar concentrations of DPI are highly toxic (Aldieri et al. 2008). Thus, DPI has not been considered an ideal NOX2 inhibitor and has been precluded from clinical trials.
We recently reported that pretreatment with DPI at subpicomolar concentrations (10−13 to 10−14 M) was capable of inhibiting lipopolysaccharide (LPS)-induced superoxide production and protecting dopaminergic neurons in primary midbrain neuron-glia cultures (Qian et al. 2007). These findings led us to further investigate whether subpicomolar DPI could be a specific NOX2 inhibitor and a potential therapeutic agent for PD. In this study, we first addressed the issues of toxicity and enzyme specificity. We found that DPI at concentrations of 10−13 and 10−14 M specifically inhibited NOX2 activation in primary midbrain neuron-glia cultures without any observed cytotoxicity. We then determined the potential of DPI as a therapeutic agent using a post-treatment regimen, which is more clinically relevant, in three in vitro rodent PD models. We found that post-administration of subpicomolar DPI exhibited neuroprotection against LPS-, l-methyl-4-phenylpyridinium (MPP+)- and rotenone-induced dopaminergic neurodegeneration. Our findings suggest that DPI at subpicomolar concentrations could be a useful tool as a specific inhibitor of microglial NOX2. Furthermore, the lack of toxicity and the potent neuroprotection indicate that ultra-low doses of DPI have high therapeutic promise for future in vivo and clinical studies in neurodegenerative diseases.
MATERIALS AND METHODS
Primary midbrain neuron-glia cultures
Primary neuron/glia cultures were prepared as described previously (Chen et al. 2013). Briefly, dissociated cells were seeded at densities of 5 × 105 cells/well and 1 × 105 cells/well in poly-D-lysine-coated 24- and 96-well plates, respectively. The cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air and were grown in minimum essential medium containing 10% heat-inactivated fetal bovine serum, 10% heat-inactivated horse serum (Invitrogen™, Grand Island, NY, USA), 1 g/L glucose, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 μM nonessential amino acids, 50 U/ml penicillin and 50 μg/ml streptomycin. Seven days later, the cultures were used for the drug treatments.
[3H]-dopamine (DA) uptake assay
Uptake assays were performed by incubating the cultures with 1 μM [3H]-DA (PerkinElmer Life Sciences, Santa Clara, CA, USA) for 20 min at 37°C, as previously described (Gao et al. 2002). Nonspecific uptake was determined in the presence of 10 μM mazindol (Sigma-Aldrich, St. Louis, MO, USA).
Immunocytochemistry and cell counting in mesencephalic neuron-glia cultures
Immunostaining was performed as previously described (Qin et al. 2004) with antibodies against tyrosine hydroxylase (TH; 1:5,000; EMD Millipore Corporation, Billerica, MA, USA), ionized calcium binding adaptor molecule 1 (Iba1; 1:5,000; Wako Chemicals, Richmond, VA, USA) and glial fibrillary acidic protein (GFAP; 1:10,000; Wako Chemicals, Richmond, VA, USA). Images were recorded using a CCD camera and the MetaMorph software (Molecular Devices, Sunnyvale, CA, USA). To quantitative cell numbers, the total number of TH-immunoreactive (THir) neurons in a well of a 24-well plate was counted. For each experiment, two to six wells were used per treatment condition, and the results from three to four independent experiments were obtained.
Measurement of superoxide and nitrite
The production of superoxide was assessed by measuring the SOD-inhibitable reduction of the tetrazolium salt WST-1, as described previously (Wang et al. 2012). Briefly, primary neuron-glia cultures were pre-treated with LPS or phorbol myristate acetate (PMA) for 12 h, then washed twice with Hanks’ balanced salt solution without phenol red. After 30 mins of DPI incubation, 50 μl of WST-1 (1 mM) with and without SOD (50 U/ml) was added to each well. The absorbance at 450 nm was read using a SpectraMax Plus microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). The absorbance difference observed between the cultures in the presence and absence of SOD represented the amount of superoxide produced. The production of nitrite was determined using Griess reagent.
