

Abstract
Global cerebral ischemia following cardiac arrest and cardiopulmonary resuscitation (CA/CPR) causes injury to hippocampal CA1 pyramidal neurons and impairs cognition. SK2 channels, expressed in CA1 pyramidal neurons, have been implicated as potential protective targets. Here we show that in mice, hippocampal long-term potentiation (LTP) is impaired as early as 3 hrs after recovery from CA/CPR and that LTP remains impaired for at least 30 days. Treatment with the SK2 channel agonist, 1-EBIO 30 minutes after CA provided sustained protection from plasticity deficits, with LTP being maintained at control levels at 30 days after recovery from CA/CPR. Minimal changes in glutamate release probability were observed at delayed times after CA/CPR, implicating post-synaptic mechanisms. Real-time quantitative RT-PCR indicates that CA/CPR does not cause a loss of NMDA receptor mRNA 7 or 30 days after CA/CPR. Similarly, no change in synaptic NMDA receptor protein levels were observed 7 or 30 days after CA/CPR. Further, patch-clamp experiments demonstrate no change in functional synaptic NMDA receptors 7 or 30 days after CA/CPR. Electrophysiology recordings showed that synaptic SK channel activity is reduced for the duration of experiments performed (up to 30 days) and that surprisingly, treatment with 1-EBIO did not prevent CA/CPR-induced loss of synaptic SK channel function. We conclude that CA/CPR causes alterations in post-synaptic signaling that are prevented by treatment with the SK2 agonist 1-EBIO, indicating that activators of SK2 channels may be useful therapeutic agents to prevent ischemic injury and cognitive impairments.
Keywords: mouse, hippocampus LTP, cardiac arrest, global cerebral ischemia, SK channels
Introduction
Cardiac arrest occurs in approximately 600,000 people each year in the US, and is a major cause of mortality and morbidity (Roger et al., 2012). Cardiac arrest is accompanied by global ischemia that results in neuronal death and injury to surviving neurons, with consequent long-term cognitive deficits including motor coordination, executive function, and learning and memory impairments (Nunes et al., 2003; Khot & Tirschwell, 2006; Peskine et al., 2010; Neumann et al., 2013). CA1 pyramidal cells in the hippocampus play an important role in learning and memory, and are particularly sensitive to ischemia, exhibiting delayed neuronal cell death in the CA1 area of the hippocampus at autopsy in people and following experimental cerebral ischemia (Pulsinelli et al., 1982; Pulsinelli, 1985; Petito et al., 1987; Schmidt-Kastner & Freund, 1991; Horn & Schlote, 1992). This injury and cell death is thought to arise from excessive excitatory glutamatergic neurotransmission that causes excitotoxic Ca2+ influx through postsynaptic ionotropic glutamate receptors (Arundine & Tymianski, 2004; Waxman & Lynch, 2005; Szydlowska & Tymianski, 2010). Indeed, inhibitors of glutamate receptors have demonstrated robust neuroprotection following experimental brain ischemia. However, these agents have failed to translate to the clinic, making it important to identify alternative approaches to improve functional recovery following cerebral ischemia (Ikonomidou & Turski, 2002; Muir, 2006; Ginsberg, 2008).
Dendritic spines are the major postsynaptic sites of glutamatergic neurotransmission. The excitatory postsynaptic potential (EPSP) reflects, in part, the depolarizing activity of postsynaptic ionotropic glutamate receptors, AMPA and NMDA receptors. Calcium entry through NMDA receptors is a critical initiator of activity-dependent enhancement in synaptic strength, termed long term potentiation (LTP) and is considered a cellular substrate for learning and memory (Malenka & Nicoll, 1993). Small conductance Ca2+-activated K channels (SK2) are expressed in the postsynaptic membrane of CA1 hippocampal neurons, near the NMDA receptor, and are activated by Ca2+ entering through the NMDA receptors (Ngo-Anh et al., 2005). Their repolarizing current reduces depolarization and attenuates Ca2+ influx through NMDA receptors by promoting Mg2+ block, and in this way, synaptic SK2 channel activity modulates the induction of NMDA-receptor dependent synaptic plasticity (Stackman et al., 2002; Hammond et al., 2006; Lin et al., 2008). These findings suggest that increasing SK2 channel activity would provide neuroprotection following ischemia by minimizing NMDA receptor activity. Indeed, we recently demonstrated that transgenic overexpression of SK2 channels, or pharmacologically increasing their activity by administering the SK channel agonist, 1-EBIO, protects CA1 pyramidal neurons against CA/CPR-induced cell death and enhances behavioral recovery (Allen et al., 2011). Moreover, synaptically evoked SK channel activity was absent for the first 24 hours following CA/CPR similar to that observed following LTP (Lin et al., 2008). Interestingly, the increased glutamatergic neurotransmission seen following CA/CPR-induced ischemia may also result in a form of LTP, ischemic LTP (iLTP). While the extent to which this iLTP mimics synaptically evoked LTP is unclear, both forms of potentiation are NMDAr-dependent (Crepel et al., 1993; Di et al., 2008). Therefore, we examined the effects of CA/CPR-induced ischemia on the subsequent induction of synaptically evoked LTP and the role of SK channels in modulating these responses.
Materials and Methods
Experimental Animals
All experimental protocols were approved by the Institutional Animal Care and Use Committee and conformed to the National Institutes of Health guidelines for the care and use of animals in research. Adult (20–25g) male C57Bl/6 mice (Charles River Laboratory) were used for this study. All mice were housed in standard 12 hr light dark cycle with free access to food and water.
