

Abstract
The limited effectiveness of therapy for patients with advanced stage Head and Neck Squamous Cell Carcinoma (HNSCC) or recurrent disease is a reflection of an incomplete understanding of the molecular basis of HNSCC pathogenesis. MUC4, a high molecular weight glycoprotein, is differentially overexpressed in many human cancers and implicated in cancer progression and resistance to several chemotherapies. However its clinical relevance and the molecular mechanisms through which it mediates HNSCC progression are not well understood. The present study revealed a significant up-regulation of MUC4 in 78% (68/87) of HNSCC tissues compared to 10% (1/10) in benign samples [p= 0.006, OR (95% C.I) = 10.74 (2.0 - 57.56)]. MUC4 knockdown (KD) in SCC1 and SCC10B HNSCC cell lines resulted in significant inhibition of growth in vitro and in vivo, increased senescence as indicated by an increase in the number of flat, enlarged and senescence-associated β-galactosidase (SA-β-Gal) positive cells. Decreased cellular proliferation was associated with G0/G1 cell cycle arrest and decrease expression of cell cycle regulatory proteins like cyclin E, cyclin D1 and decrease in BrdU incorporation. Mechanistic studies revealed upregulation of p16, pRb dephosphorylation and its interaction with HDAC1/2. This resulted in decreased histone acetylation (H3K9) at Cyclin E promoter leading to its downregulation. Orthotropic implantation of MUC4 KD SCC1 cells into the floor of the mouth of nude mice resulted in the formation of significantly small tumors (170±18.30 mg) compared to bigger tumors (375 ±17.29 mg) formed by control cells (p= 0.00007). In conclusion, our findings showed that MUC4 overexpression plays a critical role by regulating proliferation and cellular senescence of HNSCC cells. Downregulation of MUC4 may be a promising therapeutic approach for treating HNSCC patients.
Keywords: MUC4, Head and Neck squamous cell Carcinoma, Senescence, cell cycle arrest
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the 6th most common cancer with approximately 650,000 incidences and 350,000 deaths worldwide annually.1 In the USA, 41,380 new cases and 7,850 HNSCC related deaths are expected in 2013.2 Despite multiple treatment modalities including surgery, chemotherapy and /or radiotherapy, the 5 year survival rate has not improved beyond 40-50% in last three decades3 and can be attributed to the incomplete understanding of the molecular basis of HNSCC pathogenesis. Therefore, unraveling the cellular pathways for improved understanding of HNSCC pathogenesis is urgently needed.
Mucins are a family of heavily O-glycosylated proteins protecting epithelial cell surfaces under normal physiological conditions. Several studies from our lab and others have identified mucins as potential tumor markers and attractive therapeutic targets.4, 5 MUC4, a membrane-bound mucin is expressed in several normal tissues, but its expression is elevated in malignancies of ovaries, thyroid, pancreas and breast (reviewed in6. Studies from our lab have shown that ectopic overexpression of MUC4 induces neoplastic transformation of fibroblasts suggesting the oncogenic potential of MUC4. 7 MUC4 activates Src/Focal adhesion kinase (FAK) signaling by physical interaction and stabilization of HER28, 9 thereby promoting survival, invasion and metastasis.8, 9 More recently, we have shown that MUC4 stabilizes N-Cadherin expression and promotes epithelial to mechenchymal transition (EMT) in pancreatic cancer cells.10
We studied its expression in human HNSCC tissues and investigated its functional role in HNSCC cell lines. We observed significant upregulation of MUC4 in HNSCC tissues compared to normal tissues. Knockdown (KD) of MUC4 in HNSCC cells reduced proliferation; led to cell cycle arrest and induced cellular senescence by modulating p16/Rb tumor suppressor pathway. Furthermore, MUC4 KD decreased pRb and HDAC1/2 mediated histone acetylation (H3K9 acetylation) at cyclin E promoter resulting in transcriptional silencing of cyclin E. The present study is the first to propose that MUC4 KD triggers a senescence response by regulating p16, cyclin D1 and cyclin E expression.
Results
MUC4 is over expressed in HNSCC tissues and cell lines
Immunohistochemical (IHC) analysis showed overexpression of MUC4 in 78% of the tumor samples compared to only 10% in adjacent normal tissues [p= 0.006, OR (95% C.I.) = 10.74 (2.0 - 57.56)] (Figure 1a). No significant association of MUC4 expression was observed with differentiation status, nodal metastasis, tumor size, age, gender and cigarette smoking (Table 1). HNSCC tissues showed a median composite score of 4 (range 0-12) for MUC4 as compared to the benign tissues with a median composite score of 0 (p= 0.01), as shown in the box-plot analysis in Figure 1b. Receiver Operating Characteristic (ROC) curve analysis was used to evaluate the potential of MUC4 protein as a biomarker for differentiating HNSCC from normal oral tissues. The value of area-under-the-curve (AUC) calculated on the basis of total score obtained for immune-staining of MUC4 protein were 0.78, with respect to normal tissues (Supplementary Figure 1a). These results suggest that MUC4 is deregulated in HNSCC tumors and raises the possibility that overexpression of MUC4 may contribute to pathogenesis of HNSCC.
Figure 1.

Over expression of MUC4 in HNSCC tissues and HNSCC cell lines. (a) Immunohistochemical analysis of MUC4 paraffin embedded sections of normal tissues (n = 10) and HNSCC tissue (n = 87) using anti-MUC4 Mab 8G7. Anti-KLH Mab K2G6 was used as negative control. Pancreatic cancer tissue sample was used as positive control for MUC4 expression. Magnification × 100. (b) Box plots showing distribution of composite scores based on IHC of MUC4 protein. Increased expression of MUC4 was observed in HNSCC with a median score of 4 (range 0–12), as compared to the histologically normal oral tissues with a median score of 0. (c) MUC4 expression in a panel of HNSCC cell lines was determined at protein level by immunoblotting while mRNA was analyzed by RT-PCR. β-actin was used as loading control for both western blot and RT-PCR analysis. (d) Silencing of MUC4 expression in SCC1 and SCC10B cells. Cells were infected with lentiviruses carrying shRNA against MUC4 in pLKO.1 hairpin vector or with scrambled sequence containing vector and used as control. Transfected cells were selected using Puromycin and positive clones were checked for MUC4 expression using 2% agarose SDS gel. β- actin was used as a loading control.
Table 1.
Correlation of MUC4 expression with clinicopathological characteristics of patients
| Patient characteristics | MUC4 Negative (n=19) N (%) | MUC4 Positive (n=68) N (%) | p-value |
|---|---|---|---|
| Age (median 59.5, range 42-95) | |||
| <58 years | 8 (42%) | 31 (46%) | 0.75 |
| ≥58 years | 11 (58%) | 36 (54%) | |
| Race | |||
| African American | 7 (37%) | 20 (29%) | 0.83 |
| White | 11 (58%) | 44 (65%) | |
| Unknown | 1 (5%) | 4 (6%) | |
| Sex | |||
| Male | 12 (92%) | 49 (73%) | 0.40 |
| Female | 7 (8%) | 18 (27%) | |
| Tobacco | |||
| Never | 5 (28%) | 12 (18%) | |
| Current | 12 (67%) | 37 (56%) | 0.23 |
| Former | 1 (6%) | 17 (26%) | |
| Site Group 1 | |||
| Oral cavity | 6 (32%) | 13 (19%) | |
| Oropharyngeal | 13 (68%) | 55 (81%) | 0.25 |
| Site Group 2 | |||
| Oral cavity | 8 (42%) | 13 (19%) | |
| Base of tongue | 1 (5%) | 18 (26%) | |
| Tonsil | 5 (26%) | 28 (41%) | N/A |
| Pharynx | 5 (26%) | 9 (13%) | |
| Grade | |||
| Well differentiated | 1 (6%) | 6 (9%) | |
| Moderately differentiated | 9 (5O%) | 38 (59%) | 0.56 |
| Poorly differentiated | 8 (44%) | 20 (31%) | |
| T-stage | |||
| T1/T2 | 7 (41%) | 32 (51%) | |
| T3/T4 | 10 (59%) | 31 (49%) | 0.48 |
| N-stage | |||
| NO | 5 (29%) | 17 (27%) | |
| N1-N3 | 12 (71%) | 46 (73%) | 0.84 |
N/A - analysis not conducted because of unstable OR estimates due to small sample size.
