

Abstract
The positron emitting (PET) 11C-labeled Pittsburgh Compound B (PIB) ligand is used to image β-amyloid (Aβ) deposits in the brains of living subjects with the intent of detecting early stages of Alzheimer’s disease (AD). However, deposits of human-sequence Aβ in APP transgenic mice and nonhuman primates bind very little PIB. The high stoichiometry of PIB:Aβ binding in human AD suggests that the PIB binding site may represent a particularly pathogenic entity and/or report local pathologic conditions. In this study, 3H-PIB was employed to track purification of the PIB binding site in > 90% yield from frontal cortical tissue of autopsy-diagnosed AD subjects. The purified PIB binding site comprises a distinct, highly insoluble subfraction of the Aβ in AD brain with low buoyant density due to an SDS-resistant association with a limited subset of brain proteins and lipids with physical properties similar to lipid rafts and to a ganglioside:Aβ complex in AD and Down Syndrome brain. Both the protein and lipid components are required for PIB binding. Elucidation of human-specific biological components and pathways will be important in guiding improvement of the animal models for AD and in identifying new potential therapeutic avenues.
Keywords: amyloid, lipids, plaque, ELISA, radioreceptor assay, tau
Introduction
In Alzheimer’s disease, the brain contains profuse high-affinity binding sites for the positron emitting 11C isotope–labeled benzothiazole ligand Pittsburgh Compound B (PIB) [6-OH-[2,4-N-methyl-phenylbenzothiazole], which binds selectively to fibrillar deposits of β-amyloid (Aβ) in senile plaques and cerebral Aβ angiopathy (Cohen et al. 2012). The ability to selectively image β-amyloid deposition in living patients was a landmark diagnostic breakthrough (Klunk et al. 2003a). It allowed detection of the disease-defining Aβ deposits (without binding significantly to tau pathology) in living subjects, and paved the way for 18F-PET-labeled amyloid ligands such as Eli Lilly’s Amyvid™ (Florbetapir), Piramal Imaging’s Neuraceq™ (Florbetaben), and GE Healthcare’s Vizamyl™ (flutemetamol). Potentially just as important for understanding the connection between Aβ pathology and the disease process was the recent observation that this intense binding was a characteristic of the AD brain (Klunk et al. 2005, Svedberg et al. 2009); PIB binding was not readily detectable in cognitively normal aged human brain, or in genetically modified (Snellman et al. 2013, Klunk et al. 2005, Toyama et al. 2005) or natural animal models of Aβ pathology (Rosen et al. 2011, Fast et al. 2013). These observations provide a potentially valuable clue to why only humans develop AD. Elucidation of the structural and physiological basis for the difference may lead to modifications of current animal models to better recapitulate the human disease and possibly reveal novel therapeutic opportunities.
There are theoretical and practical implications to whether the lesions in the human brain contain individual fibrils that consist solely of high- or low-PIB binding intensity, or whether each fibril displays a patchwork of high- and low-binding regions. Mechanisms of fibril formation, biological processes influencing pathology development, and thus the disease process will differ depending on the structural organization of the binding site. The conformational selectivity and fidelity of Aβ fibril templating observed in vitro (O’Nuallain et al. 2004) suggests that the structural state of individual fibrils can be relatively homogeneous. Isolation of a separate fraction of Aβ that contains a high density of PIB binding sites linked to early stage disease and progression would be suggestive evidence for a distinct population of Aβ fibrils that could yield clues to its genesis. We therefore adopted a strategy for purification of high-density PIB binding sites from AD brain, guided by the binding of 3H-PIB, to determine the composition of the PIB binding entity. We obtained nearly quantitative recovery of PIB binding from the AD brain, along with an approximately stoichiometric amount of Aβ peptide with distinct physical properties, including differential solubility in detergent and low buoyant density. A limited set of proteins were identified by mass spectrometric proteomic analysis in the low buoyant density PIB binding fraction. Four of these proteins were unique to the AD brain samples and were not identified in the corresponding detergent-resistant low buoyant density fraction in control brain samples from age-matched, cognitively normal humans.
Notably, only a portion of the total insoluble Aβ from the AD brain binds 3H-PIB with high affinity, and the two insoluble Aβ populations can be separated on the basis of their physical properties. These observations support the idea that high-density PIB binding resides in a discrete population of Aβ fibril-like assemblies rather than in individual fibrils bearing a mixture of high and low binding segments. The unique biological environment that gives rise to a high density of PIB binding and a 11C-PIB PET imaging signal in living human subjects may be related to key processes heralding neurodegenerative changes closely linked to the clinical symptoms of cognitive decline.