Cardiac Arrest and Cardiopulmonary Resuscitation Model (CA/CPR)
The experimental procedure was performed as described by our group previously (Kofler et al., 2004; Allen et al., 2011; Hutchens et al., 2011). Briefly, anesthesia was induced with 3% isoflurane and maintained with 1–1.5% isoflurane in oxygen-enriched air using a facemask. Temperature probes were placed into the left ear canal and the rectum to monitor brain and body temperature simultaneously. Rectal temperature was controlled at near 37°C during surgery. Mice were endotracheally intubated using a 22G intravenous catheter and connected to a mouse ventilator (Minivent, Hugh Sachs Elektronik, Germany). A PE-10 catheter was inserted into the right jugular vein for drug administration. Electrocardiogram was monitored throughout the experimental procedure. Cardiac arrest (CA) was induced by the injection of 50 μl of 0.5 M KCl via the jugular catheter. The endotracheal tube was disconnected from the ventilator and anesthesia was stopped. During cardiac arrest, the body temperature was controlled above 35°C. Brain temperature was maintained at 37.5°C by a heated water-filled coil. Cardiopulmonary resuscitation (CPR) was begun 8 min after induction of CA by injection of 0.5–1.0 mL of epinephrine solution (16μg/mL in 0.9% saline), chest compressions at a rate of 300/min, and ventilation with 100% oxygen at a respiratory rate of 200/min. At the initiation of CPR, the body was rewarmed using a heat lamp and pad until 36.5°C. Cardiac compressions were stopped when spontaneous circulation was restored (ROSC), defined as electrocardiographic activity and visible cardiac contractions. EBIO (16 mg/kg) was administered ip 30min and 6 hrs after CPR. Catheter and temperature probes were removed and skin wounds closed, endotracheal tube removed and the mouse was returned to their housing cage on a circulating warm water blanket.
Hippocampal Slice preparation
Hippocampal slices were prepared from young adult male C57Bl/6 mice at varying times after recovery from CA/CPR. Animals were anesthetized with 3% isoflurane in an O2 enriched chamber. Mice were decapitated and the brains quickly extracted and placed in ice-cold (2–5 °C) oxygenated (95% O2/5% CO2) artificial cerebral spinal fluid (aCSF) composed of the following (in mmol/L): 125 NaCl, 2.5. KCl, 25 NaHCO3, 1.25 NaH2PO4, 2.0 CaCl2, 1.0 MgCl2, 12 glucose). Transverse hippocampal slices (300 – 350 μm thick) were cut with a Vibratome 1000 (Leica) and transferred to a holding chamber containing ACSF for 1.5–2 hrs before recording.
Electrophysiology
For synaptically evoked field potentials, hippocampal slices were placed on a temperature controlled (32±1 °C) interface chamber perfused with oxygenated aCSF at a rate of 1.5 ml/min. Extracellular field recordings were performed by stimulating the Schaffer collaterals and recording the field excitatory post synaptic potential (fEPSP). fEPSPs were adjusted to 50% of the maximum slope and test pulses were evoked at a rate of 0.05 Hz. Paired pulse responses (PPR) were recorded from the CA1 pyramidal cells using a 50 ms inter-pulse interval (20 Hz) and expressed as a ratio of the second pulse over the first pulse, P2/P1. A 20 min stable baseline period was established before delivering a theta burst stimulation (TBS) train (4 pulses delivered at 100 Hz in 30 ms bursts, repeated ten (or 75) times with 200 ms inter-burst-intervals). Following TBS, the fEPSP was recorded for 60min. Analog fEPSPs were amplified (1000×) and filtered through a pre-amplifier (Grass Instruments, Model P511) at 0.03 Hz–1.0 kHz, digitized at 10 kHz and stored on computer for later off-line analyses. The derivative (dV/dT) of the initial 2–3 ms onset of the fEPSP was measured (fEPSP slope), and the amount of potentiation calculated as the percent change from baseline (the averaged 10 min slope value from 50–60 min post-TBS divided by the averaged slope value 10 min prior to TBS). For the time course graphs, normalized fEPSP slope values were averaged and plotted as the percent change from baseline and referenced to 100%.
For whole-cell voltage clamp and current clamp experiments, patch pipettes (2–3 MΩ) were filled with a solution containing (in mmol/L): 135 K-gluconate, 8 NaCl, 1 MgCl2, 10 HEPES, 0.05 EGTA, 4 MgATP, 0.3 Na2GTP (and 10 phosphocreatine for current clamp experiments), pH 7.3. For voltage clamp experiments, series resistance was electronically compensated ≥ 70% (Axon MultiClamp 700B) and cells with series resistance changes of >20% during the recording were excluded. Cells were clamped at −60 mV and perfused with Mg2+-free aCSF containing picrotoxin (100 μm) and glycine (25 μm) prior to stimulating Shaffer collaterals to obtain mixed AMPAr and NMDAr EPSCs. Application of the AMPA-specific antagonist NBQX (10 μM) and the NMDA receptor specific antagonist D-APV (50 μM) were used to generate the AMPA/NMDA ratios. For current clamp experiments, no electronic series resistance compensation was performed. All recordings were from cells with a resting membrane potential between −75 and −55 mV and a stable input resistance and series resistance. A bias current was applied to maintain the membrane potential at −60 mV throughout all recordings. Excitatory post-synaptic potential (EPSPs) were recorded in whole-cell current clamp mode and presynaptic axons in the stratum radiatum were stimulated using a glass capillary pipette filled with ACSF connected to an Iso-Flex stimulus isolation unit. Stimulation was consistently done approximately 100 μm away from the cell being recorded. Picrotoxin (100 μm) and CGP 55845 (100 nM) was present to inhibit GABAA and GABAB receptor activity, respectively.
Quantitative Reverse Transcriptase-PCR (qPCR)
For quantitative PCR measurement of small conductance calcium-activated potassium (SK2) channel and NMDA receptor (NR1, 2a, 2b) transcripts, hippocampi were harvested at various times after CA/CPR. Total RNA was isolated using the RNAqueous-4 PCR kit (Ambion, Austin, TX, USA) per the manufacturer's instructions. Briefly, approximately 1–3 mg of tissue was lysed in lysis buffer and total RNA was isolated and eluted from a column with 50μL RNase-free elution buffer, and further treated with Turbo DNase (Ambion, Austin, TX, USA,). First strand cDNA was reverse transcribed from 500ng total RNA with High Capacity cDNA archive Kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR reactions using SsoFast PCR mastermix was performed on BioRad CFX connect detection system in duplicate using 50ng cDNA. Primers used to detect SK2, NR1, NR2a, NR2b were synthesized by Invitrogen. The housekeeping gene 18s was also assayed for each sample using 5ng of cDNA. Cycle parameters used were 95°C for 10min followed by 40 cycles of 95°C for 15s and 60°C for 30s. Expression levels were calculated as the ratio of the target gene to 18S.