We analyzed the expression of MUC4 in a panel of HNSCC cell lines and observed expression of MUC4 in 88% (7/8) of the cell lines at both RNA and protein level (Figure 1c). SCC1, SCC10B, SCC11B, SCC38, SCC47 and SCC74A showed robust expression of MUC4, while SCC23 cells showed mild expression of MUC4 at both RNA and protein level. SCC104 cells were negative for MUC4 expression (Figure 1c). MUC4 expression in HNSCC cell lines was independent of their site or origin. Cell lines derived from oral cavity (SCCI, SCC47 and SCC74A) and larynx (SCC10B and SCC11B) expressed similar levels of MUC4. Further, cell lines derived from both primary (SCC1, SCC38, SCC47, SCC74) and recurrent metastatic tumors (SCC10B and SCC11B) showed similar MUC4 expression. We also did not observe any association between MUC4 expression and aggressiveness of the cells as very aggressive SCC1 and non-aggressive SCC10B cells11 express equal amount of MUC4. In addition, highly aggressive SCC104 cells12 did not express MUC4.
MUC4 silencing decreased cell proliferation and induced G0/G1 cell cycle arrest in vitro
To dissect its functional role in HNSCC pathogenesis, MUC4 was stably knocked down in SCC1 and SCC10B cells as previously described.10 These two cell lines were selected, because SCC1 cells is derived from the primary tumor in oral cavity with wild type with no expression of p53 and while SCC10B is derived from recurrent metastatic tissue in larynx with mutated and over expressed p53 (Figure 3b). Validation of MUC4 KD revealed a significant down-regulation of MUC4 in shMUC4 SCC1 and SCC10B cells (Figure 1d). Further, Real time PCR and immunofluorescence analysis confirmed the MUC4 knockdown (Supplementary Figure 2a and b).
Figure 3.


MUC4 knockdown induces cell senescence by modulating cell cycle regulators proteins in HNSCC cells. (a) HNSCC cells were subjected to cellular morphological observation, SA-β-gal and DAPI staining and observed under microscopy. MUC4 KD caused SCC1 and SCC10B cells are large, flattened and vacuolated, characteristics of senescent cell. SA-β-gal positive cells for senescence and DAPI for SAHF formation cells were observed under microscope and quantified. (b) Protein lysates from control and MUC4 KD HNSCC cells were analyzed for p16, p21, p27, p53, cyclin D1, cyclin E, cyclin A PTEN, phosphorylated and total Akt, ERK1/2 proteins. Western blot analysis showed upregulation of p16 and downregulation of cyclin D1, cyclin E, pAkt and pERK1/2 with no change in expression of total Akt and ERK1/2. β-actin was used as a loading control. (c-d) p16 depletion partially rescues senescent phenotype and induces cyclin E expression. MUC4 KD SCC1 and SCC10B cells were transfected with p16 siRNA or with control siRNA. After 72 hrs, proteins lysates were analyzed for p16, cyclin E expression and stained for SA-β-gal positive cells. β-actin was used as a loading control for western blot analysis.
To analyze the effect of MUC4 KD on proliferation of HNSCC cells, growth kinetics studies were performed. A significant decrease in growth rates of MUC4 KD SCC1 and SCC10B cells was observed (p = 0.004 and p = 0.015 respectively) (Figure 2a) indicating the role of MUC4 in cell proliferation. The doubling time increased from 26 h to 36 h and 34 h to 42 h in SCC1 and SCC10B cells respectively following MUC4 silencing. Similarly, MUC4 KD also notably reduced BrdU incorporation by 56% and 53% in MUC4 KD SCC1 and SCC10B cells respectively (Supplementary Figure 2c). Furthermore, MUC4 KD reduced clonogenic potential (inhibiting colony formation) in both SCC1 and SCC10B cells. The number of colonies was reduced from 327 to 151 (54 % decrease) and 233 to 125 (46% decrease) in SCC1 and SCC10B cells respectively (Figure 2b). The number of larger colonies was reduced from 301/327 (92%) and 201/233 (86%) to 97/151 (64%) and 27/125 (21.6%); in SCC1 and SCC10B MUC4 KD cells respectively.
Figure 2.

MUC4 knockdown decreases cell proliferation and induces cell cycle arrest in HNSCC cells. (a) Growth kinetics analysis showing that MUC4 KD in HNSCC SCC1 and SCC10B cells has lower proliferative activity in comparison with control cells. (b) SCC1 and SCC10B MUC4 KD and control cells (1 × 103) were seeded in triplicate in 10% DMEM in a 12-well plate, and allowed them to grow in 10% DMEM for 2 weeks. Colonies formed were washed with PBS, fixed in methanol and then stained with 0.1% crystal violet in PBS. The number of colonies (per well) were counted with the automatic colony counting tool of the Quantity One Imaging software. The graphs represent the mean (±SE) number of colonies. (c-d) SCC1 and SCC10B KD and control cells were synchronized with double thymidine block. After synchronization, cells were stained with propidium iodide (PI) and analyzed by fluorescence-activated cell sorting to evaluate the number of cells in different stages of cell cycles.
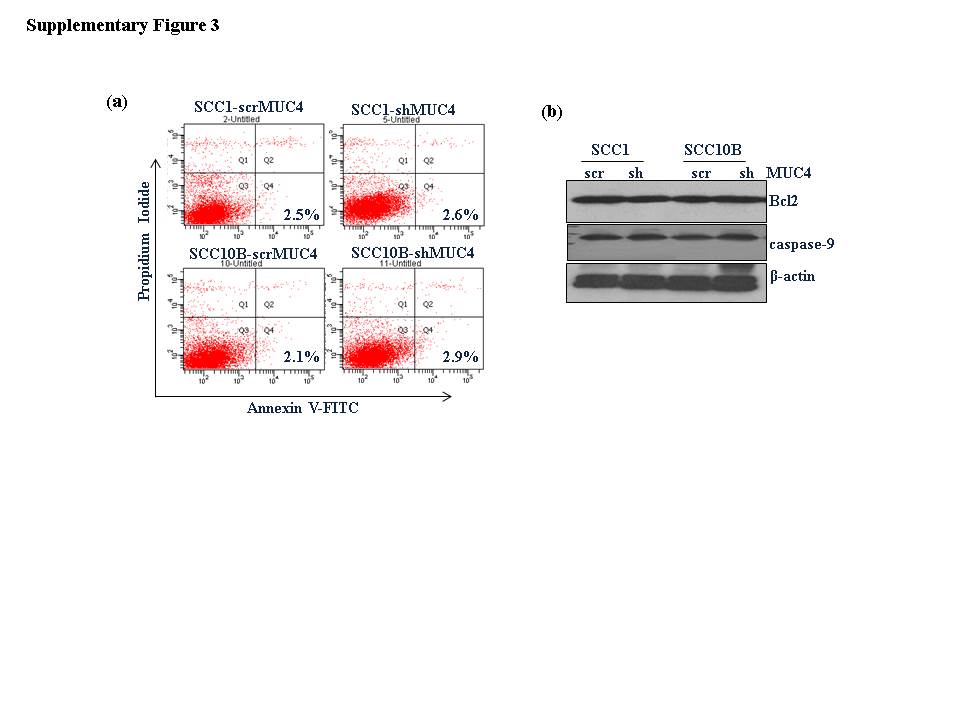
To investigate the mechanism underlying the growth suppression of HNSCC cells following MUC4 KD, we performed cell cycle analysis of the MUC4 KD cells by FACS analysis. As shown in Figure 2c and d, MUC4 KD induced the G1 arrest; the percentage of cells in G1 phase increased from 70.9% to 81% (p=0.05) and 69.9% to 82% (p=0.02) and the percentage of cells in S phase decreased from 20% to 14.8% (p=0.04) and 17.89% to 13.54% (p=0.012) in SCC1 and SCC10B cells respectively. However, MUC4 KD did not induce apoptosis in SCC1 and SCC10B cells, as demonstrated by the lack of sub-G1 peak and Annexin–V positive cells (Figure 2c and d) and (Supplementary Figure 3a and b) and absence of any change in expression of Bcl2 and caspase-9 expression (Supplementary Figure 3b).
