

Abstract
The γ-aminobutyric acid type A (GABAA) receptor is one of the three main classes of receptors activated by GABA, the principal inhibitory neurotransmitter in the central nervous system. Mutations in genes encoding various subunits of this receptor (GABRA1, GABRA2, GABRA4, GABRA5, GABRA6, GABRB1, GABRB3, GABRG1, GABRG2, GABRG3, and GABRD) are implicated in a number of neurological and developmental disorders, including epilepsy and autism. To date, no human genetics studies have implicated mutations in GABRB2, encoding the β2 subunit of the GABAA receptor, with neurodevelopmental disorders. Here we present a 12-year-old girl with intellectual disability and epilepsy, who was discovered by whole exome sequencing to have a de novo heterozygous missense variant in exon 4 of GABRB2 (c.236T>C; p.M79T). This variant is likely pathogenic, based on in silico analyses, as well as the fact that it results in the non-conservative substitution of a non-polar amino acid with a polar amino acid at a position that is evolutionarily conserved across multiple species. Our findings underscore the need for further investigation into the mechanisms by which mutations in GABRB2 contribute to neurological and developmental dysfunction.
Keywords: Epilepsy, intellectual disability, GABRB2, GABAA receptor
INTRODUCTION
The γ-aminobutyric acid type A (GABAA) receptor is one of the three main classes of receptors activated by GABA, the principal inhibitory neurotransmitter in the central nervous system. This receptor is an ionotropic, ligand-gated chloride ion channel that is usually situated in the post synaptic membrane of neurons. When activated, the ion channel becomes permeable to chloride anions, and under normal cell conditions in most mature neurons, the increased chloride conductance hyperpolarizes the resting potential of the membrane, preventing the generation of action potentials. Thus, the main effect of GABAA receptor activation is synaptic inhibition (Jacob et al., 2008).
Not surprisingly, GABAA receptors play an important role in the brain, and genetic defects affecting their composition, assembly, and trafficking are implicated in a number of neurological and developmental disorders. Mutations in genes encoding some of the GABAA receptors subunits (GABRA1, GABRA6, GABRB3, GABRG2, and GABRD) underlie various forms of epilepsy (Dibbens et al., 2009; Macdonald et al., 2010). Copy number variations of 15q11-q13, a region containing the GABA A receptors subunit genes GABRA5, GABRB3, and GABRG3, are associated with some instances of autism (Hogart et al., 2010), while duplications spanning 4p13-p12, containing the GABAA receptors subunit genes GABRA2, GABRA4, GABRB1, and GABRG1, are associated with a spectrum of neurodevelopmental disorders, including autism, developmental delay, and learning disabilities (Polan et al., 2014).
To date, no human genetics studies have implicated mutations in GABRB2, encoding the β2 subunit of the GABAA receptor, with childhood epilepsy, intellectual disability, or other neurodevelopmental disorders. Here we present a 12-year-old girl with intellectual disability and epilepsy, who was discovered to have a potentially pathogenic novel missense variant in GABRB2. We present details of her clinical history and molecular testing, and we review some of the clinical consequences of GABAA receptor dysfunction.
CLINICAL REPORT
The proband is a 12-year-old girl from Spain who was referred to the Neurogenetics Clinic at the Kennedy Krieger Institute for an etiological evaluation of her intellectual disability and epilepsy.
She had an unremarkable birth history. The family history was negative for other individuals with epilepsy or neurodevelopmental disorders. Both parents as well as two older siblings were healthy.
She was doing well as a young infant until the age of 9 months, when she developed febrile seizures associated with the onset of chickenpox. She continued to have clusters of febrile seizures but eventually developed primarily non-febrile seizures. These were initially generalized tonic clonic convulsions that over the years evolved into more subtle spells associated with headache, dizziness, paleness, loss of consciousness, and certain emotions. She currently takes lamotrigine and clobazam for seizure control. Of note, the addition of clobazam had made significant improvement in the frequency of seizures.
Gradually, her development slowed over the years; she currently carries the diagnosis of intellectual disability. Although she attends regular school, she receives special tutoring half the day; she attends the other half mainly for socialization.