Postsynaptic Densities Preparation
Hippocampi from all conditions were harvested, and postsynaptic densities were isolated as previously described (Davies et al., 2007; Davies et al., 2008). Whole hippocampi were homogenized in ice-cold homogenization buffer (1.2 mL per hippocampi) containing 320 mM sucrose, 10 mM Tris (pH 7.4), 50 mM NaF, 10 mM EDTA, 10 mM EGTA and Roche complete protease inhibitor cocktail. Then the homogenate was spun at 1000×g for 10 minutes and discarded the pellet (P1), which contains nuclei and incompletely homogenized cells. The supernatant (S1) was then spun at 10,000×g for 15 minutes. The pellet from this spin (P2) was resuspended in homogenization buffer containing 0.5% Triton X-100, homogenized with a motor pestle, incubated in 4 °C cold room for 20 minutes, and then spun at 32,000xg for 20 minutes. The pellet from this spin is the PSD-enriched fraction and was resuspended in homogenization buffer sonicated and boiled in 1%SDS, 1 mM EDTA, 10 mM Tris (pH 8) for 5 min and stored at −20°C. All of the above spins were conducted at 4°C to inhibit enzyme action.
Western Blot Analysis
The total protein content of PSD samples was measured by using a microplate bicinchoninic acid (BCA) assay (Thermo Scientific, Rockford, IL), and 2 μg protein from each fraction were resolved via electrophoresis on 7.5% SDS-PAGE gels for 1 hour at 150V. Protein was transferred to a polyvinylidene difluoride (PVDF) membrane for 1 hour at 100V, and incubated at room temperature with gentle rocking in 5% milk in Tris-buffered saline with Tween (TTBS: 140 mM NaCl, 20 mM Tris (pH 7.4), 0.1% Tween 20). Blots were incubated with anti-PSD-95 (1:2000 dilution in 1% milk, from Millipore, Billerica, MA), anti-NMDA receptor NR1-subunit (1:1000 in 1% milk, from Phosphosolutions, Aurora, CO), or anti-β-actin (1:1000 in 1% milk, from Sigma-Aldrich, St. Louis, MO) overnight at 4 °C, washed five times for 5 minutes each in TTBS, followed by a 1 h incubation with a horseradish peroxidase-conjugated goat anti-mouse (1:4,000, from Thermo Scientific) for membranes treated with anti-PSD-95, anti- NMDA receptor NR1-subunit or anti-β-actin at room temperature. Blots were then washed in TTBS five times for 5 minutes each, and bands were detected using SuperSignal® chemiluminescent substrate kits (Thermo Scientific) and ChemiDoc™ MP Imaging System (Bio-Rad, Hercules, CA). Quantitation of Western blots was performed using Image Lab software version 4.0 (Bio-Rad).
Statistical Analysis
All data are presented as mean±SEM. Statistical analysis of all data was determined using the Student's t-test for two group comparisons and one-way analysis of variance (ANOVA) and post-hoc Dunnett's test for comparison of multiple groups. Differences considered statistically significant with P<0.05.
Results
Ischemia occludes synaptically evoked LTP
To determine the effect of ischemia induced by CA/CPR on synaptically evoked LTP, extracellular field recordings of CA1 neurons were performed in acute hippocampal slices prepared at varying times following CA/CPR and compared to slices prepared from sham-operated control mice. Hippocampal slices were placed on an interface chamber and the stimulating electrodes were positioned in the stratum radiatum, close to the CA1/CA3 border, while the recording electrodes were placed in the dendritic area, thus recording Schaffer collateral to CA1 field excitatory postsynaptic potentials (fEPSPs). In sham control slices, a brief theta burst stimulation (40 pulse TBS) resulted in LTP that increased the fEPSP slope to 164 ± 9.8% (n=7, p < 0.05) of baseline after 60 min (Figure 1A, B). In contrast, recordings obtained during the acute recovery phase from CA/CPR (3, 24 hrs) demonstrated the absence of LTP (3 hrs: 115 ± 9.8% (n=9) and 24 hrs: 113 ± 8.5% (n=7), both p<0.05 compared to sham and not different than baseline; Figure 1A). This impairment in LTP caused by CA/CPR persisted for up to 30 days (Figure 1C). Thus, fEPSP after TBS was 106 ± 15.9% (n=4) at 7 days and 109 ± 13.9% (n=7) at 30 days (p<0.05 1-Way ANOVA compared to sham). Figure 1D shows the summary of these experiments, illustrating that CA/CPR causes reduced LTP at all time-points tested (P<0.05 with 1-Way ANOVA compared with sham controls). These results demonstrate that CA/CPR-induced ischemia significantly reduces LTP and this effect persists for at least 30 days after insult.
Figure 1.

Ischemia impairs synaptic plasticity in the hippocampus. A) Example fEPSPs from sham operated control and 30 day after CA/CPR mice before (black) and after (grey) TBS. B) Time course of fEPSP slope (mean ± SEM) from sham (solid circles) and mice 3 hrs (open circles) or 24 hrs (solid squares) after cardiac arrest and cardiopulmonary resuscitation (CA/CPR). Arrow indicates timing of theta burst stimulus (TBS; 40 pulses). C) Time course of fEPSP slope (mean ± SEM) from sham (solid circles) and mice 7 days (open circles) or 30 days (solid squares) after cardiac arrest and cardiopulmonary resuscitation (CA/CPR). Arrow indicates timing of theta burst stimulus (TBS; 40 pulses) D) Quantification of change in fEPSP slope following TBS. Average fEPSP slope (mean ± SEM) 60 minutes after TBS (in grey box in B normalized to baseline (black box in B). * P < 0.05 compared to sham controls.
