

Abstract
The microcephaly-lymphedema-chorioretinal dysplasia (MLCRD) syndrome is a distinct microcephaly syndrome. The hallmark features, microcephaly, chorioretinopathy, and lymphedema, are frequently recognized at birth. Another clinical entity, the chorioretinal dysplasia, microcephaly and mental retardation syndrome (CDMMR) is a highly overlapping syndrome characterized by more variable lymphedema. Recently, heterozygous mutations in KIF11, a gene encoding a critical spindle motor protein of the Kinesin family, have been reported in individuals with MLCRD, and in individuals with CDMMR. This finding is suggestive of a single clinically variable spectrum. Here, we report on de novo novel mutations of KIF11 in five individuals with severe microcephaly, marked simplification of the gyral pattern on neuroimaging, bilateral chorioretinopathy and developmental delay. Three patients had congenital lymphedema, and one had congenital bilateral sensorineural hearing loss. This report therefore further expands the clinical and molecular spectrum of KIF11-associated microcephaly.
Keywords: Microcephaly, chorioretinopathy, lymphedema, KIF11
Introduction
Congenital microcephaly (MIC), lymphedema, and ocular abnormalities are three features variably present in two previously reported and highly recognizable microcephaly syndromes. The first is microcephaly, primary lymphedema and chorioretinal dysplasia (MLCRD) [Feingold and Bartoshesky, 1992], and the second is chorioretinal dysplasia, microcephaly and mental retardation syndrome (CDMMR) [Fryns et al., 1995]. Unlike most congenital MIC syndromes, both these disorders are predominantly inherited in an autosomal dominant fashion. Given their clinical, and recently identified molecular overlap, they now appear to constitute a single distinct MIC syndrome with wide clinical variability.
Heterozygous mutations in KIF11, a gene encoding a spindle motor protein (EG5) of the Kinesin family, have been identified in 43 individuals from 25 families [Ostergaard et al., 2012; Hazan et al., 2012; Jones et al., 2013]. The most consistent features of mutation-positive individuals are congenital MIC, ranging from mild to severe, and chorioretinal abnormalities. Identified mutations include nonsense, missense, frameshift, and splice site mutations. EG5 is known to contribute to spindle assembly and function, localizing to spindle microtubules during mitosis [Valentine et al., 2006]. Aberrations in spindle and centrosome formation and function are the most common causes of congenital MIC, placing KIF11 in a growing family of genes that account for this distinct group of MIC syndromes, such as ASPM, MCPH1, CENPJ, CDK5RAP2, STIL, CEP152, among others [Thornton and Woods, 2009; Mahmood et al., 2011].
We screened a cohort of 12 sporadic individuals with congenital MIC, chorioretinopathy with or without lymphedema, for KIF11 mutations and identified 5 novel de novo mutations in 5 individuals. Two patients had normal or mildly small occipito-frontal circumference (OFC) at birth, but all became severely microcephalic later on, with OFCs of 4 to 7 standard deviations (SDs) below the mean. All 5 individuals had bilateral chorioretinopathy, while other ocular abnormalities were also observed in some of the patients. Transient congenital lymphedema was present in three individuals. Neuroimaging showed MIC with simplified gyral pattern, mildly enlarged extra-axial space and thin corpus callosum. Our cohort therefore further expands the clinical and molecular spectrum of KIF11-associated MIC.
Materials And Methods
Phenotyping
In our database of subjects with congenital brain abnormalities, 12 subjects had microcephaly, chorioretinopathy with or without lymphedema, documented by review of clinical data, clinical photographs, and radiographic images. Brain imaging studies were reviewed by the investigators. This study was approved by Seattle Children's Institutional Review Board.
Molecular Methods
Genomic DNA was extracted from peripheral blood samples using the Puregene kit® following the manufacturers recommendations. PCR amplification was performed with 50ng of genomic DNA using Taq DNA polymerase (Applied Biosystems®). Primers used to amplify the coding and flanking noncoding regions of KIF11 were designed using Primer 3 software (http://frodo.wi.mit.edu/primer3/input.htm) (Supplementary Table SI). The coding regions of KIF11 (exons 1-22) were targeted for mutation analysis performed by direct Sanger sequencing. Double-stranded DNA sequence analysis was performed using the Big Dye Terminator chemistry (Applied Biosystems ®), and reactions were run on the ABI 3730xl Genetic Analyzer (Applied Biosystems ®). Sequence chromatograms were analyzed using the Mutation Surveyor software version 3.30. Sequences were compared with normal control samples and the reference sequences for KIF11 (NCBI reference number NM_004523.3).
Results
Phenotypic features
We identified five children in our database with microcephaly and chorioretinopathy harboring KIF11 mutations. The clinical, neuroimaging, demographic and molecular findings of mutation-positive individuals are summarized in Table I. Facial and eye clinical photographs are shown in Figures 1 and 2, respectively. We did not identify any mutations in KIF11 in the remaining seven individuals with this phenotype in our research program.