Extraction of membrane fractions and Western blot analysis
Membrane fractions of HAPI microglia were prepared as described previously (Wang et al. 2012). Briefly, HAPI microglia were lysed in hypotonic lysis buffer (1 mM Tris, 1 mM KCl, 1 mM EGTA, 1 mM EDTA, 0.1 mM DTT, 1 mM PMSF and 10 μg/ml cocktail protease inhibitor) and subjected to Dounce homogenization (20–25 stokes, tight pestle A). The lysates were centrifuged at 1,600 × g for 15 min, and the supernatant was centrifuged at 100,000 × g for 30 min. The pellets were solubilized in 1% Nonidet P-40 hypotonic lysis buffer, separated using a 4–12% Bis-Tris Nu-PAGE gel and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% non-fat milk and incubated with a rabbit antibody (1:1,000) against p47phox and gp91phox (BD Transduction Laboratories, San Jose, CA, USA) and HRP-linked anti-rabbit or anti-mouse IgG (1:3,000) for 2 h. ECL reagents (Amersham Biosciences Corp, Piscataway, NJ, USA) were used as a detection system.
INOS activity assay
The iNOS activity in neuron-glia cultures was measured according to previous reports, with minor modifications (Chang et al. 2009). iNOS was first induced in neuron-glia cultures by treatment with LPS for 12 h; the media was then changed, and different concentrations of DPI (10−13 and 10−14 M) were added. Twenty-four hours later, the nitrite levels in the supernatant were measured as an index of iNOS activity.
The effects of DPI on purified iNOS activity were measured as described previously (Chang et al. 2009). Briefly, purified iNOS enzyme was incubated with or without the indicated concentrations of DPI for 3 h at 37°C in reaction buffer (50 mM Tris-HCl, pH 7.6, 2 mM NADPH, 2 mM L-arginine, 20 μM FAD, 20 μM tetrahydrobiopterin and 1 mM DTT). The iNOS activity was expressed as μmol nitrite/ml enzyme.
Xanthine oxidase activity assay
Xanthine oxidase activity was measured in the neuron-glia cultures using a commercial xanthine oxidase assay kit (Cayman Chemical, Ann Arbor, MI, USA).
The effects of DPI on purified xanthine oxidase (Sigma- Aldrich, St. Louis, MO, USA) were determined according to the manufacturer’s protocol.
NADH-ubiquinone oxidoreductase activity assay
The activity of NADH-ubiquinone oxidoreductase was measured using a commercial assay kit (MitoSciences, Eugene, OR, USA).
Cytochrome P450 reductase activity assay
Cytochrome P450 reductase activity in neuron-glia cultures was measured using a commercial cytochrome P450 reductase assay kit (Sigma- Aldrich, St. Louis, MO, USA).
The activity of purified cytochrome P450 reductase was measured by determining the initial rate of cytochrome c reduction by purified cytochrome P450 reductase enzyme in 300 mM phosphate buffer containing 100 μM NADPH, 0.1 mM EDTA, 1 mM cytochrome c oxidase inhibitor and 20 μM cytochrome c, unless otherwise indicated (Hallstrom et al. 2004). One unit of cytochrome P450 reductase activity was defined as the amount of 1 μM cytochrome c reduced by NADPH per minute and is expressed as U/ml enzyme.
Thioredoxin reductase activity assay
Thioredoxin reductase activity was measured in the neuron-glia cultures using a commercial thioredoxin reductase assay kit (Sigma- Aldrich, St. Louis, MO, USA).
The activity of purified thioredoxin reductase was measured by determining the initial rate of DTNB reduction by purified thioredoxin reductase enzyme at 25°C in reaction buffer containing 100 mM phosphate buffer, 10 mM EDTA, 1 mM DTNB, 0.2 mg/ml BSA and 0.5 mM NADPH (Leitsch et al. 2010). The activity of thioredoxin reductase was expressed as U/ml enzyme.
Statistical analysis
All values are expressed as the mean ± SEM. Differences between the means were analyzed using one-way analysis of variance (ANOVA) with treatment as the independent factor. When the ANOVA resulted in significant differences, pairwise comparisons between the means were tested using Bonferroni’s post hoc testing. In all analyses, a value of p < 0.05 was considered statistically significant.
RESULTS
Subpicomolar DPI displays no obvious cytotoxicity
Because the high cytotoxicity of DPI at micromolar concentrations has been well described, we examined the viability of midbrain neuron-glia cultures exposed to 10−13 and 10−14 M DPI. MTT assays revealed no changes in cell viability after the cultures were treated with subpicomolar concentrations of DPI for 48 h. In contrast, DPI at micromolar concentrations (10−6 and 10−7 M) reduced the cell viability by more than 90% compared with the controls (Fig. 1A). Immunocytochemical (ICC) staining with an antibody against Neu-N, a nuclear protein expressed selectively by neurons, failed to reveal any differences in the staining intensity and distribution of Neu-N-positive cells between the controls and subpicomolar DPI-treated cultures (Fig. 1B). Among these neurons, dopaminergic neurons are the most sensitive to oxidative stress-induced damage (Surmeier et al. 2012). Thus, the functional status of dopaminergic neurons was determined using an [3H]-DA uptake assay. Consistent with the ICC results, 10−13 and 10−14 M DPI showed no difference in DA uptake capacity after 48 h of exposure compared with the controls (Fig. 1C).