The effects of ischemia are postsynaptic
To determine whether ischemia alters presynaptic or postsynaptic functions we first examined paired-pulse ratio (PPR), a measure of presynaptic transmitter release probability, Pr (Debanne et al., 1996; Sudhof, 2004). Paired pulse responses were recorded from the CA1 pyramidal cells using a 50 ms inter-pulse interval (20 Hz) applied to the Shaffer collateral pathway. Figure 2A is a representative PPR trace from control CA1 cells, demonstrating increased amplitude of the second pulse. Minimal differences in PPR were observed, that likely are not physiologically relevant. Figure 2B shows that analysis of PPR showed small, but significant, reduction 3 hrs after CA/CPR compared to sham controls, 1.40 ± 0.07 (n=7) in control versus 1.17 ± 0.06 in 3 hrs (n=6) mice (P < 0.05, 1-Way ANOVA). No differences (1-Way ANOVA compared to sham control) were observed at later time-points: 1.21 ± 0.02 (n=5) at 7 days and 1.30 ± 0.05 (n=7) at 30 days (Figure 2). These data suggest that changes in presynaptic glutamate release are unlikely to account for the impaired LTP observed after CA/CPR at delayed time points.
Figure 2.
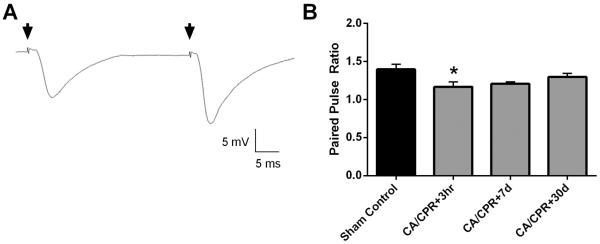
CA/CPR does not affect the probability of release. A) Example traces illustrating increased amplitude and slope of fEPSP following a second stimulus administered 50 ms after the first. B) Quantification of paired-pulse ratio of sham control mice and at each time point after CA/CPR. * P < 0.05 compared to sham control. Data presented as mean ± SEM.
As a measure of CA3 axonal intrinsic excitability and synaptic transmission, input-output (IO) functions of fEPSP vs stimulation amplitude were established at the beginning of each recording, prior to TBS. The slope of each recording was measured individually and compared to sham control slices. Figure 3 shows that analysis of IO at each time-point following CA/CPR revealed a small, but significant, transient increase in the IO slope at 7 days (Figure 3B), which was not apparent in the acute phase (3 hrs) or the chronic phase, at 30 days. The slope of control slices was 1.44 ± 0.05 (n=8) compared to 1.67 ± 0.09 (n=6) at 3 hrs, 1.79 ± 0.11 (n=5; P<0.05) at 7 days and 1.47 ± 0.06 (n=8) at 30 days (1-Way ANOVA failed to show significant differences in any group compared to sham control slices).
Figure 3.
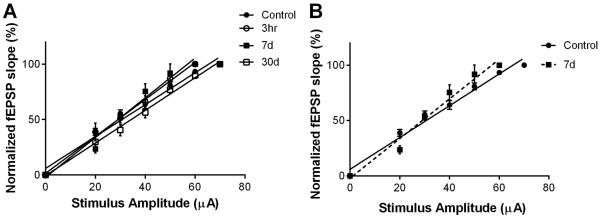
CA/CPR transiently increases slope of input-output curve (IO). A) Normalized fEPSP slope is plotted against stimulus intensity (fEPSP slope normalized to max of each recording). B) Expanded view showing increased slope at 7 days after CA/CPR compared to sham controls. No difference in IO slope was observed at 3 hrs or 30 days after CA/CPR.
Ischemia does not alter the NMDA receptor contribution to synaptic transmission
To determine whether the relative expression of functional NMDA receptors was altered by CA/CPR-induced ischemia, we used whole cell voltage-clamp recordings to determine the AMPA:NMDA current ratio from synaptically evoked EPSCs. CA1 pyramidal neurons were voltage clamped at −60 mV and synaptically evoked EPSCs were recorded in the absence of external Mg2+. Application of NBQX was used to determine the AMPA receptor contribution to EPSCs (Figure 4A-red traces), and the remaining current that was blocked by subsequent addition of D-AP5 (Figure 4A-blue traces) yielded the NMDA receptor component. The ratio was calculated as the NBQX sensitive current/D-AP5 sensitive current. The results show that the AMPA:NMDA ratio in sham control slices was 1.03 ± 0.48 (n=6), 0.936 ± 0.27 (n=6, P>0.05 1-Way ANOVA compared to sham control) at 7 days and 1.04 ± 0.193 (n=7, P>0.05 1-Way ANOVA compared to sham control) in slices obtained from mice 30 days after CA/CPR (Figure 4C). These data indicate no changes in functional NMDA receptors relative to AMPA receptors following CA/CPR (1-Way ANOVA failed to show significant differences in any group compared to sham control slices). To determine whether the CA/CPR-induced deficit in LTP reflects reduced NMDA receptor expression we performed quantitative real-time RT-PCR on hippocampal tissue from sham, 7d and 30d post-CA/CPR mice. No significant change in NMDA receptor mRNA (GluN1, 2A and 2B) expression was observed (n=6 in sham and 5 in each CA/CPR group; P>0.05 using 1-Way ANOVA to compare sham to 7 and 30 days after CA/CPR for each subunit; Figure 4C). To confirm the lack of effect of CA/CPR on NMDA receptor expression during the chronic phase of recovery, synaptic NR1 levels were quantified using Western blot of synaptic membrane preparations obtained from the hippocampus of sham control animals and 7 or 30 days after recovery from CA/CPR. No significant change in NR1 protein (NR1/PSD95 ratio), was observed, 2.64±0.23 (n=6) in sham compared to 3.55±0.28 (n=6) in 7 day and 3.30±0.64 (n=6) in 30 day (P>0.05, 1-Way ANOVA compared to sham control)
Figure 4.

Ischemia does not alter AMPA/NMDA ratio. A) Representative EPSCs from sham control. Application of NBQX (red trace) inhibits the AMPA component of the initial EPSC (black trace). Subsequent application of D-APV inhibits to remaining NMDA component (blue trace). B) Quantification of AMPA to NMDA ratio for all cells recorded demonstrates no difference of CA/CPR. C) Quantification of mRNA expression of the 3 predominant isoforms of NMDA receptor expressed in the hippocampus, GluN1, 2A and 2B. D) Representative Western blot analysis of NR1, PSD95 and β-actin in synaptic membrane preparations from hippocampi obtained at either 7 or 30 days after CA/CPR, or sham control mice. Quantification (bottom) of NR1:PSD95 ratio shows not changes in response to CA/CPR. Data presented as mean ± SEM.