Table I. Clinical, neuroimaging and molecular data of subjects with KIF11 mutations (n=5).
| DB# | LR05-145 | LR07-122 | LR07-133 | LR07-147 | LR07-226 |
|---|---|---|---|---|---|
| Gender | Female | Male | Male | Female | Male |
| Ethnicity | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian |
| Age last assessed | 8 years | 7 years | 13 years | 8 years | 7 years |
| Birth OFC – SD | −2 | −5 to −6 | −6 | −1.5 | −4 |
| Last OFC – SD (age) | −4 (7 years) | −5 (7 years) | −5.5 (13 years) | −5 (3 years) | −7 (2.5 years) |
| Chorioretinopathy | + | + | + | + | + |
| Eye findings | Bilateral chorioretinal dysplasia: retinal lacunae | Bilateral chorioretinal dysplasia | Bilateral chorioretinopathy, bilateral optic nerve hypoplasia with atrophic changes, nystagmus | Bilateral chorioretinal lacunae, macular pigment stippling and absent foveal light reflex, small corneas (10 mm), high hyperopia, borderline small optic nerves, visual acuity 20/70 (R) and 20/80 (L) | Bilateral chorioretinal lacunae; total retinal detachment; angle-closure glaucoma, cataract |
| Lymphedema | – | + (neonatal, resolved) | + (neonatal, resolved) | – | + (neonatal, resolved) |
| Neurologic exam | Generalized hypotonia | Hypertonia of lower extremities, constant non-purposeful movements of the hands | Generalized hypotonia, hand flapping | Normal | Normal |
| Seizures | No | No | Yes | No? | No |
| Neuroimaging findings (age) | MSG, mildly prominent XAX, mildly thin CC (6 months) | Severe generalized MSG (15 months) | MSG, foreshortened FL (10 years) | MSG, mildly increased XAX, mildly thin CC (2.5 months) | MSG, foreshortened FL, partial ACC, mildly small CBL vermis, mild CM (12 months) |
| ID | Mild-mod | Mod-severe | Mod-severe | Mild-mod ID | Mod-severe |
| Other clinical features | ADHD (treated with dextroamphetamine and methylphenidate), mild joint laxity | GER, agitation | Sleeping difficulties | Severe congenital bilateral SNHL (cochlear implants at 11m) | ADHD on medication |
| KIF11 Mutation | |||||
| cDNA change | c.2299_2301het_delTT | c.1C>T | c.790-1G>A | c.729C>T | c.1029G>A |
| Amino acid change | p.Phe767Serfs*8 | p.Met1Thr | Splice donor site | p.His244Tyr | p.Glu344Lys |
| Exon | 18 | 1 | 7/8 | ||
| Type | Frameshift | Missense | Splice site | Missense | Missense |
| Zygosity | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
| Inheritance | De novo | De novo | De novo | De novo | De novo |
| Paternal/maternal ages (years)* | 38/34 | 42/33 | 37/28 | 35/31 | 35/30 |
| Conservation (Conseq score) | NA | Conserved (8) | NA | Conserved (9) | Conserved (9) |
| Polyphen-2 (score) | NA | Probably damaging (0.998) | NA | Probably damaging (1.00) | Probably damaging (1.00) |
| SIFT | NA | Damaging | NA | Tolerated | Damaging |
| PROVEAN | NA | Neutral | NA | Deleterious | Deleterious |
| Domain | Motor | Motor | Motor | Motor | Motor |
| Previously reported | No | No | No | No | No |
Abbreviations: ACC, agenesis of the corpus callosum; ADHD, attention-deficit hyperactivity disorder; CC, corpus callosum; CM, cisterna magna; FL, frontal lobe; GER, gastro-esophageal reflux; ID, intellectual disability; MSG, microcephaly with simplified gyri; NA, not applicable; PROVEAN, Protein Variation Effect Analyzer; SNHL, sensorineural hearing loss; XAX, extra-axial space.
Note: Patient LR07-147 had a normal endocrine evaluation with no evidence of pituitary abnormalities.
Parental ages at conception
Figure I. Clinical photographs of two KIF11 mutation-positive patients.