MUC4 knockdown reduces motility/invasive potential in HNSCC cells
MUC4 KD has been shown to decrease cell motility and invasion in various cancer cells. 8-10 We analyzed the motility of MUC4 KD SCC1 and SCC10B cells by Transwell migration (Boyden chamber) and scratch assay and observed a significant decrease in motility (P < 0.001 and P < 0.002) of MUC4 KD cells compared to control cells (Supplementary Figure 4a and b) respectively. We also observed a concordant decrease in invasive capability of MUC4 KD cells compared to control cells (P < 0.005 and P < 0.004, respectively) (Supplementary Figure 4c).
FAK, a non-receptor protein-tyrosine kinase is involved in cellular motility and invasion. Src-mediated phosphorylation and activation of FAK is required for focal adhesion assembly and cell spreading. Therefore, we determined if the decreased motility and invasion following MUC4 KD is mediated through Src/FAK signaling. We observed decreased phosphorylated Src and FAK in both SCC1 and SCC10B MUC4 KD cells compared to control cells (SupplementaryFigure 4d) signifying the importance of Src/FAK pathway in MUC4 mediated increased cellular motility and invasion.
MUC4 silencing induces senescence in HNSCC cells
Cell cycle arrest is the central feature of senescent cells in vitro and in vivo. Morphologically, MUC4-KD SCC1 and SCC10B cells were vacuolated, flattened and much larger in size as compared to control cells (Figure 3a), a characteristic phenotype of senescent cells.13 To test whether MUC4 KD induced cell cycle arrest was accompanied by senescence induction; we examined the expression of SA-β-gal activity and SAHF, which are markers for cellular senescence, in MUC4-silenced and control cells. Our SA-β-gal staining showed 37±4% and 27±6% positive in MUC4 KD SCC1 and SCC10B cells compared to 3±2% and 4±1% in control cells respectively (Figure 3a). Furthermore, 4, 6-diamidino-2-phenylindole (DAPI) stained nuclei in MUC4 KD cells showed distinct senescence-associated heterochromatin foci compared to control cells as observed by immunofluorescence microscopy (Figure 3a). Overall these results indicate that MUC4 KD leads to cell cycle arrest and senescence.
MUC4 modulates PI3K/Akt and ERK1/2 signaling pathways in several cancers.9, 10, 14 and these pathways are important in regulating cellular senescence.15, 16 Our results revealed decreased levels of pAkt at Thr-308/Ser-473 and pERK1/2 in MUC4 KD SCC1 and SCC10B cells compared to control cells (Figure 3b) suggesting that MUC4 depletion induces cellular senescence by inactivation of PI3K/Akt or ERK1/2 signaling pathways. No change in expression of total Akt and ERK1/2 was observed.
MUC4 KD induces senescence through p16/pRb pathway
p16/pRb and p53/p21 pathways are master regulators of cell cycle progression and cell senescence17. The effect of MUC4 KD on delayed G1/S progression and induction of senescence prompted us to examine the expression of key molecules controlling G1/S transition. We observed significant upregulation of p16 protein and downregulation of cyclin E, cyclin D1, decreased phosphorylation of pRb in MUC4 KD cells compared to control cells (Figure 3b). However, no changes in the expression of p21 and p27 were observed (Figure 3b).
Upregulation of p16 in MUC4 KD cells suggested p16-mediated senescence in HNSCC cells. To directly establish the role of p16 in senescence, we silenced p16 expression by p16 specific siRNA in MUC4 KD cells. We observed a significant downregulation of p16 after 72h of transfection in MUC4 KD cells compared to control siRNA transfected cells (Figure 3c). We also observed a clear rescue of the morphological effects of MUC4 KD with a significant reduction in SA-β-Gal positive cells among siRNA transfected MUC4 KD SCC1 (9±3%) and SCC10B (8±4%) as compared to scrambled oligos transfected MUC4 KD SCC1 (39±4%) and SCC10B (32±6%) cells (Figure 3d). Importantly, the level of cyclin E, which is known to be expressed during G1/S transition and peak in the S phase and was noted to be downregulated in MUC4 silenced cells, was fully restored in p16 and MUC4 double KD (Figure 3c) SCC1 and SCC10B cells. These results suggest that MUC4 KD induces a p16/Rb dependent cellular senescence to suppress the growth of HNSCC cells.
MUC4 KD induces chromatin remodeling to down regulate cyclin E expression
SAHF formation during cellular senescence is accompanied by loss of heterochromatin markers like Lys9 tri-methyl (H3K9me3), H3K27me3 of histone H3 and exclusion of euchromatic markers histone H3K9 acetyl, H3K4me2 and H3K4me3 on promoters of various cell cycle genes. 18 Histone acetylation/deacetylation on promoter has been shown to regulate cyclin E expression. 19 MUC4 KD resulted in decreased acetylated H3K9 and H3K4me2 with concomitant increase in H3K27me3 compared to control cells (Figure 4a). However, no change in acetylated H2BK5 and H2AK5 were observed in MUC4 KD cells as compared to control cells. Furthermore, the ratio of H3K4me2 to H3K27me3 shifted more towards the inactive chromatin in MUC4 KD cells with respect to control cells (Supplementary Figure 5a) suggesting that deregulation of cell cycle regulated genes particularly cyclin E may be due to heterochromatin formation.
Figure 4.


MUC4 knockdown induces histone modification to modulate cyclin E expression. (a) Western blot analysis of lysates from control and MUC4 KD SCC1 and SCC10B cells using specific antibodies recognizing acetylated and methylated histones. (b) Enhanced interaction between pRb and HDAC1/2 in MUC4 KD cells. Association of pRb and HDAC1/2 was studied in the lysates of control and MUC4 KD SCC1 and SCC10B cells by immunoprecipitation and western blotting as described in Materials and Methods. HDAC1 and 2 were immunoprecipitated using specific antibodies and the bound levels of pRb co-precipitated with HDAC1/2 were determined by western blot analysis. (c) Trichostatin A (TSA) treatment induces cyclin E expression and increases SA-β-gal stained cells. MUC4 KD SCC1 and SCC10B cells were treated with TSA (5μΜ) and 48 h and protein lysates were analyzed by western blot analysis and probed with cyclin E, cyclin D1 and acetylated H3K9 specific antibodies. β-actin was used as a loading control. Similarly, TSA treated cells were analyzed for SA-β-gal staining and senescent cells were observed and counted. (d) Decreased H3K9 acetylation at cyclin E promoter. Cross-linked, sheared chromatin was prepared from control and MUC4 KD SCC1 and SCC10B cells and subjected to immunoprecipitation with the H3K9 acetylated antibody. The immunoprecipitated complexes were subjected to PCR analysis using primer pairs spanning the human cyclin E promoter. Isotypic igG Ab was used as negative control, whereas chromatin obtained before immunoprecipitation was used as internal control.