Ischemia impairs SK2 channel dependent plasticity
To determine whether ischemia altered the SK2 channel effect on LTP, the SK channel blocker, apamin (100 nM) was applied prior to TBS. In slices from sham controls, LTP was significantly increased in the presence of apamin with the fEPSP slope increasing to 219 ± 18.7% (n=6; P<0.05) in apamin, compared to 164 ± 9.8% (n=7) of baseline under control conditions (Figure 5A,B). In striking contrast, Figure 5B–D illustrates the complete lack of effect of apamin on the magnitude of TBS-induced LTP following CA/CPR (110 ± 8.9% (n=5; P=0.77, Student's t-test compared to CA/CPR at 3 hrs) at 3 hrs; 111 ± 5.3% (n=5; P=0.86, Student's t-test compared to CA/CPR at 24 hrs) at 24 hrs; 108 ± 7.8% (n=6; P=0.91, Student's t-test compared to CA/CPR at 7 days) at 7 days; 112 ± 17.7% (n=6; P=0.92, Student's t-test compared to CA/CPR at 30 days) at 30 days).
Figure 5.

Ischemia impairs SK2 channel dependent plasticity. A) Example fEPSPs from sham operated control and 30 days after CA/CPR mice in the presence of apamin before (black) and after (grey) TBS. B) Time course of fEPSP slope (mean ± SEM) from sham mice under control conditions (solid circles) and in the presence of 100 nM apamin (diamonds). Time course of fEPSP slope recorded in the presence of 100 nM apamin from mice 3 hrs (open squares) or 24 hrs (solid squares) after cardiac arrest and cardiopulmonary resuscitation (CA/CPR). Arrow indicates timing of theta burst stimulus (TBS; 40 pulses). C) Time course of fEPSP slope (mean ± SEM) from sham (solid circles) and in the presence of apamin in sham (diamond), mice 7 days (open squares) or 30 days (solid squares) after cardiac arrest and cardiopulmonary resuscitation (CA/CPR). Arrow indicates timing of theta burst stimulus (TBS; 40 pulses) CD) Quantification of change in fEPSP slope following TBS. Average fEPSP slope (mean ± SEM) 60 minutes after TBS (in grey box in B normalized to baseline (black box in B). * P < 0.05 compared to sham controls.
Quantitative real-time RT-PCR demonstrates that CA/CPR results in a significant loss in expression of SK2 mRNA at 24 hrs, 7 days and 30 days, reducing to 53±6% (n=4; P<0.05 1-Way ANOVA compared to control), 66±8% (n=5; P<0.05 1-Way ANOVA compared to control) and 60±11% (n=5; P<0.05 1-Way ANOVA compared to control) of sham control levels, respectively (Figure 6A), indicating the possibility that the inability of apamin to enhance LTP in post-ischemic cells is due in part to loss of SK2 channel expression. To determine whether ischemia causes prolonged changes in synaptic SK channel activity, whole-cell current clamp recordings were obtained from acute hippocampal slices at 7 or 30 days after CA/CPR and compared to sham-operated control mice. As described previously (Allen et al., 2011), subthreshold EPSPs were stimulated every 20 seconds and after obtaining a 5 minute stable baseline, apamin (100 nM) was applied and the amplitude of EPSP was analyzed after 30 minutes. In sham control mice, apamin increased the amplitude of EPSP by 116±32% (n=6) of baseline (n=6, P<0.05 compared to baseline) (Figure 6B and 6C). In contrast, the effect of apamin was significantly reduced at both 7 days and 30 days after CA/CPR, increasing the EPSP by 14±19% (n=6, P>0.05 1-Way ANOVA compared to control) and 31±14% (n=9, P>0.05 1-Way ANOVA compared to control), respectively. These data demonstrate that global ischemia induced by CA/CPR results in a sustained reduction of synaptic SK channel function. To assess the possibility that CA/CPR causes a decrease in Ca2+-sensitivity during the induction of LTP, we increased the intensity of TBS stimuli from 40 pulses to 300. In sham control slices we observed that this increased stimulus intensity increase the magnitude of LTP; 218 ± 17.7% (n=5) compared to 164 ± 9.8% (n=7; P=0.016 Student's t-test) using 40 pulse TBS stimulus. However, increased stimulus intensity does not reveal LTP in slices from mice 30 days after CA/CPR, 83.5 ± 4.3% (n=4; P=0.20 Student's t-test compared to CA/CPR at 30 days; Figure 7C).
Figure 6.

Ischemia causes sustained loss of synaptic SK channel activity in CA1 neurons. A) Quantification of mRNA expression of the SK2 channels at 24 hr, 7 and 30 days after CA/CPR. B) Time course of EPSP amplitude (mean ± SEM) from sham control mice and 7 days after CA/CPR before (black rectangle) and after (grey rectangle) a 30 minute application of 100 nM apamin. C) Quantification of the effect of apamin on EPSP amplitude. EPSP amplitude 30 minutes after application of apamin (grey square in B) normalized to baseline (black square in B). Example EPSPs from sham operated (D), 7 days after CA/CPR (E) and 30 days after CA/CPR (F) before (black) and after (red) application of apamin. *P<0.05 compared with sham controls and data presented as mean ± SEM.
Figure 7.