Figure 1.

Effects of DPI on cell viability and DA uptake capacity in primary midbrain neuron-glia cultures incubated with different concentrations of DPI for 48 h. (A) Cell viability was evaluated by MTT assays. (B) Representative images of cells immunostained with Neu-N, Iba-1 and GFAP antibodies indicate lesions on the neurons, microglia and astroglia after micromolar, but not subpicomolar, DPI exposure. The inserts show amplified microglia in each group. (C) [3H]-DA uptake analysis revealed a decrease in neurotransmitter uptake capacity after micromolar, but not subpicomolar, DPI exposure. The results are expressed as a percentage of the controls (mean ± SEM) from three experiments performed in duplicate and were analyzed using one-way ANOVA, followed by Bonferroni’s post hoc multiple comparison test. **p < 0 .01; Bar = 50 μm.
Microglia and astroglia were also evaluated by ICC staining using cell-specific antibodies against Iba1 and GFAP, respectively. No significant alterations to microglia and astroglia were observed in the neuron-glia cultures treated with 10−13 M DPI for 48 h (Fig. 1B). In contrast, the addition of micromolar (10−6 M) DPI significantly reduced the number of astroglia and damaged the microglia, as demonstrated by the cell size enlargement and vacuolation observed in the cytoplasm. Collectively, these findings indicate that DPI at subpicomolar concentrations has no significant cytotoxicity in midbrain neuron-glia cultures.
Subpicomolar DPI inhibits NOX2 activation with high specificity
To address the issue of enzyme specificity, the activities of a list of flavoprotein-containing enzymes were compared in the presence of DPI at either micromolar (10−5 M) or subpicomolar (10−13 and 10−14 M) concentrations. The flavoprotein-containing enzymes studied were iNOS, xanthine oxidase, NADH-ubiquinone oxidoreductase, cytochrome P450 reductase and thioredoxin reductase. Micromolar DPI concentrations inhibit these enzymes (Aldieri et al. 2008). We first used commercially available purified enzymes to test the specificity of DPI. Similar to previous reports, micromolar DPI did inhibit the activities of four flavoprotein enzymes, including iNOS, xanthine oxidase, cytochrome P450 reductase and thioredoxin reductase. In contrast, subpicomolar DPI did not affect the activities of these four enzymes (Fig. 2A–D). NOX2 and NADH-ubiquinone oxidoreductase were not included in this experiment because these two purified enzymes are not commercially available.
Figure 2.

Subpicomolar DPI displays specificity for NOX2. (A–D) DPI at 10−13 and 10−14 M fails to inhibit commercially purified (A) iNOS (nitrite production as an index), (B) xanthine oxidase, (C) cytochrome P450 reductase and (D) thioredoxin reductase, although DPI at micromolar concentrations decreases these enzyme activities. Data are expressed as the mean ± SEM from three to four experiments performed in duplicate. (E–J) The effects of DPI on the enzymatic activities of NOX2, iNOS, xanthine oxidase, cytochrome P450 reductase, thioredoxin reductase and NADH-ubiquinone oxidoreductase in neuron-glia cultures. (E) Cellular NOX2 activation was induced by PMA in neuron-glia cultures. Superoxide production was used as an index of NOX2 activity. The addition of 10−13 or 10−14 M DPI inhibits NOX2-generated superoxide as efficiently as micromolar concentrations, indicating NOX2 inhibition. Data are expressed as a percentage of the PMA group (mean ± SEM) from three to four experiments performed in duplicate. (F) Cellular iNOS was induced in neuron-glia cultures by incubation with LPS for 12 h. Unlike micromolar concentrations, DPI at 10−13 and 10−14 M fails to reduce the generation of iNOS-generated nitrite. (G–J) DPI at 10−5 M, but not 10−13 and 10−14 M, inhibits xanthine oxidase, cytochrome P450 reductase, thioredoxin reductase and NADH-ubiquinone oxidoreductase in neuron-glia cultures. Data are expressed as the mean ± SEM from three to four experiments performed in duplicate. The results were analyzed using one-way ANOVA, followed by Bonferroni’s post hoc multiple comparison test. **p < 0.01. XO, xanthine oxidase; CPR, cytochrome P450 reductase; TR, thioredoxin reductase (TR); NUO, NADH-ubiquinone oxidoreductase.