Differential binding of Histone acetyl transferases (HATs) and (Histone deacetylases) HDACs regulate cyclin E promoter activity.20 Hypophosphorylated Rb interacts with HDAC 1, 2, and 3 and recruits them to the promoters of E2F target genes leading to repression of their expression. 18 To investigate the role of HDAC1/2 and Rb in senescence, we immunoprecipitated Rb complexes from MUC4 KD SCC1, SCC10B and observed increased levels of HDAC1/2 bound to unphosphorylated Rb in MUC4 KD cells compared to control cells (Figure 4b) indicating the involvement of HDAC 1 and 2 mediated cyclin E downregulation. More pronounced interaction between Rb and HDAC2 was observed in MUC4 KD cells as compared between HDAC1 and Rb. To further confirm the involvement of HDACs, we treated MUC4 KD cells with 5μM Trichostatin A (TSA), an HDAC inhibitor for 48 h and observed the re-expression of cyclin E compared to untreated MUC4 KD cells (Figure 4c). But on the contrary, we observed increased SA-β-gal stained cells (Figure 4c). Therefore, we investigated if MUC4 mediated cyclin E down regulation was due to loss of acetylated chromatin on its promoter by performing ChIP assay using anti-acetylated histone H3 antibody. The H3 occupancy at E2F consensus binding sites of cyclin E promoter was analyzed using specific primers spanning the nucleotides between 1098 and 1795 bp on cyclin E promoter.21 Less acetylated histone H3 was observed on the cyclin E promoter in both SCC1 and SCC10B MUC4 KD cells as compared to the control cells (Figure 4d) confirming that cyclin E downregulation upon MUC4 KD is mediated by histone deacetylation and heterochromatin formation.
MUC4 knockdown suppresses tumorigenicity in vivo
MUC4 KD HNSCC cells were less proliferative, migratory and invasive in vitro; we thus tested if our in vitro observations impacted tumorigenicity and metastasis in vivo. We orthotopically implanted (2.5×105 in 50μl of PBS) either SCC1 MUC4 KD or control cells/animal into the floor of mouth of nude mice (7 mice per group). Because of the large tumors formed by controls cells, animals were euthanized after 15 days of implantation, whereas animals implanted with MUC4 KD cells formed relatively smaller tumors and were euthanized after 30 days post implantation. Tumors formed by control cells were significantly larger (median weight of 375±17.29 mg) than MUC4 KD cells of only (170±18.30 mg) (p= 0.00007) (Figure 5a) indicating that KD of MUC4 in HNSCC cancer cells decreased tumorigenicity. IHC analysis of the harvested tumors revealed decreased MUC4 expression compared to control animals, indicating that MUC4 KD was maintained in vivo (Figure 5b). Furthermore, reduced Ki-67 positive cells were observed in tumors from MUC4 KD implanted animals compared to control cells (Figure 5b). Similar to in vitro observations, we also observed increased p16 expression and decreased cyclin E expression in tumors from MUC4 KD cells implanted animals compared to control cells (Figure 5b). Further, the percentage of SA-β-gal positive cells was higher (~70%) in tumors from MUC4 KD cells as compared to control cells (~15%) (Figure 5c), strongly indicating cellular senescence in vivo is driven by MUC4 KD. Overall, our results suggest that MUC4 KD significantly suppressed tumor size by inhibiting proliferation and inducing cellular senescence in vivo.
Figure 5.


MUC4 knockdown inhibits HNSCC cell tumorigenicity. (a) Box-plot showing the distribution of tumor weight isolated from mice injected with MUC4 KD cells versus control cells. Error bars represent SE for n = 7 mice. (b) Immunohistochemistry of MUC4, P16, cyclin E and Ki-67 in tumor sections obtained from mice injected with MUC4 KD HNSCC cells (bottom) and control cells (top) along with corresponding H&E sections. (c) Showing SA-β-gal stained cells in tumor sections obtained from mice injected with MUC4 KD SCC1 cells (right) and control cells (left). (d) Proposed model for MUC4-mediated cellular senescence. MUC4 KD induces p16 expression which in turn decreased phosphorylated pRb. Under-phosphorylated pRb recruits HDAC1/2 to gene promoters like cyclin E, induces histone de acetylation and heterochromatin formation. Decreased cell cycle proliferating genes like cyclin E as a result of heterochromatin formation leads to cell cycle arrest and finally cell senescence. MUC4 via physical interaction and subsequent stabilization of HER2/ErbB2 leads to activation of Src/FAK, PI3K/Akt and ERK signaling pathways for enhanced motility, viability and increased cell proliferation.
Discussion
MUC4 has recently emerged as a useful diagnostic marker and potential target for therapeutic intervention in several malignancies due to its functional involvement in promoting cell proliferation, invasion, metastasis and inhibition of apoptosis.9, 14, 22-24 Several studies have reported aberrant expression of mucins (MUC1, MUC2, MUC4 and MUC5AC), but no functional study has yet been reported in HNSCC.25-29 Using 1G8 antibody, MUC4 over expression has been reported in HNSCC (oral cavity, oropharynx, larynx, and hypopharynx) and associated with a worse prognosis.27 However, head-to-head comparison of 1G8 with Mab 8G7 which recognizes human MUC4 has conclusively demonstrated that 1G8 neither recognizes human MUC4, nor exhibits staining pattern similar to human MUC4 antibody.30, 31 A recent study using MAb 8G7 reported MUC4 overexpression (58%) in OSCC and its association with higher T classification, positive nodal metastasis, advanced tumor stage, diffuse invasion of cancer cells and bad prognosis.29 In accordance with this report, we also observed a significant upregulation of MUC4 (68%) in HNSCC tissues compared to benign samples (P= 0.006), but contrary we did not observe any significant association of MUC4 overexpression with any clinical or pathological characteristics of patients (Figure 1 and Table 1). The major difference between the two studies was that we analyzed tissues samples of HNSCC which includes oral cavity, oropharynx, larynx, and hypopharynx, whereas the previous study used tissues from oral cavity only. Therefore lack of comprehensive knowledge about mucins prompted us to analyze the clinical and functional significance of mucins particularly MUC4 in HNSCC.
Our functional analyses using gene silencing approach indicated that MUC4 is involved in the regulation of cell growth, motility and invasion of HNSCC cells. We have previously demonstrated that MUC4 upregulates N-Cadherin expression which in turn interacts and stabilizes FGFR1 thereby promoting epithelial to mesenchymal transition (EMT) and therefore aggressive metastatic behavior of pancreatic cancer cells.10 Further, previous studies in breast, ovarian and pancreatic cancers have established that MUC4 interacts with and stabilizes human epidermal growth factor receptor 2 (HER2), also known as ErbB2 leading to Src, FAK, Akt and ERK activation implicated in cell survival, cell motility/invasion and metastasis.8, 10, 14, 32, 33 Similarly, in the present study, MUC4 KD in HNSCC cells resulted in decreased phosphorylation of Src, FAK, Akt and ERK and resulted in decreased proliferation, motility and invasion (Supplementary Figures 4a-d). Further we observed that MUC4 augments proliferation by modulating cell cycle regulatory proteins like p16, cyclin E and cyclin D1. MUC4 silencing suppressed growth via a unique mechanism involving G0/G1 cell cycle arrest. Interestingly, MUC4 silencing in HNSCC cell lines resulted in cellular senescence as suggested by large and flat cell morphology, increased SA-β-galactosidase stained cells and SAHF formation (Figure 3a) which are considered to be characteristics of senescent cells.34 This is the first report demonstrating that MUC4 expression augments senescence in cancer cells.