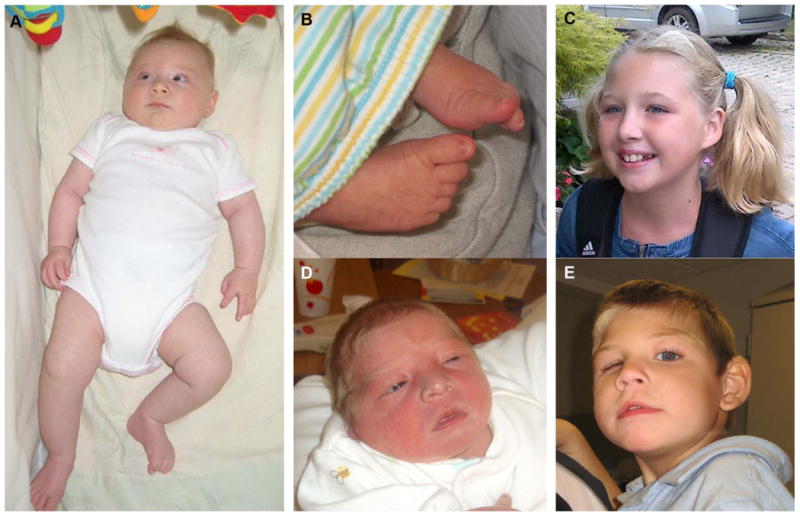
Photographs of Patient LR05-145 shortly after birth (A, B) and at ∼10 years of age (C). Note apparent microcephaly (C), and mild lymphedema of the feet at birth (B). Photographs of Patient LR07-226 shortly after birth (D) and at 3-4 years of age (E) following ocular surgery of the right eye. Note apparent microcephaly.
Figure 2. Eye photographs of three KIF11 mutation-positive patients.
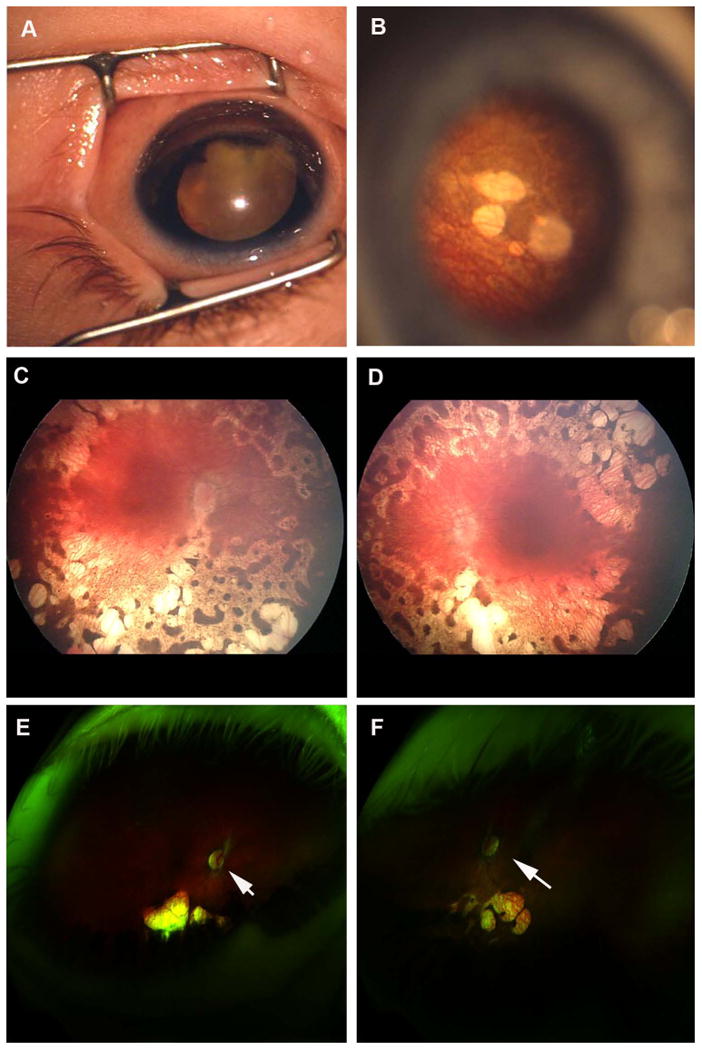
A, Anterior segment photo of the right eye of Patient LR07-226 showing leukocoria secondary to retinal detachment and cataract. B, Fundus photo of the left eye of Patient LR07-226 showing retinal lacunae outside of macula. C and D, Retinal photos of the right (C) and left (D) eyes of Patient LR07-147 showing symmetric widespread chorioretinal atrophy with lacunae and spiculated pigmentation. The maculae are relatively spared, but there are mild macular pigmentary changes and loss of the foveal reflexes bilaterally. The optic nerves are slightly pale, and the retinal vessels are attenuated. E and F, Retinal photos of the right (E) and left (F) eyes of Patient LR05-145 showing symmetric chorioretinal lacunae outside of the maculae with optic nerve pallor (indicated by arrows).
Patient LR05-145
This girl is the second born child to healthy non-consanguineous parents of mixed European ancestry. Birth weight was 2.6 kg (5-10th centile for age and gender), length 45.7 cm (5-10th centile) and OFC 32 cm (2 standard deviations, SD, below the mean). She was noted to have MIC and diffuse hypotonia at birth. Neonatal course was uncomplicated except for mild jaundice that spontaneously resolved. She fed vigorously without difficulties. Head growth continued at 2 SD below the mean until 6 months of age when head growth velocity declined further. Weight and linear growth were appropriate throughout childhood. She never had lymphedema.