On her initial evaluation, when she was 12 years old, her growth parameters were unremarkable. She was non-dysmorphic, and the rest of her general exam was normal. On neurological exam, she appeared somewhat anxious, but she warmed up as the encounter proceeded. She spoke short 2–3 word sentences in Spanish without any dysarthria but with significant perseveration. She followed two step commands. Overall, cognitively she appeared at the level of a 3–4 year-old. Her cranial nerves appeared normal. She had normal muscle tone and strength but was clumsy with fine motor movements. Sensation was grossly intact. Finger nose finger testing was normal; there was no ataxia. She was able to walk on her tiptoes but had difficulty walking on her heels or along a line, and seemed to be unable to replicate the examiner’s movements. Stress gait assessment was deferred but there was bilateral flexure posturing of elbows when asked to run. Tendon reflexes were brisk at 3+ throughout, there was no clonus, and Babinski sign was negative.
She had a complete genetic workup that was unrevealing. Prior testing included karyotype, single-nucleotide polymorphism chromosome microarray, Fragile X trinucleotide repeat analysis, plasma amino acids, urine organic acids, very long chain fatty acids, carbohydrate deficient transferrin analysis for congenital disorders of glycosylation, methylation analysis for Angelman syndrome, and a 38-gene “Infantile Epilepsy” NextGen sequencing panel performed at GeneDx Laboratory. Her brain MRI was normal. She had several EEGs in the past, showing bursts of spike and polyspikes against an otherwise normal background (see Figure 1). Given the unrevealing nature of her prior workup, she underwent whole exome sequencing.
Figure 1.

Sample EEG recording from the proband.
METHODS
Whole exome sequencing was performed by GeneDx Laboratory on genomic DNA obtained from the proband, mother, and father. Exonic regions were targeted with the Agilent SureSelect XT2 All Exon V4 kit. Targeted regions were sequenced with the Illumina HiSeq 2000 sequencing system. Sequenced DNA was compared with the reference human genome assembly, GRCh37 hg19. A sequence variant was discovered in GABRB2. It was confirmed by dideoxy sequencing.
Several approaches were used to analyze the pathogenicity of the GABRB2 variant. The novelty of the variant was based on its absence from the Human Gene Mutation Database (http://www.hgmd.org/), National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server (http://evs.gs.washington.edu/EVS/), and Pubmed (http://www.ncbi.nlm.nih.gov/pubmed/). The primary protein structure of the variant was analyzed to see if the variant resulted in a non-conservative substitution, and if the variant occurred at a residue that was evolutionarily conserved, using sequence homologs from HomoloGene (http://www.ncbi.nlm.nih.gov/homologene/) and multiple sequence alignment with MAFFT (http://mafft.cbrc.jp/alignment/software/). Disruption of protein function by the variant was determined with pathogenicity prediction software, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml), which uses a Naïve Bayes classifier trained using supervised machine-learning and classifies variants as benign, possibly damaging, or probably damaging. Finally, the structural and functional importance of the variant was predicted by analyzing a related protein structure based on protein structural alignment using Protein Basic Local Alignment Search Tool (BLAST) (http://blast.ncbi.nlm.nih.gov) against the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do)
RESULTS
Whole exome sequencing revealed a de novo heterozygous mutation in exon 4 of GABRB2 (c.236T>C; p.M79T). This variant is not found in the Human Gene Mutation Database or NHLBI Exome Variant Server, nor has it been reported in the literature. The variant resulted in the non-conservative substitution of methionine, a non-polar amino acid, with threonine, a polar amino acid. This position is evolutionarily conserved across multiple species (see Figure 2a). The variant was predicted to be possibly damaging based on analysis with PolyPhen-2.
Figure 2.