Enzyme specificity was further determined in the cell cultures. Using a post-treatment regimen, we induced NOX2 activation in neuron-glia cultures by treating cells with PMA, a classic activator of NOX2, for 12 h prior to DPI treatment. Interestingly, DPI significantly inhibited NOX2-generated superoxide at 10−13 and 10−14 M as efficiently as it did at micromolar concentrations (Fig. 2E). To investigate the specificity of subpicomolar DPI concentrations for NOX2, we evaluated the effects of DPI (10−13 and 10−14 M) on the enzymatic activities of cellular iNOS, xanthine oxidase, NADH-ubiquinone oxidoreductase, cytochrome P450 reductase and thioredoxin reductase. Cellular iNOS was induced in neuron-glia cultures by incubation with LPS for 12 h. The levels of nitrites in the supernatant were measured as an index of iNOS activity. Unlike micromolar concentrations, subpicomolar DPI failed to reduce the generation of nitrites (Fig. 2F), suggesting that subpicomolar DPI has no effect on iNOS activity. Furthermore, DPI at 10−13 and 10−14 M failed to inhibit the activities of cellular xanthine oxidase, NADH-ubiquinone oxidoreductase, cytochrome P450 reductase and thioredoxin reductase in neuron-glia cultures, although micromolar DPI potently inhibited these enzymes (Fig. 2G–J).
Post-treatment with subpicomolar DPI protects dopaminergic neurons
To determine whether subpicomolar DPI can be a potential therapeutic agent for inflammation-related neurodegenerative diseases in clinical studies, neuroprotection in a post-treatment regimen must be demonstrated. Therefore, we first investigated the ability of subpicomolar DPI to rescue dopaminergic neurons in midbrain neuron-glia cultures 12 h after LPS challenge (Fig. 3A). Microglia stimulation by LPS induced neuroinflammation and resulted in subsequent collateral dopaminergic neurodegeneration. Post-treatment with subpicomolar DPI significantly attenuated the LPS-mediated dopaminergic neurodegeneration (Fig. 3B) and preserved the THir cells and their processes (TH, a marker for dopaminergic neurons; Fig. 3C, D).
Figure 3.

Dopaminergic neuroprotection by post-treatment with subpicomolar DPI 12 h after inflammatory challenge in primary neuron-glia cultures. (A) Experimental designs. Midbrain neuron-glia cultures were pre-treated with LPS (20 ng/ml) for 12 h, followed by DPI (10−14 or 10− 13 M) treatment. (B) [3H]-DA uptake assay and (C) THir neuron count analysis revealed significant dopaminergic protection 7 days after DPI treatment. (D) Representative cell images of TH immunostaining 7 days after DPI treatment indicate prominent protection of the dopaminergic neuronal cell bodies and dendrites. The results of DA uptake are expressed as a percentage of the controls and are the mean ± SEM from three to four experiments performed in duplicate. The results of THir cell counts are expressed as the mean ± SEM from three to four experiments performed in duplicate. Data were analyzed using one-way ANOVA, followed by Bonferroni’s post hoc multiple comparison test. **p < 0.01; Bar = 50 μm.
To further evaluate whether post-treatment with subpicomolar DPI is effective in other rodent in vitro PD models, we compared other commonly used toxins, MPP+ and rotenone, with LPS. In this study, post-treatment with DPI was performed 24 h after pretreating the cultures with toxins because up to 24 h was required for both MPP+ and rotenone to produce sufficient neuronal damage. Dopaminergic neuroprotection by post-treatment with 10−13 and 10−14 M DPI was still observed 24 h after LPS insult (Fig. 4B). Although MPP+ and rotenone directly damage dopaminergic neurons, previous reports have indicated that reactive microgliosis resulting from the toxic substances released from damaged neurons could initiate neuroinflammation, thus causing further neuronal damage (Gao et al. 2002; Liberatore et al. 1999). Subpicomolar DPI was able to suppress this immune-mediated neurotoxicity to achieve neuroprotection, as indicated by the restored [3H]-DA uptake capacity 24 h after MPP+ or rotenone-induced dopaminergic degeneration (Fig. 4C, D).
Figure 4.