On last assessment at 7 years of age, her weight was 23.9 kg (50-75th centile), height 124 cm (50-75nd centile), and OFC 46 cm (4 SD below the mean; at the 50th centile for 14 months of age). She had obvious MIC with sloping forehead but no specific dysmorphic facial features, skin lesions or birthmarks. She had mild right 2-3 toe syndactyly. Neurologic examination revealed mild hypotonia but good muscle strength, normal deep tendon reflexes and intact cranial nerves. Ophthalmologic evaluation revealed bilateral chorioretinopathy with retinal lacunae, a “mothy” or “lacy” depigmentation in the retina with normal optic nerves.
Developmentally, she had mildly delayed language comprehension and significant issues with attention and distractibility. Formal developmental behavioral testing using the Beery-Buktenica Developmental Test of Visual -Motor Integration (Beery VMI; Fifth Edition) at 7 years assessed her developmental age to be 4 years and 3 months. Word reading and spelling were K-2 grade equivalent, whereas her math computation was below K grade level. She was also formally diagnosed with attention deficit hyperactivity disorder (ADHD).
Patient LR07-122
This boy was born full term to healthy nonconsanguineous parents. MIC and intrauterine growth restriction were identified on prenatal ultrasounds. Birth weight was 2.4 kg (2 to 3 SD below the mean), and OFC 28 cm (6 SD below the mean). Minor dysmorphic facial features including low-nasal bridge, micrognathia, full cheeks and anteriorly placed hair whorl were noted. He had mild congenital lymphedema of the lower legs and feet that later resolved. TORCH screen was negative.
Ophthalmologic evaluations revealed widespread diffuse retinal pigment epithelial degeneration and retinal atrophy mixed with pigment clumping and spiculing with relative sparing of macular retina and bilaterally pale optic discs.
On last assessment at 7 years of age, his developmental age was assessed to be 2 years. His OFC was 5 SD below the mean. He was able to walk with assistance but was non-verbal with poor expressive language, communicating mostly via assistive communication devices. He had constant non-purposeful movements of the hands that were disrupted by myoclonic type jerks. He also had episodes of agitation and aggression that were treated with Risperidone, as well as tactile sensitivity. Repeat neuropsychiatric evaluation at age 10 years determined he was functioning at 24 months. Other medical issues include ocular motility defects, spastic quadriparesis, bilateral tight foot contractures requiring ankle foot orthotics (AFOs), and gastroesophageal reflux. His hearing is normal.
Patient LR07-133
This boy was born at 36 weeks of gestation to Caucasian nonconsanguineous parents. His mother took antidepressants during pregnancy and smoked 1-2 cigarettes per day. Prenatal ultrasounds at 12 and 36 weeks of gestation were normal. Birth weight was 2 kg (5-10th centile for age, gender, and gestational age), length 44.5 cm (10th centile) and OFC 28 cm (2-3 SD below the mean). He walked at age 2 years and said his first words between 10 to 11 years of age. On assessment at 13 years, his weight was 30.8 kg (5th centile), height 160 cm (90-95th centile) and OFC 46.3 cm (5 to 6 SD below the mean). He was a thin boy with obvious MIC, and disproportionately large ears and nose. Neurologic exam revealed global hypotonia. He occasionally had episodes of aggression. His sleeping pattern was abnormal, characterized by periods of agitation that were partially relieved by Clonidine. He began having complex partial and generalized epilepsy at 4 years of age and was treated with Divalproex sodium.
Ophthalmologic assessment revealed bilateral chorioretinopathy, bilateral optic nerve hypoplasia with atrophic changes. On assessment at 16 years, he had been seizure-free for the past two years but remained on anti-epileptic medication. At that time, pupils were small and poorly reactive and there was intermittent nystagmus. Motor exam showed diffuse muscle weakness and generalized hypotonia, including facial hypotonia. Deep tendon reflexes were decreased and toes were down-going bilaterally, with an ataxic and dysmetric gait. Developmentally, he was functioning at the level of a 3-year-old, speaking in short sentences, and following simple commands.
Patient LR07-147
This girl is the first child born to healthy nonconsanguineous parents. She was small for gestational age on prenatal ultrasound at 20-24 weeks of gestation. She was delivered full term via normal vaginal delivery. Birth weight was 2.6 kg (5-10th centile for age and gender), length 48.2 cm (25-50th centile), and OFC 32 cm (2 SD below the mean). She failed her initial hearing tests and was subsequently found to have severe bilateral sensorineural hearing loss for which cochlear implants were placed. Cytomegalovirus (CMV) testing was negative. By 2 months of age, it was evident she had MIC as well as poor visual tracking and nystagmus. Her head growth rate decreased as her OFC became 4 SD below the mean by age 6 months.