Cellular senescence is a potent tumor suppressor mechanism preventing unregulated growth and malignant transformation. p53 and p16/Rb signal transduction cascades are master regulators for cell cycle and promotion of cellular senescence.35 Often lost in a variety of malignancies, p16 acts as an allosteric inhibitor of cdk4/6 complex to prevent its interaction with cyclin D1, inducing the cell cycle arrest and senescence by activating Rb pathway. Cdk4/6-cyclin D1 complex mediated phosphorylation and inactivation of Rb allows the transcription of E2F-dependent various cell cycle regulatory genes including cyclin E. MUC4 silencing induced cellular senescence in HNSCC cells in a p16 dependent manner as indicated by: (a) increased p16 expression in MUC4 KD cells (b) abrogation of MUC4 silencing-induced senescence phenotype following p16 knockdown (Figure 3c-d). Our studies further indicated that senescence induction in MUC4 KD cells involved pRb dephosphorylation and chromatin remodeling to regulate cell cycle regulating protein cyclin E (Figure 3b and Figure 4a-d). Both P53 and p16/Rb signaling pathways are almost universally disrupted in 60-70% of HNSCC patients either by mutation, gene disruption or by promoter hypermethylation.36, 37 Even though the involvement of p16 in cellular senescence and its downregulation in HNSCC is well established, there is still lack of a comprehensive study of its role in HNSCC senescence. Overexpression of p16 and p53 induced growth arrest of HNSCC cells38, suggesting that p53 or p16 restoration would be enough to decrease cell proliferation and tumor growth. Intriguingly, MUC4 silencing-induced senescence seemed to occur in a p53 independent manner as MUC4 KD induced growth arrest and senescence in both SCC1 and SCC10B cells (Figure 3b). Furthermore, western blot analysis revealed no difference in expression level of p53 between MUC4 knockdown and control shRNA transfected cells (Figure 3b). Besides the p53 and p16/Rb pathway, PTEN is also involved in the decision making and maintenance of oncogene-driven senescence; however no change in PTEN expression in MUC4 KD cells suggested the involvement of only p16/Rb pathway in senescence induction on MUC4 KD (Figure 3b).
Increased proliferation is mostly driven by altered cell cycle progression.32 In HNSCC, cyclin E overexpression39 has been shown to promote G1 to S phase transition and induce escape from Ras induced senescence in mouse embryonic fibroblasts.40 Conversely, a decrease in cyclin E-Cdk2 activity results in inhibition of the G1 to S phase transition and induction of senescence.19 Silencing of MUC4 resulted in decreased proliferation and G0/G1 cell cycle arrest, providing evidence that MUC4 is a modulator of the cell cycle (Figure 2a-d). The decreased proliferation and cell cycle arrest was associated with downregulation of cell cycle regulatory proteins such as cyclin E and cyclin D1 (Figure 3b). Previously we have shown G0/G1 cell cycle arrest and decreased proliferation upon MUC4 KD in breast and pancreatic cancer cells.14, 41 In contrast to oncogenic role of MUC4 in PC, ovarian and breast, our recent work has shown a tumor suppressor role of MUC4 in lung cancer with decreased proliferation and induction of G2/M cell cycle arrest upon MUC4 over expression 42 along with downregulation of cyclin A, cyclin D1 and upregulation of cyclin dependent kinase inhibitors p21 and p27, suggesting that MUC4 has the potential to regulate (i.e. either promote or suppress) proliferation by altering cell cycle regulatory proteins.42 In accordance with all these observations, we also observed increased expression of p16 and decreased expression of pRb, cyclin D1 and cyclin E expression upon KD of MUC4 in HNSCC cells and it appears that cyclin E/D down-regulation appears to be a direct consequence of MUC4 depletion (Figure 3b).
Local perturbations of chromatin structure could alter the accessibility and/or function of transcriptional regulatory proteins that bind DNA sequences in the region where histone acetylation or de-acetylation occurs.8, 43 previous studies have shown that p300 downregulation decreased cyclin E expression through decreased histone acetylation and increased methylation of cyclin E promoter at the E2F binding site 19 leading to chromatin rearrangement accompanied by senescence associated hetero-chromatin foci (SAHF).8, 18 As seen by DAPI staining, we also observed numerous SAHF in MUC4 KD cells compared to control cells (Figure 3a) suggesting that heterochromatin modification may be involved in cyclin E downregulation. These SAHF were shown to be enriched with heterochromatin markers like Lys9 tri-methyl (K9me3), H3K27me3 and heterochromatin protein 1 (HP1) of histone H3, and excluded euchromatic markers, such as histone H3K9 acetyl and H3K4me3.18 Acetylation and methylation of H3K9 are mutually exclusive and therefore, de-acetylation of H3K9 by HDAC, which leads to repression, also allows for H3K9 methylation by methyl-transferases like Suv39H.44 Similarly, our results revealed that MUC4 KD in HNSCC cells resulted in decreased expression of acetylated H3K9 and increased expression of H3K27me3 compared to control cells (Figure 4a) with no global changes in expression of acetylated H2BK5 and H2AK5. Ratios of H3K4me3/H3K27me3 were also decreased in MUC4 KD cells than the control cells. Similar to this observation, an earlier study has also shown the differential gene expression upon either H3K4me3/H3K27me3 methylation.45 Histone modification at cyclin E promoter and its associated downregulation was further confirmed by CHIP assay using H3K9 acetylated antibody. In accordance with the results obtained by Bandyopadhyay et al19 in human melanocytes that decreased acetylated histone at the cyclin E promoter resulted in decreased cyclin E expression, we also observed cyclin E downregulation by MUC4 mediated reduced H3K9 acetylation (Figure 4d). Under-phosphorylated pRb can recruit histone de-acetylases (HDAC 1, 2, and 3) to E2F dependent promoters like cyclin E, de-acetylating nearby histones and repressing gene expression.46, 47 So we investigated whether HDAC may be involved in deacetylating H3K9 at cyclin E promoter. Co-immunoprecipitation and immublot analysis showed increased association between Rb and HDAC1 and HDAC2 in MUC4 KD cells compared to control cells with no change in total HDACs (Figure 4b). HDAC inhibitor Trichostatin A (TSA) has been shown to abolish Rb repression on some E2F target promoters like cyclin E.19 Although TSA treatment to the MUC4 KD cells induced cyclin E expression comparable to control cells (Figure. 4c), we observed increased SA-β-gal staining cells (Figure 4c). A likely explanation could be the necessity of the cells to maintain a critical balance between acetylated and deacetylated chromatin domains to sustain proliferation.19 While hyper-acetylation caused by loss of HDAC activity could activate anti-proliferative genes, loss of acetylation may shift the balance toward repressive heterochromatin, causing silencing of genes associated with cell cycle progression.
HPV is an important etiological factor for oropharyngeal cancers among head and neck cancers.48 These tumors have distinct molecular profile and significantly better prognosis than the traditional HPV negative and tobacco and alcohol related cancers.49 Recently exome sequencing, revealed several mutations in MUC4 as well as in other mucins including MUC6 and MUC16 in HPV positive tumors compared to HPV negative tumors.50, 51 Although, functional impact of these mutations remain undetermined, it is possible that these mutations may affect mucin expression, stability and post-translational modifications. While it is of interest to determine MUC4 expression in the context of HPV infections particularly in oropharyngeal cancer, due to small sample size and unknown HPV status such correlations could not be investigated in the present study. However, we plan to study this association in a large cohort of oropharyngeal patients in future.