1EBIO prevents CA/CPR loss of SK2 channel activity in synaptic plasticity. A) Time course of fEPSP slope (mean ± SEM) from sham (solid circles) and mice 30 days (open circles) after cardiac arrest and cardiopulmonary resuscitation (CA/CPR) and mice treated with 1-EBIO early after CA/CPR and time course recorded 30 days later (solid square). Arrow indicates timing of theta burst stimulus (TBS; 40 pulses). B) Time course of fEPSP slope (mean ± SEM) from 1-EBIO treated mice 30 days after CA/CPR in the presence (open square) and absence (solid square) of 100 nM apamin. Arrow indicates timing of theta burst stimuls (TBS; 40 pulses) C) Time course of fEPSP slope (mean ± SEM) from sham control mice stimulated with standard 40 pulse TBS (solid circle) or 300 pulse TBS stimulus (open square) and mice 30 days after CA/CPR stimulated with 300 pulse TBS. Arrow indicates timing of theta burst stimulus (TBS; 40 or 300 pulses) D) Quantification of change in fEPSP slope following TBS. Average fEPSP slope (mean ± SEM) 60 minutes after TBS (in grey box in A normalized to baseline (black box in A). * P < 0.05. E) Time course of EPSP amplitude (mean ± SEM) from sham control mice and 30 days after CA/CPR mice treated with 1-EBIO before (black rectangle) and after (grey rectangle) a 30 minute application of 100 nM apamin. F) Quantification of the effect of apamin on EPSP amplitude. EPSP amplitude 30 minutes after application of apamin (grey square in E) normalized to baseline (black square in E).
Increasing SK channel activity prevents the ischemia-induced occlusion of LTP
Previously, we showed that systemic administration of the SK channel agonist, 1-EBIO, to mice following CA/CPR protects CA1 neurons from ischemia-induced excitotoxic cell death by preserving synaptic SK2 channel activity (Allen et al., 2011). To test whether 1-EBIO treatment would also prevent ischemia-induced loss of LTP, 1-EBIO (16 mg/kg, ip injection) was administered to mice 30 min after CA/CPR, with a second, boosting dose administered 6 hrs after CA/CPR. Figure 7A illustrates that using this regimen, 1-EBIO provided sustained protection against ischemia-induced loss of LTP in CA1 pyramidal cells when assessed 30 days after CA/CPR, with the fEPSP slope increasing to 157 ± 8.0% (n=6; P=0.017 Student's t-test compared to CA/CPR at 30 days). Interestingly, while 1-EBIO administration preserved TBS-induced LTP 30 days after CA/CPR, apamin no longer increased the magnitude of LTP (Figure 7B, D). Consistent with the lack of apamin effect on LTP in 1-EBIO treated mice, the effect of apamin on EPSP amplitude was lost in mice treated with 1-EBIO and current clamp recordings performed 30 days after CA/CPR, apamin only increased EPSP amplitude by 3.5±9.3% (n=7, P=0.047 Student's t-test compared to controls) (Figure 7E).
Discussion
The results presented here show that global cerebral ischemia induced by cardiac arrest and cardiopulmonary resuscitation (CA/CPR) causes impairment in long-term potentiation within 3 hours of resuscitation that is sustained for at least 30 days in the CA1 region of the hippocampus. Our data further shows that SK2 channel activity is lost at all time-points analyzed following CA/CPR, as inhibition of SK channels with apamin failed to enhance LTP or EPSP amplitude. In contrast, treatment of mice 30 minutes after CA/CPR with the SK2 agonist 1-EBIO prevented the CA/CPR-induced loss of synaptic plasticity observed 30 days after CA/CPR. Finally, CA/CPR-induced loss of LTP appears to be post-synaptic, however is not associated with loss of NMDAr function, implicating alternative mechanisms for ischemia-induced impairments.
We observe rapid and sustained loss of synaptic plasticity (LTP) in the CA1 synapses of mice following cardiac arrest-induced cerebral ischemia, consistent with the extensive literature indicating that global cerebral ischemia causes selective CA1 injury. Data remains unclear regarding the effects of global cerebral ischemia on hippocampal synaptic plasticity, with studies demonstrating no impairment (Li et al., 2013) and several studies reporting impaired LTP during the first week following global ischemia (Mori et al., 1998; Gillardon et al., 1999; Kiprianova et al., 1999; Dai et al., 2007; Nagy et al., 2011). Our data is consistent with the large number of reports demonstrating deficits in synaptic plasticity following global cerebral ischemia and provides important new information regarding the timing of onset and duration of impaired synaptic plasticity. We observed a deficit in LTP remarkably early (within 3 hours) that is sustained for the duration of our study (30 days). It is likely that the sustained impairment in LTP observed in our study contributes to cognitive deficits observed following global cerebral ischemia, as we and others have demonstrated (Kofler et al., 2004; Allen et al., 2011). The mechanism of ischemia-induced loss of LTP remains unknown, although several studies have implicated loss of NMDA receptor expression and function (Zhang et al., 1997; Hsu et al., 1998; Dos-Anjos et al., 2009; Liu et al., 2010). However, unlike previous studies, our data demonstrates the presence of functional NMDA receptors 7 and 30 days after CA/CPR. It is not clear why our study fails to observe ischemia-induced loss of NMDA receptors. One possibility is that few studies have utilized patch-clamp electrophysiology to directly measure NMDA receptor current, thereby previous studies have interpreted decreased protein or mRNA levels as loss of function. We used synaptic membrane preparations to quantify possible ischemia-induced changes in NMDA receptor function, unlike more commonly used whole brain protein preparations. Additionally, we utilize the novel mouse model of CA/CPR to induce global cerebral ischemia, which is likely different than the more commonly used model of occlusion of vessels in the brain. An alternative possibility to explain the CA/CPR-induced loss of LTP in the presence of functional NMDA receptors would be decreased calcium-sensitivity of the various signaling cascades engaged to induce and sustain LTP. For example, decreased expression or activation of CaMKII, MAPK, proteases, among others (Lisman et al., 2002; MacDonald et al., 2006; Rosenkranz et al., 2009; Sanhueza & Lisman, 2013), would result in impaired LTP, regardless of NMDA receptor activity. While our data does not completely resolve this possibility, our experiments using increased stimulus intensity imply that increasing calcium influx is not sufficient to rescue LTP, implicating changes in the signaling cascade downstream of NMDA receptor activation.