On last examination at age 3 years, weight was 11.2 kg (5-10th centile), height 87.2 cm (5-10th centile) and OFC 40.5 cm (5-6 SD below the mean). Her craniofacial appearance was significant for a small round head, deep-set eyes, flat philtrum and mildly small jaw. Eye movements were full with normal pupils, and face was asymmetric. Motor examination revealed mildly decreased muscle bulk with normal tone, strength and deep tendon reflexes. Her coordination was borderline clumsy for age.
Developmentally, she first sat at age 5 months, crawled at 12.5 months, and began to walk at 15 months. She began speaking words at 16 months, and her vocabulary increased in an age-appropriate manner. At 35 months of age, Battelle developmental inventory testing showed 20-30% delays, Maxfield-Buchholz testing showed a social quotient (SQ) of 109 with 56% delays, the Oregon Project test identified 56% of her skills in the 2-3 year age range, and the Rossetti Infant Toddler Language Scale identified delays in the range of 30-33 months for play and 21-24 months for pure language.
Ophthalmologic assessment revealed small corneas (10 mm horizontal diameter), widespread symmetrical chorioretinal atrophy and pigmentation in the mid-periphery, mild bilateral macular pigment stippling with loss of the foveal reflex, bilaterally small optic nerves, high hyperopia, and intermittent nystagmus. An electroretinogram identified markedly reduced rod and cone function. At age 8 years, the best-corrected visual acuity was 20/70 in the right eye and 20/80 in the left eye, and the retinal exam had remained stable since infancy.
Patient LR07-226
This boy was born full term to healthy nonconsanguineous parents. His birth weight was 3.1 kg (10-25th centile), and OFC 28.5 cm (4 SD below the mean). Exam at birth revealed an asymmetric head shape due to overlapping coronal sutures. On last assessment at age 23 months, his OFC was 38.5 cm (6 SD below the mean). He had no specific dysmorphic facial features. His digital, skin and neurologic examinations were normal.
Developmentally, he had mild to moderate delays. He walked at 21 months. At 23 months, he said a few words with many vocalizations, and was social and interactive. He overall did not have any gross motor or fine motor issues. He had early speech delay. At 8 years of age, he said 7-8 word sentences that were understandable to non-family members. He required assistance with learning but was making progress. He had ADHD and was on medication. He never had seizures.
Nystagmus was noted at birth. Ophthalmologic assessment at 2 months revealed no light perception and leukocoria with a cataract, infantile closed angle glaucoma, and complete retinal detachment of the right eye, with hyaline and fibrous metaplasia of the retinal pigment epithelium. He underwent cyclodestructive surgery for uncontrolled glaucoma in the right eye at 5 months of age. In the left eye, retinal lacunae were noted along the inferotemporal retinal arcade and nasal to the optic disc. He developed some functional vision in the left eye and was able to ambulate and identify objects.
Neuroimaging features (Fig 3)
Figure 3. Brain MRI images of three KIF11 mutation-positive patients.
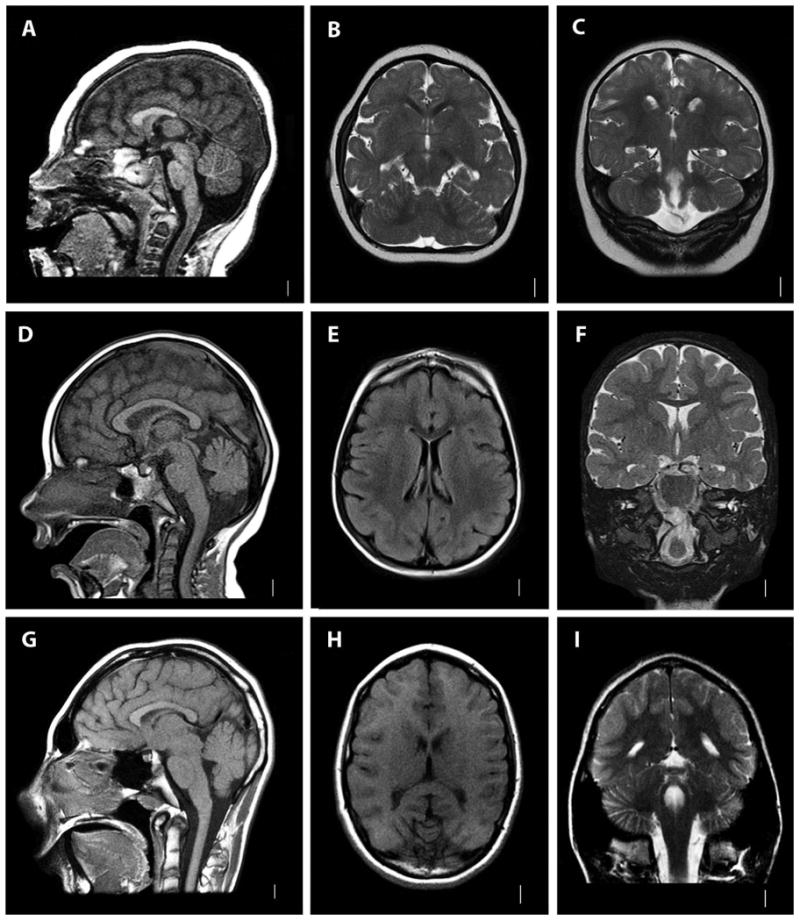
A-C, T1-weighted sagittal (A), and T2-weighted coronal (B, C) images of Patient LR07-226. D-F, T1-weighted sagittal (D) and axial (E) and T2-weighted coronal (F) images of Patient LR07-147. G-I, T1-weighted sagittal (G) and axial (H) and T2-weighted coronal (I) images of Patient LR07-133. Note microcephaly with simplified gyri in all patients. Patient LR07-226 also has partial agenesis of the corpus callosum, mildly small cerebellar vermis and a mild cisterna magna (A).