Overall, we have shown that MUC4 KD inhibits cell proliferation both in vitro and in vivo through induction of senescence programming pathways (Figure 5d). Several studies have shown that loss of oncogene function induces cellular senescence.19, 52 Knockdown of MUC4 is crucial for ensuring the irreversibility of the senescence arrest even in p53 mutant cells. Our findings not only offer new perspectives in the modulation of senescence by MUC4 but also provide strong evidence for MUC4 based therapeutic interventions in HNSCC patients.
Materials and methods
Chemicals and antibodies
Trichostatin A (TSA) was purchased from Cayman Chemicals (Ann Arbor, MI, USA). The protein assay kit was from Bio-Rad (Hercules, CA, USA). Annexin-V conjugated AlexaFluor488 Apoptosis Detection Kit from Molecular Probes, Inc., (Eugene, OR). p16 siRNA was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). MUC4 monoclonal antibody (8G7) was developed in our laboratory.53 Horseradish peroxidase conjugated anti-mouse and anti-rabbit IgG were procured from GE Healthcare Biosciences (Uppsala, Sweden) and FITC-conjugated anti-mouse IgG was obtained from Invitrogen (California, USA). All other antibodies used are listed in supplementary table 1.
Cell culture and MUC4 knockdown
Head and Neck Cancer cell lines UMSCC1, UMSCC10B, UMSCC11B, UMSCC23, UMSCC38, UMSCC47, UMSCC74 and UMSCC104 were a kind gift from Dr. Thomas Carey (University of Michigan, USA) and maintained as described in previous reports.54 Stable MUC4 KD HNSCC cells were generated as described previously.8 Stable MUC4 shRNA or scramble transfected clones were selected, pooled and tested for MUC4 expression by real time PCR10, confocal microscopy33 and by western blot analysis.54 Co-immunoprecipitation (co-IP) assay was done as previously described.55
Cell proliferation, colony formation and BrdU incorporation
For growth rate analysis, 1 × 103 cells/well were grown in 1% αMEM and viable cell numbers were determined every 24 h for 8 days using a Vi-Cell XR instrument (Beckman Coulter, Fullerton, CA). Cell proliferation was also determined by measuring the incorporation of BrdU using a BrdU In-Situ Detection Kit (BD Pharmingen™, USA). In brief, control and MUC4 KD (10 × 103) cells were incubated overnight with 10μM BrdU for 24 h at 37°C. The incorporated BrdU was detected with an HRP conjugated anti-BrdU antibody and using tetramethylbenzidine (TMB) as a substrate. After stopping the reaction with H2SO4, plate was read at 450 nm. Effect of MUC4 KD on cell cycle, in vitro colony formation, cell motility and invasion was done as described by us previously.42, 54
Quantitative in situ SA-β-gal assay
Senescence associated-β-galactosidase (SA-β-gal) activity was measured according to the manufacturer's instructions (Cell Signaling Technology, Beverly, MA, USA). SA-β-Gal activity was detected using X-gal (5-bromo-4-chloro-3-indolyl β-D-galactoside) staining at pH 6.0. Using light microscope several representative fields (n=10) were randomly selected for the quantification of the percentage of SA-β-gal positive cells. Similarly, frozen tumor tissues from mice were stained for SA-β-gal activity as above.
Tumorigenicity assay
Control and MUC4 KD SCC1 cells of >97% viability (2.5 × 105 cells/50 μl) were orthotopically implanted at the base of tongue (seven animals/group).56 Athymic nude mice were purchased from the Animal Production Area of the National Cancer Institute-Frederick Cancer Research and Development Center (Frederick, MD, USA.). The mice were treated in accordance with Institutional Animal Care and Use Committee (IACUC) of UNMC guidelines. Based on the size of tumor, mice bearing control cells were euthanized after 15 days of implantation whereas MUC4 KD implanted mice were euthanized after 30 days. Tumors were excised, weighed, measured and formalin fixed. A part of the tumor tissue was also snap-frozen for SA-β-gal analysis. Experiment was repeated twice.
Immunohistochemistry
The study was approved by the institutional review board of University of Nebraska Medical Center, USA. Eighty seven primary HNSCC samples and 10 benign tissues were analyzed by immunohistochemical (IHC) analysis for MUC4 as described previously.14 Tumors from orthotopically implanted animals were also analyzed for MUC4, p16, cyclin E and Ki-67 expression. Replacement of the primary antibody with isotype IgG was used as negative control. Slide were analyzed by two pathologists (SL and SJ) blinded to the study. Intensity of expression and percentage of cells stained were analyzed and overall staining was then represented by a composite score as done by us earlier.14
Statistical analysis
The data was analyzed using the Medcalc software (version 9.6.4.0) for windows. The relationship between MUC4 protein expression and clinicopathological parameters was tested using Chi-Square and Fischer's test. Mean tumor weights were compared between groups using a two-tailed independent sample student t-test. The criterion for statistical significance was *P < 0.05, **P < 0.01 and ***P < 0.005.
Supplementary Material
Supplementary figure 1. ROC curves analysis. (a) ROC curves were plotted to determining the potential of MUC4 protein as a marker to distinguish HNSCC from normal oral tissues. Y-axis of the plot shows True-positive fraction and X-axis shows the false positive fraction for Normal vs HNSCC.
{kind=link}
Supplementary figure 2. (a-b) Real time PCR and confocal microscopic analysis showing a decreased expression of MUC4 in SCC1 and SCC10B KD cells compared to control transfected cells. (C) BrdU incorporation assay showing decreased proliferation of MUC4 KD in HNSCC SCC1 and SCC10B in comparison with control cells.
{kind=link}
Supplementary figure 3. MUC4 KD does not induce apoptosis in HNSCC cells. (a) The quantification of apoptotic or necrotic cells was done using the dual staining with Annexin-V and PI. (b) Western blot analysis showing expression of Bcl2 and Caspase-9 in lysates from MUC4 KD and control SCC1 and SCC10B cells. β- actin was used as loading control.
{kind=link}
Supplementary figure 4. MUC4 knockdown decreases motility and invasive behavior of SCC1 and SCC10B cells. Serum free media containing cells (5 × 105 for motility and 106 for invasion) were seeded on non-coated for motility (a) or Matrigel-coated membranes for invasion; c, After 24 h, cells migrated into the lower chamber containing 10% FBS were fixed, stained and photographed in 10 random fields under bright-field microscopy (magnification X10). Significantly decreased motility and invasion was observed in MUC4 KD SCC1 and SCC10B cells compared to scramble controls (p< 0.001). (b) 106 cells were plated in a 10 cm dish and allowed to grow until they formed a confluent monolayer. A uniform scratch was drawn across the center of the monolayer with a 100μl sterile pipette tip. The cells were carefully washed with 10% DMEM to remove the unattached cells. Images of the scratch wound were taken immediately (t=0 hours) and after incubation for 24 hours and 48 hours. The distance migrated was calculated as follows: width of scratch at time t=24 width at time t=0 h.
{kind=link}
Supplementary figure 5. (a) Bar graph showing the ratio of H3K4me2/H3K27me3. The band intensities were measured as integrated density values using Alpha Ease FC Software and the ratios calculated and plotted.
{kind=link}
Acknowledgements
The authors acknowledge the invaluable technical support from Ms. Kavita Mallya and Hanan I. Farghaly. We also thank Janice A. Taylor and James R. Talaska of the confocal laser scanning microscope core facility at UNMC for their support.