In vitro ischemia causes synaptic potentiation (iLTP) that mimics many of the features of physiological LTP utilized in the current study (Crepel et al., 1993; Di et al., 2008). It is likely therefore that following our in vivo model of CA/CPR-induced ischemia causes iLTP in the injured hippocampus. Indeed, the loss of LTP observed within 3 hours of CA/CPR is consistent with the rapid onset of iLTP observed in vitro. Similarly, our previous study demonstrated rapid internalization of SK2 channels in CA1 synapses following CA/CPR, similar to that observed following synaptically evoked LTP (Lin et al., 2008; Lin et al., 2010) again consistent with iLTP. The presence of iLTP is likely to occlude further potentiation and thus would represent a mechanism of CA/CPR-induced loss of synaptic plasticity. However, it is unclear whether iLTP would be maintained for the extended duration of the current study (30 days). Further studies are needed to confirm the induction of iLTP following CA/CPR and to resolve the mechanism of ischemia-induced impairment of synaptic plasticity in the hippocampus.
We observed that neuroprotective doses of 1-EBIO prevented CA/CPR-induced loss of synaptic plasticity, leading to the prediction that 1-EBIO treatment would result in improved functional recovery. Indeed, we previously reported that 1-EBIO improves performance following CA/CPR in the hippocampal-dependent task novel object recognition compared to vehicle treated mice (Allen et al., 2011). Therefore, our data indicates that in addition to direct neuroprotection, rescue of synaptic function is an important therapeutic target to improve long term functional recovery. The mechanism of 1-EBIO protection of synaptic function is unclear. One likely mechanism would be to prevent the ischemia-induced loss of SK2 channels, as observed in the acute phase following CA/CPR in our previous study (Allen et al., 2011). However, our data does not indicate sustained SK2 channel function at 30 days after resuscitation. Recordings from the hippocampus of mice treated with 1-EBIO 30 minutes after CA/CPR showed intact LTP that was not further increased with the SK2 channel inhibitor apamin, in contrast to control mice which exhibited increased LTP in the presence of apamin. Similarly, we found that apamin failed to increase EPSP amplitude in acute brain slices obtained from 1-EBIO treated mice 30 days after CA/CPR, indicating loss of functional SK channels. Coupled with our previous observation that 1-EBIO treatment prevents acute SK2 internalization following ischemia, this data implicates additional mechanisms that result in the loss of SK channels in the later, chronic phase of recovery following CA/CPR. Further, these data provide evidence for separate mechanisms regulating synaptic SK channel function and synaptic plasticity following cerebral ischemia.
In conclusion, the current study has thoroughly characterized the effects of CA/CPR on hippocampal synaptic plasticity, demonstrating a rapid and sustained impairment. Further, the ability of the SK2 agonist 1-EBIO to prevent CA/CPR-induced plasticity impairment was observed and demonstrates a role for these channels in neuroprotection and sustained functional benefits. The CA/CPR-induced loss of synaptic plasticity is not due to reduced NMDA receptor function or obvious perturbations in pre-synaptic neurotransmitter release. As such, the current study provides important insights into the kinetics of ischemia-induced synaptic changes and begins to reveal mechanisms that may be targeted to prevent impairments.
Acknowledgements
Project funded by NIH grants NS065855 and NS080851, Walter S. and Lucienne Driskill Foundation grant, AHA-Philips Resuscitation Fellowship 12POST11930031
References
- Allen D, Nakayama S, Kuroiwa M, Nakano T, Palmateer J, Kosaka Y, Ballesteros C, Watanabe M, Bond CT, Lujan R, Maylie J, Adelman JP, Herson PS. SK2 channels are neuroprotective for ischemia-induced neuronal cell death. J Cereb Blood Flow Metab. 2011;31:2302–2312. doi: 10.1038/jcbfm.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol.Life Sci. 2004;61:657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepel V, Hammond C, Krnjevic K, Chinestra P, Ben-Ari Y. Anoxia-induced LTP of isolated NMDA receptor-mediated synaptic responses. J.Neurophysiol. 1993;69:1774–1778. doi: 10.1152/jn.1993.69.5.1774. [DOI] [PubMed] [Google Scholar]
- Dai X, Chen L, Sokabe M. Neurosteroid estradiol rescues ischemia-induced deficit in the long-term potentiation of rat hippocampal CA1 neurons. Neuropharmacology. 2007;52:1124–1138. doi: 10.1016/j.neuropharm.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Davies KD, Alvestad RM, Coultrap SJ, Browning MD. alphaCaMKII autophosphorylation levels differ depending on subcellular localization. Brain research. 2007;1158:39–49. doi: 10.1016/j.brainres.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KD, Goebel-Goody SM, Coultrap SJ, Browning MD. Long term synaptic depression that is associated with GluR1 dephosphorylation but not alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor internalization. J Biol Chem. 2008;283:33138–33146. doi: 10.1074/jbc.M803431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Guerineau NC, Gahwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. The Journal of physiology. 1996;491(Pt 1):163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di FM, Tozzi A, Costa C, Belcastro V, Tantucci M, Picconi B, Calabresi P. Plasticity and repair in the post-ischemic brain. Neuropharmacology. 2008;55:353–362. doi: 10.1016/j.neuropharm.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Dos-Anjos S, Martinez-Villayandre B, Montori S, Regueiro-Purrinos MM, Gonzalo-Orden JM, Fernandez-Lopez A. Transient global ischemia in rat brain promotes different NMDA receptor regulation depending on the brain structure studied. Neurochem.Int. 2009;54:180–185. doi: 10.1016/j.neuint.2008.09.016. [DOI] [PubMed] [Google Scholar]
- Gillardon F, Kiprianova I, Sandkuhler J, Hossmann KA, Spranger M. Inhibition of caspases prevents cell death of hippocampal CA1 neurons, but not impairment of hippocampal long-term potentiation following global ischemia. Neuroscience. 1999;93:1219–1222. doi: 10.1016/s0306-4522(99)00292-4. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. doi: 10.1016/j.neuropharm.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond RS, Bond CT, Strassmaier T, Ngo-Anh TJ, Adelman JP, Maylie J, Stackman RW. Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J.Neurosci. 2006;26:1844–1853. doi: 10.1523/JNEUROSCI.4106-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn M, Schlote W. Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathol.(Berl) 1992;85:79–87. doi: 10.1007/BF00304636. [DOI] [PubMed] [Google Scholar]