Dopaminergic neuroprotection by post-treatment with subpicomolar DPI 24 h after xenobiotic damage in primary neuron-glia cultures. (A) Experimental designs. Midbrain neuron-glia cultures were pre-treated with LPS (20 ng/ml), MPP+ (0.15 μM) or rotenone (10 nM) for 24 h, followed by DPI (10−14 or 10−13 M) treatment. (B–D) Seven days after DPI treatment, significant dopaminergic protection was observed using the [3H]-DA uptake assay. The results are expressed as a percentage of the controls and are the mean ± SEM from three to four experiments performed in duplicate. Data were analyzed using one-way ANOVA, followed by Bonferroni’s post hoc multiple comparison test. *p < 0.05, **p < 0.01.
NOX2 inhibition alone is sufficient to mediate dopaminergic neuroprotection
Previous reports have indicated that a series of proinflammatory factors, such as free radicals and cytokines, released from microglia are associated with LPS-induced neurotoxicity. This study showed that DPI at subpicomolar concentrations displayed high specificity for NOX2 inhibition without affecting other flavoprotein enzymes (Fig. 2) and exerted potent neuroprotection (Figs. 3 and 4). Therefore, we investigated whether NOX2 inhibition alone by DPI is sufficient to mediate dopaminergic neuroprotection. The production of most proinflammatory cytokines, such as tumor necrosis factor alpha (TNFα) and interleukin-1 beta (IL-1β), by microglia was completed within 12 h of LPS stimulation (Liu et al. 2003). After repeated washing, neuron-glia cultures should contain minimal amounts of cytokines. However, the production of superoxide (from NOX2), nitrite (from iNOS) and PGE2 (from cyclooxygenase 2, COX2) should continue due to their activated enzymes. DPI was added 12 h after washing, and the effects of post-treatment DPI on nitrite, PGE2 and superoxide production were determined. Subpicomolar DPI failed to inhibit nitrite (Fig. 2B) and PGE2 production (data not show) in neuron-glia cultures. Interestingly, post-treatment with subpicomolar DPI almost completely inhibited LPS-induced superoxide production (Fig. 5A). Our previous report indicated that NOX2 is the major source of extracellular superoxide in inflammation-treated neuron-glia cultures (Qin et al. 2004). Thus, these results indicate that NOX2-generated superoxide not only played a key role in sustaining the microglia-induced neuroinflammation but also could explain the fact that DPI exerted its neuroprotective effect in this post-treatment regimen by merely inhibiting superoxide production.
Figure 5.
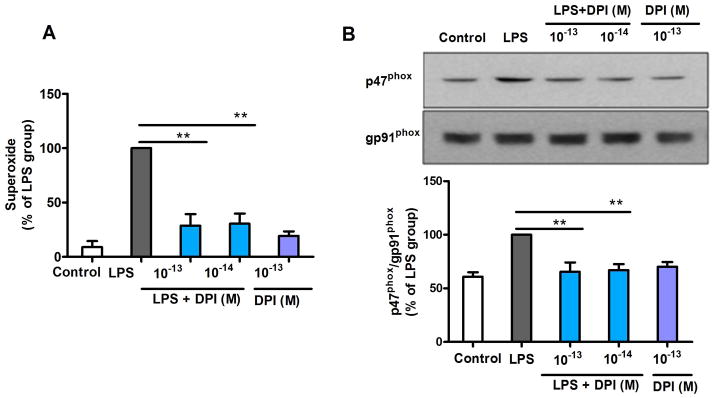
Post-treatment with subpicomolar DPI attenuates NOX2 activation induced by LPS through detachment of p47phox from the plasma membrane. (A) Midbrain neuron-glia cultures were pre-treated with LPS for 12 h, followed by DPI (10−14 or 10−13 M) treatment. Superoxide production was significantly inhibited by subpicomolar DPI post-treatment. (B) Western blot analysis revealed that subpicomolar DPI post-treatment detaches p47phox from the plasma membrane in LPS-treated HAPI microglia cells (gp91phox as an internal membrane control). The densities of the membrane p47phox signals were quantified. The results are expressed as a percentage of the LPS group (mean ± SEM) from three to four experiments performed in duplicate and were analyzed using one-way ANOVA, followed by Bonferroni’s post hoc multiple comparison test. **p < 0.01.