Multiple sequence alignment of human GABRB2 with related peptides. a) Multiple sequence alignment of vertebrate GABRB2 homologs, with focus on the region surrounding position 79 of human GABRB2. b) Multiple sequence alignment of human GABRB2 with the human β1 subunit (GABRB1) and β3 subunit (GABRB3) of the GABAA receptor, with focus on the region surrounding position 79 of human GABRB2. The arrow indicates all the conserved residues at position 79 of human GABRB2. An asterisk (‘*’) denotes a position where residues are conserved. A period (‘.’) denotes a position where there is conservation between groups of amino acids with weakly similar properties. A colon (‘:’) denotes a position where there is conservation between groups of amino acids with strongly similar properties. The National Center for Biotechnology Information (NCBI) Reference Sequence accession number for each protein is indicated in parentheses.
Sequence alignment with Protein BLAST revealed statistically significant relatedness of human GABRB2 to each subunit of GluClα, a homopentameric glutamate-gated chloride channel found in Caenorhabditis elegans (Hibbs and Gouaux, 2011) (59% of query covered by alignment to the database sequence). See Figure 3a. M79 in human GABRB2 is conserved with a residue (M47) in the β2 structure of each subunit of GluClα. In GluClα, the β1-β2 loop is an extracellular domain loop that facilitates communication between the neurotransmitter site and the transmembrane pore. M47 in each subunit of GluClα is 6 residues away from the start of loop D, a stretch of seven amino acids within the β2 structure that forms part of the neurotransmitter site (Hibbs and Gouaux, 2011). See Figure 3b.
Figure 3.

Structural comparison of human GABRB2 with the homopentameric C. elegans glutamategated chloride channel GluClα. a) Alignment of human GABRB2 to each subunit of GluClα (E = 1e-71, 42% identities, 62% positives). The arrow indicates position 79 of human GABRB2. b) Two 3D representations of GluClα. The first (left) is looking parallel to the membrane. The second (right) is looking down the pore from the intracellular side toward the extracellular side. The intracellular (“In”) and extracellular (“Out”) sides are indicated. The red circle and arrow indicates the residue (M47) within the β2 loop of each subunit of GluClα, corresponding to M79 in human GABRB2 (note that this residue is not always visible within each subunit depending on the angle of the 3D view).
DISCUSSION
The novel missense variant c.236T>C (p.M79T) in GABRB2 is likely pathogenic. The variant affects a position in the gene that is highly conserved across multiple species. Moreover, it results in the non-conservative substitution of a hydrophobic amino acid with a polar amino acid. Finally, in silico analysis with PolyPhen-2 predicts that the protein change resulting from this variant is possibly damaging.
The variant affects the β2 subunit of the structurally complex GABAA receptor. GABAA receptors are hetero-pentamers arranged from combinations of several different subunits, each with four transmembrane (TM) domains. There are 19 different subunits across eight subunit classes: α1-α6, β1-β3, γ1-γ3, δ, ε, θ, π, and ρ1-ρ3 (Olsen and Sieghart, 2008). The genes encoding these subunits are distributed across the genome, with the majority clustered on four chromosomes: chromosome 4p12 (GABRA2, GABRA4, GABRB1, GABRG1), 5q34 (GABRA1, GABRA6, GABRB2, GABRG2), 15q12 (GABRA5, GABRB3, GABRG3), and Xq28 (GABRA3, GABRE, GABRQ) (Russek, 1999). See Table I. The extensive variety of subunits may suggest an equally diverse repertoire of possible receptor subtypes; however, this heterogeneity is limited by the requirements of a fully functional GABAA receptor to have, at minimum, an α subunit, β subunit, and one other subunit type (Olsen and Sieghart, 2008).
Table I.
Subunits of the GABAA receptor and their respective gene symbols and locations. Gene locations were determined by querying National Center for Biotechnology Information (NCBI) Gene (http://www.ncbi.nlm.nih.gov/gene).