- Hsu JC, Zhang Y, Takagi N, Gurd JW, Wallace MC, Zhang L, Eubanks JH. Decreased expression and functionality of NMDA receptor complexes persist in the CA1, but not in the dentate gyrus after transient cerebral ischemia. J.Cereb.Blood Flow Metab. 1998;18:768–775. doi: 10.1097/00004647-199807000-00008. [DOI] [PubMed] [Google Scholar]
- Hutchens MP, Traystman RJ, Fujiyoshi T, Nakayama S, Herson PS. Normothermic cardiac arrest and cardiopulmonary resuscitation: a mouse model of ischemia-reperfusion injury. Journal of visualized experiments : JoVE. 2011 doi: 10.3791/3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Khot S, Tirschwell DL. Long-term neurological complications after hypoxic-ischemic encephalopathy. Seminars in neurology. 2006;26:422–431. doi: 10.1055/s-2006-948323. [DOI] [PubMed] [Google Scholar]
- Kiprianova I, Sandkuhler J, Schwab S, Hoyer S, Spranger M. Brain-derived neurotrophic factor improves long-term potentiation and cognitive functions after transient forebrain ischemia in the rat. Experimental neurology. 1999;159:511–519. doi: 10.1006/exnr.1999.7109. [DOI] [PubMed] [Google Scholar]
- Kofler J, Hattori K, Sawada M, DeVries AC, Martin LJ, Hurn PD, Traystman RJ. Histopathological and behavioral characterization of a novel model of cardiac arrest and cardiopulmonary resuscitation in mice. J.Neurosci.Methods. 2004;136:33–44. doi: 10.1016/j.jneumeth.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Li J, Sasaki H, Fujiwara H, Kato H, Kaneko K, Yamazaki Y, Fujii S. Synaptic plasticity in hippocampal CA1 neurons and learning behavior in transient ischemia-loaded gerbils. Biomed Res. 2013;34:75–85. doi: 10.2220/biomedres.34.75. [DOI] [PubMed] [Google Scholar]
- Lin MT, Lujan R, Watanabe M, Adelman JP, Maylie J. SK2 channel plasticity contributes to LTP at Schaffer collateral-CA1 synapses. Nat.Neurosci. 2008;11:170–177. doi: 10.1038/nn2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Lujan R, Watanabe M, Frerking M, Maylie J, Adelman JP. Coupled activity-dependent trafficking of synaptic SK2 channels and AMPA receptors. J.Neurosci. 2010;30:11726–11734. doi: 10.1523/JNEUROSCI.1411-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zhao W, Xu T, Pei D, Peng Y. Alterations of NMDA receptor subunits NR1, NR2A and NR2B mRNA expression and their relationship to apoptosis following transient forebrain ischemia. Brain research. 2010;1361:133–139. doi: 10.1016/j.brainres.2010.09.035. [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Jackson MF, Beazely MA. Hippocampal long-term synaptic plasticity and signal amplification of NMDA receptors. Crit Rev.Neurobiol. 2006;18:71–84. doi: 10.1615/critrevneurobiol.v18.i1-2.80. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- Mori K, Yoshioka M, Suda N, Togashi H, Matsumoto M, Ueno K, Saito H. An incomplete cerebral ischemia produced a delayed dysfunction in the rat hippocampal system. Brain research. 1998;795:221–226. doi: 10.1016/s0006-8993(98)00295-9. [DOI] [PubMed] [Google Scholar]
- Muir KW. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr.Opin.Pharmacol. 2006;6:53–60. doi: 10.1016/j.coph.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Nagy D, Kocsis K, Fuzik J, Marosi M, Kis Z, Teichberg VI, Toldi J, Farkas T. Kainate postconditioning restores LTP in ischemic hippocampal CA1: onset-dependent second pathophysiological stress. Neuropharmacology. 2011;61:1026–1032. doi: 10.1016/j.neuropharm.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Neumann JT, Cohan CH, Dave KR, Wright CB, Perez-Pinzon MA. Global cerebral ischemia: synaptic and cognitive dysfunction. Current drug targets. 2013;14:20–35. doi: 10.2174/138945013804806514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat.Neurosci. 2005;8:642–649. doi: 10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- Nunes B, Pais J, Garcia R, Magalhaes Z, Granja C, Silva MC. Cardiac arrest: long-term cognitive and imaging analysis. Resuscitation. 2003;57:287–297. doi: 10.1016/s0300-9572(03)00033-9. [DOI] [PubMed] [Google Scholar]
- Peskine A, Rosso C, Picq C, Caron E, Pradat-Diehl P. Neurological sequelae after cerebral anoxia. Brain injury : [BI] 2010;24:755–761. doi: 10.3109/02699051003709581. [DOI] [PubMed] [Google Scholar]
- Petito CK, Feldmann E, Pulsinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology. 1987;37:1281–1286. doi: 10.1212/wnl.37.8.1281. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA. Selective neuronal vulnerability: morphological and molecular characteristics. Prog.Brain Res. 1985;63:29–37. doi: 10.1016/S0079-6123(08)61973-1. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann.Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart Disease and Stroke Statistics--2012 Update: A Report From the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Frick A, Johnston D. Kinase-dependent modification of dendritic excitability after long-term potentiation. The Journal of physiology. 2009;587:115–125. doi: 10.1113/jphysiol.2008.158816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, Lisman J. The CaMKII/NMDAR complex as a molecular memory. Molecular brain. 2013;6:10. doi: 10.1186/1756-6606-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Freund TF. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience. 1991;40:599–636. doi: 10.1016/0306-4522(91)90001-5. [DOI] [PubMed] [Google Scholar]
- Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP, Tzounopoulos T. Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J.Neurosci. 2002;22:10163–10171. doi: 10.1523/JNEUROSCI.22-23-10163.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annual review of neuroscience. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Lynch DR. N-methyl-D-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist. 2005;11:37–49. doi: 10.1177/1073858404269012. [DOI] [PubMed] [Google Scholar]
- Zhang L, Hsu JC, Takagi N, Gurd JW, Wallace MC, Eubanks JH. Transient global ischemia alters NMDA receptor expression in rat hippocampus: correlation with decreased immunoreactive protein levels of the NR2A/2B subunits, and an altered NMDA receptor functionality. J.Neurochem. 1997;69:1983–1994. doi: 10.1046/j.1471-4159.1997.69051983.x. [DOI] [PubMed] [Google Scholar]