The phosphorylation and subsequent translocation of cytosolic subunit p47phox to the plasma membrane is required for the activation of NOX2 and, therefore, the production of superoxide (Lambeth 2004; Li et al. 2010; Zhu et al. 2006). We hypothesized that post-treatment with DPI was capable of inhibiting LPS-induced superoxide production by disassembling the NOX2 cytosolic/membrane subunit complex. Therefore, we investigated the DPI-induced detachment of p47phox from the plasma membrane. HAPI microglia cells were treated with LPS for 15 min to initiate p47phox membrane translocation (Supplementary Fig. 1) prior to DPI treatment. Thirty minutes after DPI treatment, the amount of p47phox in the membrane was significantly decreased (Fig. 5B), suggesting that DPI is capable of inhibiting activated NOX2 by dislodging the bound cytosolic subunits from the plasma membrane. Additionally, the resistance of gp91phox−/− mice-derived neuron-glia cultures to inflammation-mediated lesions (Supplementary Fig. 2) further supports the effectiveness of NOX2 inhibition in dopaminergic neuroprotection.
DISCUSSION
The present study demonstrated that DPI at subpicomolar concentrations inhibited NOX2 activation but did not inhibit other electron-transferring flavoprotein enzymes in primary midbrain neuron-glia cultures; additionally, this treatment did not result in any observed toxicity. Furthermore, potent dopaminergic neuroprotection by subpicomolar DPI post-treatment regimens was observed in three in vitro PD models. These findings indicate that DPI at ultra-low doses can be used experimentally as an excellent tool for specifically inhibiting microglial NOX2. Moreover, from the therapeutic point of view, results from this study also provide a convincing basis for using low-dose DPI as a potential candidate for further animal and future clinical studies.
Although DPI has been widely used as a NOX2 inhibitor, its high toxicity and lack of specificity at standard micromolar concentrations have excluded its consideration as a possible drug candidate. Our findings show that DPI at subpicomolar concentrations is a safe and highly specific NOX2 inhibitor. We verified the safety of DPI at subpicomolar concentrations using an MTT assay, ICC staining and functional neuronal analysis. No observable toxicity was detected, even after 7 days of treatment. Additionally, DPI specifically inhibited NOX2, but not other electron-transferring flavoprotein enzymes, including iNOS, xanthine oxidase, cytochrome P450 reductase, thioredoxin reductase and NADH-ubiquinone oxidoreductase. The potential mechanism underlying the selective inhibition of NOX2 by subpicomolar concentrations of DPI is attributed to its ability to interfere with the binding of cytosolic subunit p47phox to the plasma membrane, which is critical for NOX2 activation (Lambeth 2004; Li et al. 2010; Zhu et al. 2006). Our studies clearly demonstrate that DPI at subpicomolar concentrations significantly detached p47phox from the plasma membrane. It’s well known that DPI at micromolar concentrations covalently binds to gp91phox and serves as a long-acting NOX2 inhibitor (Doussiere et al. 1999; O’Donnell et al. 1993). Considering that gp91phox can recognize a variety of substances with higher affinity (Li et al. 2005; Qin et al. 2005a; Qin et al. 2005b), we speculated that DPI at subpicomolar concentrations could still bind to gp91phox, although we are not sure whether the binding site is the same with that of micromolar concentrations. Binding of DPI might change the conformation of gp91phox and subsequently decrease the affinity of binding of the cytosolic subunit (p47phox). Due to the technical limitation, we do not yet have evidence showing actual conformational change of gp91phox after DPI binding. Collectively, this study provides convincing evidence supporting the high specificity of DPI for NOX2 inhibition at subpicomolar concentrations.
Neuroinflammation is widely accepted to be associated with neurodegeneration. We and others have provided evidence indicating that free radicals are intimately linked with the initiation of neuroinflammation and subsequent neuronal damage (Block et al. 2007; Gao and Hong 2008; Minghetti 2005). Although numerous studies have been published using “antioxidant” treatments for neurodegenerative diseases, this strategy has continuously failed in clinical trials. We reason that antioxidants are unable to sufficiently halt neuroinflammation due to either their weak potency or bioavailability issues. In vitro and animal studies from our laboratory show that inhibiting NOX2-generated superoxide results in far more potent neuroprotective effects than antioxidants (Block et al. 2007; Gao et al. 2012). Supporting this notion, we and others have found that microglia-mediated neuroinflammation contributes to the progressive dopaminergic neurodegeneration in PD patients and animal models (Gao and Hong 2008; Wu et al. 2003). Mechanistic studies revealed that NOX2 is a key mediator in the maintenance of microglia-mediated neuroinflammation (Block and Hong 2005; Block et al. 2007; Brown 2007; Levesque et al. 2010). Thus, NOX2 may be an optimal target for developing neurodegenerative disease-modifying drugs. Consistent with this concept, we showed that post-treatment with 10−13 or 10−14 M DPI is neuroprotective in LPS-, MPP+- and rotenone-generated in vitro PD models (even after the onset of dopaminergic neurodegeneration). Interestingly, in the same experiment, post-treatment with subpicomolar DPI inhibited NOX2-generated superoxide but not iNOS-generated nitrite and COX2-generated PGE2 production, suggesting that inhibition of NOX2 activity alone is sufficient to mediate neuroprotection. We reason that superoxide plays a critical role in initiating and sustaining the production of other proinflammatory factors, such as TNFα and IL-1β, among others (Block and Hong 2005). In addition, DPI may prevent the formation of peroxynitrite, a highly cytotoxic oxidative radical derived from a reaction between superoxide and nitrite, even although nitrite production continues. Altogether, DPI at an ultra-low dose is a highly potent protector of dopaminergic neurons. Because the treatment regimen was performed post-insult, our findings are significant from a therapeutic point of view.