| Subunit | Gene | Location |
|---|---|---|
| α1 | GABRA1 | 5q34 |
| α2 | GABRA2 | 4p12 |
| α3 | GABRA3 | Xq28 |
| α4 | GABRA4 | 4p12 |
| α5 | GABRA5 | 15q12 |
| α6 | GABRA6 | 5q34 |
| β1 | GABRB1 | 4p12 |
| β2 | GABRB2 | 5q34 |
| β3 | GABRB3 | 15q12 |
| γ1 | GABRG1 | 4p12 |
| γ2 | GABRG2 | 5q34 |
| γ3 | GABRG3 | 15q12 |
| δ | GABRD | 1p36.3 |
| ε | GABRE | Xq28 |
| θ | GABRQ | Xq28 |
| π | GABRP | 5q35.1 |
| ρ1 | GABRR1 | 6q15 |
| ρ2 | GABRR2 | 6q15 |
| ρ3 | GABRR3 | 3q11.2 |
In mammals, the most common configuration of the GABAA receptor is a complex comprising two α subunits, two β subunits, and one γ subunit (though the δ, ε, θ, or π subunit can take the place of the γ subunit) (see Figure 4). When the receptor is assembled, the five subunits come together so that the second TM domain lines the central pore of the ion channel. The N-terminal region is the binding site for GABA, in the α/β interface. The intracellular loop between the third and fourth TM domains also plays an important role, interacting with regulatory kinases and other proteins that keep the receptor anchored in the correct position (Jacob et al., 2008).
Figure 4.
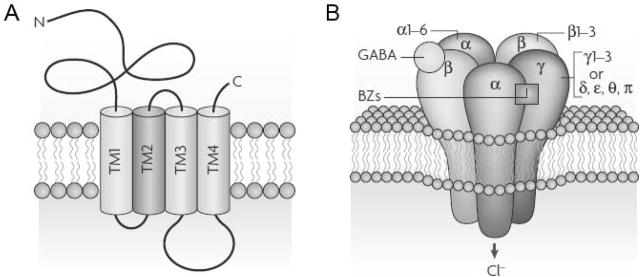
Schematic of the GABAA receptor. a) Transmembrane topology of each receptor subunit. Each subunit has a large extracellular N-terminal binding domain, four TM segments, and an extracellular Cterminal end. b) Assembly of the receptor from different classes of subunits. The receptor is composed of two α subunits, two β subunits, and one additional γ subunit (which can be replaced by a δ, ε, θ, or π subunit). The binding site for GABA is located at the interface between the α and β subunits. A binding site for benzodiazepines (BZs) is located at the interface between the α and γ subunits. Ligand binding activates the receptor, which then becomes permeable to chloride (Cl-) anions. Figure is reprinted with permission from Macmillan Publishers Ltd: Nature Reviews Neuroscience (Jacob et al., 2008), copyright 2008.
The GABRB2 variant occurring in our patient has key structural and functional implications. The M79 position of GABRB2 is conserved with the other two β subunits of the GABAA receptor (GABRB1 and GABRB3) (see Figure 2b), and GABRB2 has overall strong sequence identity to both GABRB1 (77.8% identity) and GABRB3 (79.9% identity). Therefore, alterations in equivalent regions to the M79 position in the other GABAA β subunits presumably have similar functional impacts. The position of the variant M79T lies in the large N-terminal extracellular domain, which has been implicated in GABA binding (Jacob et al., 2008). Moreover, based on structural comparisons between human GABRB2 and the C. elegans glutamate-gated chloride channel GluClα, this residue may be in close proximity to structural regions that play a role in ligand binding as well as allosteric changes between the neurotransmitter site and transmembrane pore. Given the essential role of this motif, this variant may reduce the function of the GABAA receptor, impairing fast synaptic inhibition and increasing susceptibility to seizure activity.
Mutations in GABAA receptor subunits can affect one of several steps involved in the formation of a fully functional receptor, although the precise molecular defect caused by the M79T variant is open to speculation. The steps that are necessary for this process include subunit synthesis, receptor oligomerization/assembly, and receptor trafficking. Impaired oligomerization is the postulated mechanism underlying many different GABAA receptor missense mutations associated with genetic generalized epilepsies (Macdonald and Kang, 2012), so this defect could also be present in the M79T variant. Another possible molecular explanation for the M79T variant is that subunit oligomerization proceeds normally, in spite of the defective subunit, and the receptor traffics to the membrane, but gating is somehow altered because of β2 subunit dysfunction. More data is needed to support one hypothesis over another. Moreover, it is also unclear why epileptogenesis occurs gradually, even though β2 expression is ubiquitous throughout development (Fritschy et al., 1994).