This work was supported, in part, by the grants from National Institutes of Health (RO1 CA133774, U54 CA163120, UO1 CA111294, P50 CA 127297, R21 CA156037 and P20 RR021937).
Footnotes
Conflicts of Interest
There are no potential conflicts of interest involved with this work.
References
- 1.Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet. 2008;371:1695–1709. doi: 10.1016/S0140-6736(08)60728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013. 63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Gil Z, Fliss DM. Contemporary management of head and neck cancers. Isr Med Assoc J. 2009;11:296–300. [PubMed] [Google Scholar]
- 4.Kaur S, Kumar S, Momi N, Sasson AR, Batra SK. Mucins in pancreatic cancer and its microenvironment. Nat Rev Gastroenterol Hepatol. 2013;10:607–620. doi: 10.1038/nrgastro.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9:874–885. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Torres MP, Chakraborty S, Souchek J, Batra SK. Mucin-based targeted pancreatic cancer therapy. Curr Pharm Des. 2012;18:2472–2481. doi: 10.2174/13816128112092472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bafna S, Singh AP, Moniaux N, Eudy JD, Meza JL, Batra SK. MUC4, a multifunctional transmembrane glycoprotein, induces oncogenic transformation of NIH3T3 mouse fibroblast cells. Cancer Res. 2008;68:9231–9238. doi: 10.1158/0008-5472.CAN-08-3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaturvedi P, Singh AP, Moniaux N, Senapati S, Chakraborty S, Meza JL, et al. MUC4 mucin potentiates pancreatic tumor cell proliferation, survival, and invasive properties and interferes with its interaction to extracellular matrix proteins. Mol Cancer Res. 2007;5:309–320. doi: 10.1158/1541-7786.MCR-06-0353. [DOI] [PubMed] [Google Scholar]
- 9.Ponnusamy MP, Batra SK. Ovarian cancer: emerging concept on cancer stem cells. J Ovarian Res. 2008;1:4–1. doi: 10.1186/1757-2215-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rachagani S, Macha MA, Ponnusamy MP, Haridas D, Kaur S, Jain M, et al. MUC4 potentiates invasion and metastasis of pancreatic cancer cells through stabilization of fibroblast growth factor receptor 1. Carcinogenesis. 2012;33:1953–1964. doi: 10.1093/carcin/bgs225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sano D, Xie TX, Ow TJ, Zhao M, Pickering CR, Zhou G, et al. Disruptive TP53 mutation is associated with aggressive disease characteristics in an orthotopic murine model of oral tongue cancer. Clin Cancer Res. 2011;17:6658–6670. doi: 10.1158/1078-0432.CCR-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang AL, Hauff SJ, Owen JH, Graham MP, Czerwinski MJ, Park JJ, et al. UM-SCC-104: a new human papillomavirus-16-positive cancer stem cell-containing head and neck squamous cell carcinoma cell line. Head Neck. 2012;34:1480–1491. doi: 10.1002/hed.21962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campisi J, d'Adda di FF. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 14.Mukhopadhyay P, Lakshmanan I, Ponnusamy MP, Chakraborty S, Jain M, Pai P, et al. MUC4 overexpression augments cell migration and metastasis through EGFR family proteins in triple negative breast cancer cells. PLoS One. 2013;8:e54455. doi: 10.1371/journal.pone.0054455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cammarano MS, Nekrasova T, Noel B, Minden A. Pak4 induces premature senescence via a pathway requiring p16INK4/p19ARF and mitogen-activated protein kinase signaling. Mol Cell Biol. 2005;25:9532–9542. doi: 10.1128/MCB.25.21.9532-9542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Kim J, Ruthazer R, McDevitt MA, Wazer DE, Paulson KE, et al. The HBP1 transcriptional repressor participates in RAS-induced premature senescence. Mol Cell Biol. 2006;26:8252–8266. doi: 10.1128/MCB.00604-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 19.Bandyopadhyay D, Okan NA, Bales E, Nascimento L, Cole PA, Medrano EE. Down-regulation of p300/CBP histone acetyltransferase activates a senescence checkpoint in human melanocytes. Cancer Res. 2002;62:6231–6239. [PubMed] [Google Scholar]
- 20.Harbour JW, Dean DC. Rb function in cell-cycle regulation and apoptosis. Nat Cell Biol. 2000;2:E65–E67. doi: 10.1038/35008695. [DOI] [PubMed] [Google Scholar]
- 21.Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci U S A. 1995;1992:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaturvedi P, Singh AP, Batra SK. Structure, evolution, and biology of the MUC4 mucin. FASEB J. 2008;22:966–981. doi: 10.1096/fj.07-9673rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dey P, Rachagani S, Chakraborty S, Singh PK, Zhao X, Gurumurthy CB, et al. Overexpression of ecdysoneless in pancreatic cancer and its role in oncogenesis by regulating glycolysis. Clin Cancer Res. 2012;18:6188–6198. doi: 10.1158/1078-0432.CCR-12-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rakha EA, Boyce RW, Abd El-Rehim D, Kurien T, Green AR, Paish EC, et al. Expression of mucins (MUC1, MUC2, MUC3, MUC4, MUC5AC and MUC6) and their prognostic significance in human breast cancer. Mod Pathol. 2005;18:1295–1304. doi: 10.1038/modpathol.3800445. [DOI] [PubMed] [Google Scholar]
- 25.Alos L, Lujan B, Castillo M, Nadal A, Carreras M, Caballero M, et al. Expression of membrane-bound mucins (MUC1 and MUC4) and secreted mucins (MUC2, MUC5AC, MUC5B, MUC6 and MUC7) in mucoepidermoid carcinomas of salivary glands. Am J Surg Pathol. 2005;29:806–813. doi: 10.1097/01.pas.0000155856.84553.c9. [DOI] [PubMed] [Google Scholar]
- 26.Rabassa ME, Croce MV, Pereyra A, Segal-Eiras A. MUC1 expression and anti-MUC1 serum immune response in head and neck squamous cell carcinoma (HNSCC): a multivariate analysis. BMC Cancer. 2006;6253:253. doi: 10.1186/1471-2407-6-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weed DT, Gomez-Fernandez C, Bonfante E, Lee TD, Pacheco J, Carvajal ME, et al. MUC4 (sialomucin complex) expression in salivary gland tumors and squamous cell carcinoma of the upper aerodigestive tract. Otolaryngol Head Neck Surg. 2001;124:127–141. doi: 10.1067/mhn.2001.112575. [DOI] [PubMed] [Google Scholar]
- 28.Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007;67:7713–7722. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- 29.Hamada T, Wakamatsu T, Miyahara M, Nagata S, Nomura M, Kamikawa Y, et al. MUC4: a novel prognostic factor of oral squamous cell carcinoma. Int J Cancer. 2012;130:1768–1776. doi: 10.1002/ijc.26187. [DOI] [PubMed] [Google Scholar]
- 30.Kitazono I, Higashi M, Kitamoto S, Yokoyama S, Horinouchi M, Osako M, et al. Expression of MUC4 mucin is observed mainly in the intestinal type of intraductal papillary mucinous neoplasm of the pancreas. Pancreas. 2013;42:1120–1128. doi: 10.1097/MPA.0b013e3182965915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamura Y, Higashi M, Kitamoto S, Yokoyama S, Osako M, Horinouchi M, et al. MUC4 and MUC1 expression in adenocarcinoma of the stomach correlates with vessel invasion and lymph node metastasis: an immunohistochemical study of early gastric cancer. PLoS One. 2012;7:e49251. doi: 10.1371/journal.pone.0049251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 33.Ponnusamy MP, Lakshmanan I, Jain M, Das S, Chakraborty S, Dey P, et al. MUC4 mucin-induced epithelial to mesenchymal transition: a novel mechanism for metastasis of human ovarian cancer cells. Oncogene. 2010;29:5741–5754. doi: 10.1038/onc.2010.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dimri GP, Campisi J. Molecular and cell biology of replicative senescence. Cold Spring Harb Symp Quant Biol. 1994;5967-73:67–73. doi: 10.1101/sqb.1994.059.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 36.Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. 1996;56:2488–2492. [PubMed] [Google Scholar]