Brain neuroimaging of all affected individuals revealed MIC with simplified gyral pattern and shortened frontal lobes with or without a thin corpus callosum. One child had partial agenesis of the corpus callosum, subtle cerebellar vermis hypoplasia and mildly prominent cisterna magna. There were no additional cortical brain abnormalities or cortical brain malformations in this cohort.
Molecular findings
Five novel de novo mutations in KIF11 were identified by Sanger sequencing that include three missense, one frameshift and one splicing mutation (Table II). None of these mutations are present in dbSNP137 or the National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server of 6500 exomes. The three missense mutations (p.Met1Thr, p.His244Tyr, p.Glu344Lys) affect highly conserved residues and are predicted to be pathogenic by Polyphen-2. The frameshift mutation (p.Phe767Serfs*8) leads to a prematurely truncated protein. The splicing (c.790-1G>A) mutation affects the splice donor site of exons 7-8. This mutation is expected to abolish the splice donor site according to a number of splice prediction programs including the Splice Site Finder-Like, MaxEntScan, Gene Splicer and the Human Splicing Finder (v.2.4.1).
Table II.
The identified KIF11 mutations and their inheritance.
| Patient # | Mutation type | Nucleotide variant | Exon | Protein change | Inheritance | Genomic location (hg19) |
|---|---|---|---|---|---|---|
| LR05-145 | Frameshift | c.2299_2301_delTT | 18 | p.Phe767Serfs*8 | De novo | chr10:94,405,151 |
| LR07-122 | Missense | c.1C>T | 1 | p.Met1Thr | De novo | chr10:94,353,133 |
| LR07-133 | Splice donor | c.790-1G>A | 7/8 | – | De novo | chr10:94,373,133 |
| LR07-147 | Missense | c.729C>T | 7 | p.His244Tyr | De novo | chr10:94,372,827 |
| LR07-226 | Missense | c.1029G>A | 8 | p.Glu344Lys | De novo | chr10:94,373,373 |
Discussion
The microcephaly-lymphedema-chorioretinal dysplasia (MLCRD) syndrome is a clinically recognizable MIC syndrome with a wide range of clinical severity. Heterozygous mutations in KIF11 have been recently identified in this syndrome [Ostergaard et al., 2012; Hazan et al., 2012; Jones et al., 2013]. The most consistent features are MIC and ocular abnormalities, especially bilateral chorioretinopathy (Table III). Of the 43 reported cases, 60% of the mutations were parentally inherited, with marked inter-familial variability. Indeed, three reported individuals with disease-causing mutations were clinically unaffected, suggesting incomplete penetrance [Jones et al., 2013].
Table III.
Review of the phenotypic features of KIF11-associated microcephaly.
| Features | Ostergaard et al., 2013 | Hazan et al., 2013 | Jones et al., 2013 | This report |
|---|---|---|---|---|
| Number of patients | 27 | 1 | 15 | 5 |
| Number of families | 15 | 1 | 9 | 5 |
| MIC (<2 SD) | 23/27 (85%) | 1/1 | 15/15 (100%) | 5/5 |
| Severe cMIC (<3 SD) | ND | 0 | ND | 3/5 |
| Severe pnatMIC (<4 SD) | ND | 1 | ND | 5/5 |
| Ocular abnormalities | 15/27 (55%)a | 1/1 | 14/15 (93%) | 5/5 |
| Chorioretinopathy | 11/27 (40%) | 1/1 | 6/15 (40%) | 5/5 |
| Lymphedema | 14/27 (51%)b | 1/1 | 7/15 (46%) | 2/5 |
| ID | 19/27 (70%) mild-mod | Unknown (young age) | 12/15 (80%) mild-mod | 5/5 |
| Epilepsy | ND c | 0/1 | 0/15 (0%)c | 1/5 |
Including one with diabetic retinopathy
Including one with adult onset post-traumatic lymphedema that was mild
One individual had myoclonic epilepsy, another had absence seizure and two had clinical seizures with normal EGG (Ostergaard et al., 2013, Jones et al., 2013).