In summary, we report for the first time that subpicomolar DPI exhibits specificity for NOX2 and subsequently protects dopaminergic neurons against xenobiotic-induced toxicity. The demonstrated specificity, neuroprotective potency and low toxicological profiles at ultra-low concentrations suggest that DPI may be a promising drug candidate for future clinical trials in PD patients.
Supplementary Material
Main points.
DPI at ultra-low doses displays no cytotoxicity.
DPI at ultra-low doses exhibits high specificity towards NOX2.
DPI at ultra-low doses has therapeutic potential.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Footnotes
The authors have no conflicting financial interests.
References
- Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I, Ghigo D. Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr Drug Metab. 2008;9:686–96. doi: 10.2174/138920008786049285. [DOI] [PubMed] [Google Scholar]
- Barnum CJ, Tansey MG. Modeling neuroinflammatory pathogenesis of Parkinson’s disease. Prog Brain Res. 2010;184:113–32. doi: 10.1016/S0079-6123(10)84006-3. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Brown GC. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem Soc Trans. 2007;35:1119–21. doi: 10.1042/BST0351119. [DOI] [PubMed] [Google Scholar]
- Chang LP, Lai YS, Wu CJ, Chou TC. Liquid perfluorochemical inhibits inducible nitric oxide synthase expression and nitric oxide formation in lipopolysaccharide-treated RAW 264.7 macrophages. J Pharmacol Sci. 2009;111:147–54. doi: 10.1254/jphs.09043fp. [DOI] [PubMed] [Google Scholar]
- Chen SH, Oyarzabal EA, Hong JS. Preparation of rodent primary cultures for neuron-glia, mixed glia, enriched microglia, and reconstituted cultures with microglia. Methods Mol Biol. 2013;1041:231–40. doi: 10.1007/978-1-62703-520-0_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Pagano E, Meijles DN, Pagano PJ. The Quest for Selective Nox Inhibitors and Therapeutics: Challenges, Triumphs and Pitfalls. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2013.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doussiere J, Gaillard J, Vignais PV. The heme component of the neutrophil NADPH oxidase complex is a target for aryliodonium compounds. Biochemistry. 1999;38:3694–703. doi: 10.1021/bi9823481. [DOI] [PubMed] [Google Scholar]
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–71. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Hong JS. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008;29:357–65. doi: 10.1016/j.it.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Hong JS, Zhang W, Liu B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2002;22:782–90. doi: 10.1523/JNEUROSCI.22-03-00782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Zhou H, Hong JS. NADPH oxidases: novel therapeutic targets for neurodegenerative diseases. Trends Pharmacol Sci. 2012;33:295–303. doi: 10.1016/j.tips.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Zhou H, Zhang F, Wilson BC, Kam W, Hong JS. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J Neurosci. 2011;31:1081–92. doi: 10.1523/JNEUROSCI.3732-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom CK, Gardner AM, Gardner PR. Nitric oxide metabolism in mammalian cells: substrate and inhibitor profiles of a NADPH-cytochrome P450 oxidoreductase-coupled microsomal nitric oxide dioxygenase. Free Radic Biol Med. 2004;37:216–28. doi: 10.1016/j.freeradbiomed.2004.04.031. [DOI] [PubMed] [Google Scholar]
- Hernandes MS, Britto LR. NADPH oxidase and neurodegeneration. Curr Neuropharmacol. 2012;10:321–7. doi: 10.2174/157015912804143540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–97. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S210–2. doi: 10.1016/S1353-8020(11)70065-7. [DOI] [PubMed] [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–9. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Leitsch D, Kolarich D, Duchene M. The flavin inhibitor diphenyleneiodonium renders Trichomonas vaginalis resistant to metronidazole, inhibits thioredoxin reductase and flavin reductase, and shuts off hydrogenosomal enzymatic pathways. Mol Biochem Parasitol. 2010;171:17–24. doi: 10.1016/j.molbiopara.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Levesque S, Wilson B, Gregoria V, Thorpe LB, Dallas S, Polikov VS, Hong JS, Block ML. Reactive microgliosis: extracellular micro-calpain and microglia-mediated dopaminergic neurotoxicity. Brain. 2010;133:808–21. doi: 10.1093/brain/awp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Cui G, Tzeng NS, Wei SJ, Wang T, Block ML, Hong JS. Femtomolar concentrations of dextromethorphan protect mesencephalic dopaminergic neurons from inflammatory damage. FASEB J. 2005;19:489–96. doi: 10.1096/fj.04-2555com. [DOI] [PubMed] [Google Scholar]