- 37.Forastiere A, Koch W, Trotti A, Sidransky D. Head and neck cancer. N Engl J Med. 2001;345:1890–1900. doi: 10.1056/NEJMra001375. [DOI] [PubMed] [Google Scholar]
- 38.Liggett WH, Jr., Sewell DA, Rocco J, Ahrendt SA, Koch W, Sidransky D. p16 and p16 beta are potent growth suppressors of head and neck squamous carcinoma cells in vitro. Cancer Res. 1996;56:4119–4123. [PubMed] [Google Scholar]
- 39.Cai Y, Liu YF, Li SL, Pan YX, Zhu Y, Yu YN. [Cyclin E overexpression and centrosome amplification in squamous cell carcinoma of oral cavity]. Zhonghua Bing Li Xue Za Zhi. 2007;36:375–378. [PubMed] [Google Scholar]
- 40.Peeper DS, Shvarts A, Brummelkamp T, Douma S, Koh EY, Daley GQ, et al. A functional screen identifies hDRIL1 as an oncogene that rescues RAS-induced senescence. Nat Cell Biol. 2002;4:148–153. doi: 10.1038/ncb742. [DOI] [PubMed] [Google Scholar]
- 41.Singh AP, Moniaux N, Chauhan SC, Meza JL, Batra SK. Inhibition of MUC4 expression suppresses pancreatic tumor cell growth and metastasis. Cancer Res. 2004;64:622–630. doi: 10.1158/0008-5472.can-03-2636. [DOI] [PubMed] [Google Scholar]
- 42.Majhi PD, Lakshmanan I, Ponnusamy MP, Jain M, Das S, Kaur S, et al. Pathobiological implications of MUC4 in non-small-cell lung cancer. J Thorac Oncol. 2013;8:398–407. doi: 10.1097/JTO.0b013e3182829e06. [DOI] [PubMed] [Google Scholar]
- 43.Urnov FD, Wolffe AP. Chromatin remodeling and transcriptional activation: the cast (in order of appearance). Oncogene. 2001;20:2991–3006. doi: 10.1038/sj.onc.1204323. [DOI] [PubMed] [Google Scholar]
- 44.Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 45.Ke XS, Qu Y, Rostad K, Li WC, Lin B, Halvorsen OJ, et al. Genome-wide profiling of histone h3 lysine 4 and lysine 27 trimethylation reveals an epigenetic signature in prostate carcinogenesis. PLoS One. 2009;4:e4687. doi: 10.1371/journal.pone.0004687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- 47.Ferreira R, Magnaghi-Jaulin L, Robin P, Harel-Bellan A, Trouche D. The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc Natl Acad Sci U S A. 1998;95:10493–10498. doi: 10.1073/pnas.95.18.10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hammarstedt L, Lindquist D, Dahlstrand H, Romanitan M, Dahlgren LO, Joneberg J, et al. Human papillomavirus as a risk factor for the increase in incidence of tonsillar cancer. Int J Cancer. 2006;119:2620–2623. doi: 10.1002/ijc.22177. [DOI] [PubMed] [Google Scholar]
- 49.Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008;20100:261–269. doi: 10.1093/jnci/djn011. [DOI] [PubMed] [Google Scholar]
- 50.Nichols AC, Chan-Seng-Yue M, Yoo J, Xu W, Dhaliwal S, Basmaji J, et al. A Pilot Study Comparing HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas by Whole Exome Sequencing. ISRN Oncol. 2012;2012:809370. doi: 10.5402/2012/809370. doi: 10.5402/2012/809370. Epub;2012 Dec 12.: 809370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lleonart ME, Artero-Castro A, Kondoh H. Senescence induction; a possible cancer therapy. Mol Cancer. 2009;8:3. doi: 10.1186/1476-4598-8-3. doi: 10.1186/1476-4598-8-3.: 3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moniaux N, Varshney GC, Chauhan SC, Copin MC, Jain M, Wittel UA, et al. Generation and characterization of anti-MUC4 monoclonal antibodies reactive with normal and cancer cells in humans. J Histochem Cytochem. 2004;52:253–261. doi: 10.1177/002215540405200213. [DOI] [PubMed] [Google Scholar]
- 54.Macha MA, Rachagani S, Gupta S, Pai P, Ponnusamy MP, Batra SK, et al. Guggulsterone decreases proliferation and metastatic behavior of pancreatic cancer cells by modulating JAK/STAT and Src/FAK signaling. Cancer Lett. 2013:10. doi: 10.1016/j.canlet.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ponnusamy MP, Singh AP, Jain M, Chakraborty S, Moniaux N, Batra SK. MUC4 activates HER2 signalling and enhances the motility of human ovarian cancer cells. Br J Cancer. 2008;99:520–526. doi: 10.1038/sj.bjc.6604517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chung IS, Son YI, Ko YJ, Baek CH, Cho JK, Jeong HS. Peritumor injections of purified tumstatin delay tumor growth and lymphatic metastasis in an orthotopic oral squamous cell carcinoma model. Oral Oncol. 2008;44:1118–1126. doi: 10.1016/j.oraloncology.2008.01.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1. ROC curves analysis. (a) ROC curves were plotted to determining the potential of MUC4 protein as a marker to distinguish HNSCC from normal oral tissues. Y-axis of the plot shows True-positive fraction and X-axis shows the false positive fraction for Normal vs HNSCC.
Supplementary figure 2. (a-b) Real time PCR and confocal microscopic analysis showing a decreased expression of MUC4 in SCC1 and SCC10B KD cells compared to control transfected cells. (C) BrdU incorporation assay showing decreased proliferation of MUC4 KD in HNSCC SCC1 and SCC10B in comparison with control cells.
Supplementary figure 3. MUC4 KD does not induce apoptosis in HNSCC cells. (a) The quantification of apoptotic or necrotic cells was done using the dual staining with Annexin-V and PI. (b) Western blot analysis showing expression of Bcl2 and Caspase-9 in lysates from MUC4 KD and control SCC1 and SCC10B cells. β- actin was used as loading control.
Supplementary figure 4. MUC4 knockdown decreases motility and invasive behavior of SCC1 and SCC10B cells. Serum free media containing cells (5 × 105 for motility and 106 for invasion) were seeded on non-coated for motility (a) or Matrigel-coated membranes for invasion; c, After 24 h, cells migrated into the lower chamber containing 10% FBS were fixed, stained and photographed in 10 random fields under bright-field microscopy (magnification X10). Significantly decreased motility and invasion was observed in MUC4 KD SCC1 and SCC10B cells compared to scramble controls (p< 0.001). (b) 106 cells were plated in a 10 cm dish and allowed to grow until they formed a confluent monolayer. A uniform scratch was drawn across the center of the monolayer with a 100μl sterile pipette tip. The cells were carefully washed with 10% DMEM to remove the unattached cells. Images of the scratch wound were taken immediately (t=0 hours) and after incubation for 24 hours and 48 hours. The distance migrated was calculated as follows: width of scratch at time t=24 width at time t=0 h.
Supplementary figure 5. (a) Bar graph showing the ratio of H3K4me2/H3K27me3. The band intensities were measured as integrated density values using Alpha Ease FC Software and the ratios calculated and plotted.