From a clinical standpoint, variants in the genes encoding other GABAA receptor subunits besides the β2 subunit, particularly the α1, α6, β3, γ2, and δ subunits, are the cause of a number of epilepsy syndromes (see Table II). These syndromes include childhood absence epilepsy (CAE), Dravet syndrome (DS), febrile seizures (FS), generalized epilepsy with febrile seizures plus (GEFS+), infantile spams (IS), juvenile myoclonic epilepsy (JME), and Lennox- Gastaut syndrome (LGS). In some studies, the patients were labeled as having idiopathic generalized epilepsy (IGE) which can subsume some of the aforementioned syndromes. Among the different kinds of GABAA receptor coding mutations associated with epilepsy, heterozygous missense mutations (in GABRA1, GABRA6, GABRB3, GABRG2, GABRD) are prevalent, though nonsense mutations (in GABRG2) and frameshift mutations (in GABRA1) can also occur. In some instances of epilepsy, mutations in untranslated sequences can affect the splice sites of GABRA1 and GABRG2 and the 5’ upstream promoter regions of GABRB3. IS and LGS, two childhood epileptic encephalopathy syndromes featuring severe epilepsy and neurobehavioral problems, have been associated with defects in GABRA1 (IS) and GABRB3 (IS and LGS).
Table II.
Variants in genes encoding GABAA receptor subunits underlying different epilepsy syndromes.
| Reference | Affected gene | Affected Subunit | Variant* | Mutation Type | Epilepsy syndrome |
|---|---|---|---|---|---|
| (Cossette et al., 2002) | GABRA1 | α1 | A322D | Missense | JME |
| (Maljevic et al., 2006) | GABRA1 | α1 | S326fs328X | Frameshift | CAE |
| (Lachance-Touchette et al., 2011) | GABRA1 | α1 | D219N | Missense | FS/IGE |
| (Lachance-Touchette et al., 2011) | GABRA1 | α1 | K353delins18 X | Splice site | IGE |
| (Epi4K Consortium et al., 2013) | GABRA1 | α1 | T292I | Missense | IS |
| (Dibbens et al., 2009) | GABRA6 | α6 | R46W | Missense | CAE |
| (Tanaka et al., 2008) | GABRB3 | β3 | P11S | Missense | CAE, autism |
| (Tanaka et al., 2008) | GABRB3 | β3 | S15F | Missense | CAE |
| (Tanaka et al., 2008) | GABRB3 | β3 | G32R | Missense | CAE |
| (Epi4K Consortium et al., 2013) | GABRB3 | β3 | N110D | Missense | IS |
| (Epi4K Consortium et al., 2013) | GABRB3 | β3 | Y302C | Missense | LGS |
| (Epi4K Consortium et al., 2013) | GABRB3 | β3 | D120N | Missense | LGS |
| (Epi4K Consortium et al., 2013) | GABRB3 | β3 | E180G | Missense | LGS |
| (Urak et al., 2006) | GABRB3 | β3 | exon 1a promoter haplotype 2 | Promoter | CAE |
| (Hirose, 2006) | GABRG2 | γ2 | Q40X | Nonsense | DS |
| (Wallace et al., 2001) | GABRG2 | γ2 | R82Q | Missense | CAE/FS |
| (Audenaert et al., 2006) | GABRG2 | γ2 | R177G | Missense | FS |
| (Baulac et al., 2001) | GABRG2 | γ2 | K328M | Missense | GEFS+ |
| (Harkin et al., 2002) | GABRG2 | γ2 | Q390X | Nonsense | GEFS+/DS |
| (Sun et al., 2008) | GABRG2 | γ2 | Q429X | Nonsense | GEFS+ |
| (Lachance-Touchette et al., 2011) | GABRG2 | γ2 | P83S | Missense | FS/IGE |
| (Kananura et al., 2002) | GABRG2 | γ2 | IVS6+2T→G | Splice site | CAE/FS |
| (Dibbens et al., 2004) | GABRD | δ | E1771A | Missense | GEFS+ |
| (Dibbens et al., 2004) | GABRD | δ | R220H | Missense | GEFS+ |
The variants in each subunit are designated with respect to the precursor peptide sequence, which includes a signal sequence that is normally cleaved in the mature protein (Macdonald et al., 2010).