Abbreviations: cMIC, congenital microcephaly; ID, intellectual disability; ND, no data; pnatMIC, postnatal microcephaly; SD, standard deviations.
Microcephaly in this syndrome is most often congenital. The OFCs of our patients at birth ranged from 2 to 6 SD below the mean. Later OFCs in our cohort and across the literature ranged, with mean OFCs in affected males of 4-5 SD below the mean, and in females ∼6 SD below the mean. Notably, all reported affected parents had milder MIC than their children, with mean paternal OFCs of −2.5 SD and maternal OFCs of −4 SD, suggesting that this syndrome is overall milder than most other congenital MIC syndromes (Supplementary Table SII). Neuroimaging of all reported individuals shows microcephaly with simplified gyri. Less frequently seen findings include mildly increased extra-axial space and mild thinning of the corpus callosum [Basel-Vanagaite and Dobyns, 2010; Adachi et al., 2011]. Therefore this syndrome radiologically fits within the broad group of MIC with simplified gyri.
The ocular abnormalities identified in our cohort resembled those previously reported in the literature. Bilateral chorioretinopathy with typical lacunae outside the macula is the most common eye finding in this syndrome. Two of our patients had severe eye involvement, one with widespread chorioretinal atrophy that spared the macula, bilaterally small optic nerves, and moderate vision loss. The other had a total retinal detachment, angle-closure glaucoma, and cataract of one eye and typical retinal lacunae along the inferotemporal arcade and nasal to the nerve in the other eye. Other ophthalmologic findings in our cohort included high hyperopia, small corneas, nystagmus, optic atrophy, and optic nerve hypoplasia. This is consistent with the wide range of ocular abnormalities reported in mutation-positive individuals overall that include nystagmus, leukocoria, myopia, astigmatism and other refractive errors, microcornea, microphthalmia, cataracts, optic nerve abnormalities (pallor and/or hypoplasia), retinal folds, retinal detachment, colobomas, and other macular and choroidal abnormalities [Ostergaard et al., 2012; Jones et al., 2013]. These ocular features vary widely in severity among individuals. Notably, the retinal lacunae in Aicardi syndrome appear similar, but are typically located around the optic disc with relative sparing of the periphery [Fruhman et al., 2012]. Given the variability or absence of lymphedema in this syndrome, an ophthalmologic evaluation is prudent in all children with congenital or early onset microcephaly for early detection of ocular problems.
Lymphedema involving the lower extremities is known to be the most variable of the three core features of this syndrome. It is most often noticed during the neonatal period and spontaneously resolves, as occurred in three of our patients. Interestingly, a few reported individuals had adult onset intermittent lymphedema [Jones et al., 2013].
The range of intellectual disability is wide from normal development to moderate to severe developmental handicaps (and intellectual disability when old enough to assess). Four of our patients had additional neurobehavioral findings that include ADHD, agitation, and sleeping difficulties. Autism with ADHD and behavioral problems were previously reported in one patient each, suggesting that these may be common features [Jones et al., 2013]. Notably, one of our patients had severe congenital sensorineural hearing loss requiring cochlear implants. Two other reported patients also had hearing loss, one had severe hearing loss attributed to maternal antiepileptic medication and the other due to an unspecified infection [Jones et al., 2013]. Therefore, further reports are necessary to substantiate whether hearing loss is another variable feature of this syndrome.
Most severe congenital MIC syndromes (birth OFC below −3 SD) are autosomal recessive disorders caused by mutations in centrosomal and spindle-associated proteins. This syndrome is among the few known autosomal dominant MIC syndromes, with MIC that can be either severe congenital (<−3 SD) or postnatal MIC with a borderline/low normal OFC at birth. The neurologic phenotype is overall milder than other congenital MIC syndromes, probably explaining the high parental transmission rate. Parents with mutations in KIF11 have notably milder phenotypes. Ocular features reported in parents include refractive errors more commonly (myopia, astigmatism, hypermetropia), and, less frequently, bilateral chorioretinopathy. A few parents had adult onset post-traumatic lymphedema. Some parents had mild learning disabilities [Jones et al., 2013].
Recessive mutations in TUBGCP6 have been identified in a child with microcephaly with chorioretinopathy [Puffenberger et al., 2012]. The phenotype is distinct from KIF11-associated MIC as the reported TUBGCP6-positive child had more severe cortical involvement with pachygyria and cerebellar abnormalities [Puffenberger et al., 2012]. None of the children in our cohort with microcephaly and bilateral chorioretinopathy had pachygyria or other cortical or non-cortical brain malformations. Therefore, we expect that the likelihood of identifying mutations in TUBGCP6 to be low, and suspect genetic heterogeneity in this syndrome.