- Li XJ, Marchal CC, Stull ND, Stahelin RV, Dinauer MC. p47phox Phox homology domain regulates plasma membrane but not phagosome neutrophil NADPH oxidase activation. J Biol Chem. 2010;285:35169–79. doi: 10.1074/jbc.M110.164475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–9. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Liu B, Gao HM, Hong JS. Parkinson’s disease and exposure to infectious agents and pesticides and the occurrence of brain injuries: role of neuroinflammation. Environmental health perspectives. 2003;111:1065–73. doi: 10.1289/ehp.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minghetti L. Role of inflammation in neurodegenerative diseases. Curr Opin Neurol. 2005;18:315–21. doi: 10.1097/01.wco.0000169752.54191.97. [DOI] [PubMed] [Google Scholar]
- Nunes P, Demaurex N, Dinauer MC. Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic. 2013;14:1118–31. doi: 10.1111/tra.12115. [DOI] [PubMed] [Google Scholar]
- O’Donnell BV, Tew DG, Jones OT, England PJ. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. The Biochemical journal. 1993;290 (Pt 1):41–9. doi: 10.1042/bj2900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Gao X, Pei Z, Wu X, Block M, Wilson B, Hong JS, Flood PM. NADPH oxidase inhibitor DPI is neuroprotective at femtomolar concentrations through inhibition of microglia over-activation. Parkinsonism Relat Disord. 2007;13(Suppl 3):S316–20. doi: 10.1016/S1353-8020(08)70023-3. [DOI] [PubMed] [Google Scholar]
- Qin L, Block ML, Liu Y, Bienstock RJ, Pei Z, Zhang W, Wu X, Wilson B, Burka T, Hong JS. Microglial NADPH oxidase is a novel target for femtomolar neuroprotection against oxidative stress. FASEB J. 2005a;19:550–7. doi: 10.1096/fj.04-2857com. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Hong JS, Crews FT. NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia. 2013;61:855–68. doi: 10.1002/glia.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Liu Y, Qian X, Hong JS, Block ML. Microglial NADPH oxidase mediates leucine enkephalin dopaminergic neuroprotection. Ann N Y Acad Sci. 2005b;1053:107–20. doi: 10.1196/annals.1344.009. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–21. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- Sorce S, Krause KH, Jaquet V. Targeting NOX enzymes in the central nervous system: therapeutic opportunities. Cell Mol Life Sci. 2012;69:2387–407. doi: 10.1007/s00018-012-1014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surace MJ, Block ML. Targeting microglia-mediated neurotoxicity: the potential of NOX2 inhibitors. Cell Mol Life Sci. 2012;69:2409–27. doi: 10.1007/s00018-012-1015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Guzman JN, Sanchez J, Schumacker PT. Physiological phenotype and vulnerability in Parkinson’s disease. Cold Spring Harbor perspectives in medicine. 2012;2:a009290. doi: 10.1101/cshperspect.a009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhou H, Gao H, Chen SH, Chu CH, Wilson B, Hong JS. Naloxone inhibits immune cell function by suppressing superoxide production through a direct interaction with gp91phox subunit of NADPH oxidase. J Neuroinflammation. 2012;9:32. doi: 10.1186/1742-2094-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2003;100:6145–50. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Marchal CC, Casbon AJ, Stull N, von Lohneysen K, Knaus UG, Jesaitis AJ, McCormick S, Nauseef WM, Dinauer MC. Deletion mutagenesis of p22phox subunit of flavocytochrome b558: identification of regions critical for gp91phox maturation and NADPH oxidase activity. J Biol Chem. 2006;281:30336–46. doi: 10.1074/jbc.M607191200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.