CAE = childhood absence epilepsy; DS = Dravet syndrome; FS = febrile seizures; GEFS+ = generalized epilepsy with febrile seizures plus; IGE = idiopathic generalized epilepsy; IS = infantile spams; JME = juvenile myoclonic epilepsy; LGS = Lennox-Gastaut syndrome. Portions of table adapted from (Macdonald et al., 2010).
Our patient presented with a pattern of seizures that may fall under the spectrum of GEFS+. The most common epilepsy phenotypes seen in GEFS+ are FS or FS+ (febrile seizures which persist past the age of 6 years and/or are accompanied by afebrile generalized tonic-clonic seizures) (Singh et al., 1999). Her clinical course falls into the latter: she developed febrile seizures in infancy, which resolved, but she went on to develop generalized epilepsy. There is significant phenotypic variability in GEFS+, and up to 33% of affected individuals may present with other seizure types, including absence, partial, atonic, and myoclonic seizures (Scheffer and Berkovic, 1997; Singh et al., 1999).
GEFS+ is an autosomal dominant channelopathy that can affect several members of a family or arise spontaneously from de novo mutations. Thus far, a handful of mutations have been identified as the cause of GEFS+, and these impact both sodium channels (in the case of SCN1A, SCN1B, and SCN2A mutations) and the GABAA receptor (in the case of GABRG2 mutations) (Scheffer et al., 2009). The mechanisms of pathogenicity involve either enhanced excitatory neurotransmission (in the case of sodium channel mutations) or decreased synaptic inhibition (in the case of GABAA receptor mutations), both of which contribute to neuronal hyperexcitability. The variant occurring in the proband points to the possibility of dysfunction in the β2 subunit of the GABAA receptor (previously unreported in relation with generalized epilepsy and/or febrile seizures) as another potential cause of GEFS+.
Cognitive impairment is evident in our patient, but the mechanism by which this has occurred is unclear. Animal models suggests that the α5 subunit of the GABAA receptor has an important part in learning and memory (Collinson et al., 2002; Martin et al., 2010). Surprisingly, adult mice lacking the β2 subunit of the GABAA receptor have no significant phenotypic abnormalities or spontaneous seizures (Sur et al., 2001). There are limited data in human studies about the specific effects of a defective β2 subunit on cognitive processes like memory and learning, and more research is warranted.
One potential caveat to our report is the emerging idea that variants in other ion channel genes can influence neurological phenotypes, including epilepsy. In an exome sequencing study of 237 channel genes in both unaffected controls and patients with sporadic idiopathic epilepsy, missense mutations were prevalent in both patients and the controls. In fact, 66.9% of the controls had a missense mutation in at least one ion channel gene known to cause familial human epilepsy (among the GABAA receptor genes, these include GABRA1, GABRB3, and GABRG2), versus 96.1% of the patients. In addition, 5% of controls had at least two non-synonymous single nucleotide polymorphisms in GABRB1, GABRB2, or GABRB3 versus 14.5% of the affected patients (Klassen et al., 2011). Thus, an epileptic phenotype attributed to an ion channel gene mutation may actually be the result of the complex interaction of multiple ion channel gene variants (and gene variants in other biological systems) with each other. A similar argument can be made explaining the normal phenotype of unaffected individuals who harbor deleterious ion channel mutations; the end result of this complex gene network interaction is rescuing of the deleterious effects of individual mutations. In our case, besides the single GABRB2 variant, there were no other deleterious mutations detected (including in other ion channel genes), and in light of all the aforementioned evidence, specifically that the variant had occurred de novo in a highly conserved position, we believe that the patient’s neurological phenotype is due to the effects of the M79T variant.
In summary, our case represents the first report of a missense mutation in the β2 subunit of the GABAA receptor as a cause of genetic epilepsy and intellectual disability. Our findings underscore the need for further investigation into the mechanisms by which mutations in this GABAA receptor subunit contribute to neurological and developmental dysfunction.