In summary, we report here on five individuals with novel de novo heterozygous mutations in KIF11 and a consistent phenotype characterized by microcephaly, bilateral chorioretinopathy, developmental delay and a simplified gyral pattern on neuroimaging. More variable clinical features include transient lymphedema, behavioral problems, ADHD, and sensorineural hearing loss.
Supplementary Material
Acknowledgments
We thank the families and physicians for their contribution to this project. The Dobyns laboratory is funded by the US National Institutes of Health under NINDS grant NS058721.
Footnotes
Conflict of Interest: The authors report no conflict of interest.
References
- Adachi Y, Poduri A, Kawaguch A, Yoon G, Salih MA, Yamashita F, Walsh CA, Barkovich AJ. Congenital microcephaly with a simplified gyral pattern: associated findings and their significance. AJNR Am J Neuroradiol. 2011;32:1123–1129. doi: 10.3174/ajnr.A2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basel-Vanagaite L, Dobyns WB. Clinical and brain imaging heterogeneity of severe microcephaly. Pediatr Neurol. 2010;43:7–16. doi: 10.1016/j.pediatrneurol.2010.02.015. [DOI] [PubMed] [Google Scholar]
- Feingold M, Bartoshesky L. Microcephaly, lymphedema, and chorioretinal dysplasia: a distinct syndrome? Am J Med Genet. 1992;43:1030–1031. doi: 10.1002/ajmg.1320430623. [DOI] [PubMed] [Google Scholar]
- Fruhman G, Eble TN, Gambhir N, Sutton VR, Van den Veyver IB, Lewis RA. Ophthalmologic findings in Aicardi syndrome. J AAPOS. 2012;16:238–241. doi: 10.1016/j.jaapos.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryns JP, Smeets E, Van den Berghe H. On the nosology of the “primary true microcephaly, chorioretinal dysplasia, lymphoedema” association. Clin Genet. 1995;48:131–3. doi: 10.1111/j.1399-0004.1995.tb04072.x. [DOI] [PubMed] [Google Scholar]
- Hazan F, Ostergaard P, Ozturk T, Kantekin E, Atlihan F, Jeffery S, Ozkinay F. A novel KIF11 mutation in a Turkish patient with microcephaly, lymphedema, and chorioretinal dysplasia from a consanguineous family. Am J Med Genet A. 2012;158A:1686–1689. doi: 10.1002/ajmg.a.35371. [DOI] [PubMed] [Google Scholar]
- Jones GE, Ostergaard P, Moore AT, Connell FC, Williams D, Quarrell O, Brady AF, Spier I, Hazan F, Moldovan O, Wieczorek D, Mikat B, Petit F, Coubes C, Saul RA, Brice G, Gordon K, Jeffery S, Mortimer PS, Vasudevan PC, Mansour S. Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): review of phenotype associated with KIF11 mutations. Eur J Hum Genet. 2013:1–7. doi: 10.1038/ejhg.2013.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood S, Ahmad W, Hassan MJ. Autosomal Recessive Primary Microcephaly (MCPH): clinical manifestations, genetic heterogeneity and mutation continuum. Orphanet J Rare Dis. 2011;6:39. doi: 10.1186/1750-1172-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostergaard P, Simpson MA, Mendola A, Vasudevan P, Connell FC, Van Impel A, Moore AT, Loeys BL, Ghalamkarpour A, Onoufriadis A, Martinez-Corral I, Devery S, Leroy JG, Van Laer L, Singer A, Bialer MG, McEntagart M, Quarrell O, Brice G, Trembath RC, Schulte-Merker S, Makinen T, Vikkula M, Mortimer PS, Mansour S, Jeffery S. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am J Hum Genet. 2012;90:356–362. doi: 10.1016/j.ajhg.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puffenberger EG, Jinks RN, Sougnez C, Cibulskis K, Willert RA, Achilly NP, Cassidy RP, Fiorentini CJ, Heiken KF, Lawrence JJ, Mahoney MH, Miller CJ, Nair DT, Politi KA, Worcester KN, Setton RA, Dipiazza R, Sherman EA, Eastman JT, Francklyn C, Robey-Bond S, Rider NL, Gabriel S, Morton DH, Strauss KA. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS ONE. 2012;7:e28936. doi: 10.1371/journal.pone.0028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton GK, Woods CG. Primary microcephaly: do all roads lead to Rome? Trends Genet. 2009;25:501–510. doi: 10.1016/j.tig.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine MT, Fordyce PM, Krzysiak TC, Gilbert SP, Block SM. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nat Cell Biol. 2006;8:470–476. doi: 10.1038/ncb1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.