Acknowledgments
We would like to thank the patient and her family.
Footnotes
CONFLICTS OF INTEREST
Swaroop Aradhya, Dianalee McKnight, and Elizabeth Butler are paid employees of GeneDx Laboratory.
References
- Audenaert D, Schwartz E, Claeys KG, Claes L, Deprez L, Suls A, Van Dyck T, Lagae L, Van Broeckhoven C, Macdonald RL, De Jonghe P. A novel GABRG2 mutation associated with febrile seizures. Neurology. 2006;67:687–690. doi: 10.1212/01.wnl.0000230145.73496.a2. [DOI] [PubMed] [Google Scholar]
- Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud’homme JF, Baulac M, Brice A, Bruzzone R, LeGuern E. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001;28:46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- Collinson N, Kuenzi FM, Jarolimek W, Maubach KA, Cothliff R, Sur C, Smith A, Otu FM, Howell O, Atack JR, McKernan RM, Seabrook GR, Dawson GR, Whiting PJ, Rosahl TW. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J Neurosci. 2002;22:5572–5580. doi: 10.1523/JNEUROSCI.22-13-05572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossette P, Liu L, Brisebois K, Dong H, Lortie A, Vanasse M, Saint-Hilaire J-M, Carmant L, Verner A, Lu W-Y, Wang YT, Rouleau GA. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184–189. doi: 10.1038/ng885. [DOI] [PubMed] [Google Scholar]
- Dibbens LM, Feng H-J, Richards MC, Harkin LA, Hodgson BL, Scott D, Jenkins M, Petrou S, Sutherland GR, Scheffer IE, Berkovic SF, Macdonald RL, Mulley JC. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. 2004;13:1315–1319. doi: 10.1093/hmg/ddh146. [DOI] [PubMed] [Google Scholar]
- Dibbens LM, Harkin LA, Richards M, Hodgson BL, Clarke AL, Petrou S, Scheffer IE, Berkovic SF, Mulley JC. The role of neuronal GABA(A) receptor subunit mutations in idiopathic generalized epilepsies. Neurosci Lett. 2009;453:162–165. doi: 10.1016/j.neulet.2009.02.038. [DOI] [PubMed] [Google Scholar]
- Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu Y-F, Madou MRZ, Marson AG, Mefford HC, Esmaeeli Nieh S, O’Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin Thio L, Venkat A, Vining EPG, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, Winawer MR Epi4K Consortium, Epilepsy Phenome/Genome Project. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy JM, Paysan J, Enna A, Mohler H. Switch in the expression of rat GABAA-receptor subtypes during postnatal development: an immunohistochemical study. J Neurosci. 1994;14:5302–5324. doi: 10.1523/JNEUROSCI.14-09-05302.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkin LA, Bowser DN, Dibbens LM, Singh R, Phillips F, Wallace RH, Richards MC, Williams DA, Mulley JC, Berkovic SF, Scheffer IE, Petrou S. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2002;70:530–536. doi: 10.1086/338710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez CC, Gurba KN, Hu N, Macdonald RL. The GABRA6 mutation, R46W, associated with childhood absence epilepsy, alters 6β22 and 6β2 GABA(A) receptor channel gating and expression. J Physiol. 2011;589:5857–5878. doi: 10.1113/jphysiol.2011.218883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cysloop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S. A new paradigm of channelopathy in epilepsy syndromes: intracellular trafficking abnormality of channel molecules. Epilepsy Res. 2006;70(Suppl 1):S206–217. doi: 10.1016/j.eplepsyres.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Hogart A, Wu D, LaSalle JM, Schanen NC. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis. 2010;38:181–191. doi: 10.1016/j.nbd.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci. 2008;9:331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kananura C, Haug K, Sander T, Runge U, Gu W, Hallmann K, Rebstock J, Heils A, Steinlein OK. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol. 2002;59:1137–1141. doi: 10.1001/archneur.59.7.1137. [DOI] [PubMed] [Google Scholar]
- Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, McPherson J, Bourquin T, Lewis L, Villasana D, Morgan M, Muzny D, Gibbs R, Noebels J. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance-Touchette P, Brown P, Meloche C, Kinirons P, Lapointe L, Lacasse H, Lortie A, Carmant L, Bedford F, Bowie D, Cossette P. Novel α1 and γ2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. Eur J Neurosci. 2011;34:237–249. doi: 10.1111/j.1460-9568.2011.07767.x. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Kang J-Q. mRNA surveillance and endoplasmic reticulum quality control processes alter biogenesis of mutant GABAA receptor subunits associated with genetic epilepsies. Epilepsia. 2012;53(Suppl 9):59–70. doi: 10.1111/epi.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Kang J-Q, Gallagher MJ. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol. 2010;588:1861–1869. doi: 10.1113/jphysiol.2010.186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maljevic S, Krampfl K, Cobilanschi J, Tilgen N, Beyer S, Weber YG, Schlesinger F, Ursu D, Melzer W, Cossette P, Bufler J, Lerche H, Heils A. A mutation in the GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy. Ann Neurol. 2006;59:983–987. doi: 10.1002/ana.20874. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Zurek AA, MacDonald JF, Roder JC, Jackson MF, Orser BA. Alpha5GABAA receptor activity sets the threshold for long-term potentiation and constrains hippocampus-dependent memory. J Neurosci. 2010;30:5269–5282. doi: 10.1523/JNEUROSCI.4209-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W. International Union of Pharmacology. LXX Subtypes of gammaaminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update Pharmacol Rev. 2008;60:243–260. doi: 10.1124/pr.108.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polan MB, Pastore MT, Steingass K, Hashimoto S, Thrush DL, Pyatt R, Reshmi S, Gastier-Foster JM, Astbury C, McBride KL. Neurodevelopmental disorders among individuals with duplication of 4p13 to 4p12 containing a GABAA receptor subunit gene cluster. Eur J Hum Genet. 2014;22:105–109. doi: 10.1038/ejhg.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russek SJ. Evolution of GABA(A) receptor diversity in the human genome. Gene. 1999;227:213– 222. doi: 10.1016/s0378-1119(98)00594-0. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain J Neurol. 1997;120 ( Pt 3):479–490. doi: 10.1093/brain/120.3.479. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Zhang Y-H, Jansen FE, Dibbens L. Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus? Brain Dev. 2009;31:394–400. doi: 10.1016/j.braindev.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Singh R, Scheffer IE, Crossland K, Berkovic SF. Generalized epilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Ann Neurol. 1999;45:75–81. doi: 10.1002/1531-8249(199901)45:1<75::aid-art13>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Sun H, Zhang Y, Liang J, Liu X, Ma X, Wu H, Xu K, Qin J, Qi Y, Wu X. SCN1A, SCN1B, and GABRG2 gene mutation analysis in Chinese families with generalized epilepsy with febrile seizures plus. J Hum Genet. 2008;53:769–774. doi: 10.1007/s10038-008-0306-y. [DOI] [PubMed] [Google Scholar]
- Sur C, Wafford KA, Reynolds DS, Hadingham KL, Bromidge F, Macaulay A, Collinson N, O’Meara G, Howell O, Newman R, Myers J, Atack JR, Dawson GR, McKernan RM, Whiting PJ, Rosahl TW. Loss of the major GABA(A) receptor subtype in the brain is not lethal in mice. J Neurosci. 2001;21:3409–3418. doi: 10.1523/JNEUROSCI.21-10-03409.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Olsen RW, Medina MT, Schwartz E, Alonso ME, Duron RM, Castro-Ortega R, Martinez-Juarez IE, Pascual-Castroviejo I, Machado-Salas J, Silva R, Bailey JN, Bai D, Ochoa A, Jara-Prado A, Pineda G, Macdonald RL, Delgado-Escueta AV. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am J Hum Genet. 2008;82:1249–1261. doi: 10.1016/j.ajhg.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urak L, Feucht M, Fathi N, Hornik K, Fuchs K. A GABRB3 promoter haplotype associated with childhood absence epilepsy impairs transcriptional activity. Hum Mol Genet. 2006;15:2533–2541. doi: 10.1093/hmg/ddl174. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]