Methods and Materials
Materials
Thermo Fisher Scientific (Pittsburgh, PA) supplied BudgetSolve scintillation fluid, plastic-backed thin layer (200 μm) Silica Gel 60 plates, Maxisorp ELISA plates, NH4OH, HCl, and acetic acid. Tris base and Tris-HCl, were from Research Organics (Cleveland, OH). NuPAGE Mes gels, buffer, and Protein G-Dynabeads were purchased from Life Technologies (Carlsbad, CA). Streptavidin-conjugated to horseradish peroxidase was from Rockland Immunochemicals (Gilbertsville, PA). Nitrocellulose membranes (0.22 μm pore size) and SDS were obtained from BioRad (Hercules, CA). BTA-1 was from Calbiochem/EMD-Millipore (Billerica, CA). CNBr was from Alfa Aesar (Ward Hill, MA). Other reagents without a source noted were obtained from Sigma-Aldrich (St. Louis, MO).
Methods
Tissue homogenization and preparation of the PIB binding site (PIB BS)
Brain tissue was frozen in liquid nitrogen and pulverized to a fine powder in a Biopulverizer (Biospec Products, Inc., Bartlesville, OK) cooled thoroughly with liquid nitrogen. In order to ensure sufficient disease tissue for study, the frozen powder from samples of frontal cortical brain regions from six different AD subjects was thoroughly mixed and stored as aliquots of 150–200 mg in 1.5 ml polypropylene Eppendorf centrifuge tubes pre-chilled in liquid nitrogen, and then stored at −75ºC. Frontal cortical samples from control subjects were processed individually and stored separately.
Five volumes of homogenization buffer, 50 mM Tris-HCl, 150 mM NaCl, pH 7.5, (HB) supplemented with 1X complete protease inhibitor cocktail (sc-29130 Santa Cruz Biochemicals, (Santa Cruz, CA)) were added to a known wet weight of frontal cortex to give a concentration of 167 mg weight of original wet tissue in one milliliter of solution.
Dounce homogenization
Powdered tissue suspended in HB in a 2 ml glass/glass Dounce homogenizer (Bellco Glass, Vineland, NJ) was processed with 15 strokes on ice with a B pestle.
Sonication
Powdered tissue suspended in HB or HB + 2% w/v SDS was subjected to three rounds of pulse sonication with a microtip (0.125 inch diameter) at 25% power with a 500 watt Model 500 Sonic Dismembrator, (Fisher Scientific) at room temperature, 15 sec each, in 1.5 ml polypropylene Eppendorf tubes. One round consisted of 15 pulses of 0.5 sec sonication with 0.5 sec pause between pulses. The tube was incubated for 15 sec on ice between rounds of sonication.
Differential Centrifugation
Crude homogenate was centrifuged at 15,000 × g for 10 min at 4ºC (8ºC for SDS-extracted material) in a Biofuge Fresco refrigerated microfuge (Kendro Laboratory Products, Newtown, CT). The cloudy supernatant and soft pellet were combined and transferred to fresh tubes. The dark hard pellet of cellular debris contained no PIB binding activity and was discarded.
The combined 15, 000 × g supernatant and soft pellet were transferred to 1.5 ml polyallomer tubes and subjected to centrifugation at 100,000 × g in a TLA-55 rotor for 1 h at 8ºC in an Optima TLX Ultracentrifuge, (Beckman-Coulter, Fullerton, CA). For SDS-extracted material the 100,000 × g pellet was resuspended in 1 ml of 10 mM sodium phosphate, 150 mM NaCl, pH 7.4 (PBS), and re-centrifuged to remove excess SDS. Each final pellet was resuspended in a volume of PBS equal to the original volume of crude homogenate of brain tissue and stored in aliquots at −75ºC.
Equilibrium sucrose gradient buoyant density separation
Fractionation of PIB BS by buoyant density was performed on a discontinuous sucrose gradient in 5 ml Ultra-Clear tubes at 100,000 × g for 18 h in an SW 55.1 rotor (Beckman-Coulter). Sucrose solutions were prepared fresh from solid sucrose on the day of use. The sucrose gradient consisted of six layers of sucrose in 1 mM NaHCO3, 2.4 M (1.2 ml)/1.5 M (0.8 ml)/1.2 M (0.8 ml)/1 M (0.8 ml)/0.85 M (0.8 ml)/0.35 M (0.4 ml). Samples were applied on the top of the gradient (150 μl in PBS). Alternatively, 150 μl of extract in 1.5 ml polyallomer tubes was centrifuged at 100,000 × g in a TLA-55 rotor for 1 h at 16ºC, the pellet resuspended in 1.2 ml of 2.4 M sucrose in 1 mM NaHCO3, and applied on the bottom of the gradient with 150 μL of PBS layered on top of the gradient. Applying the sample at the top or bottom of the gradient did not affect the results, indicating that the samples had equilibrated at their buoyant density. Fractions (0.4 ml) were collected from the top of the gradient.
Subjects
We used postmortem brain tissue from six different female subjects with end-stage AD and three cognitively normal control female subjects. Demographics and clinical information are compiled in Table 1. Blocks of unfixed frontal cortical tissue were fresh-frozen at autopsy and stored at −80 °C. The human tissue was obtained from the University of Kentucky Center on Aging Brain Bank of the Alzheimer’s Disease Center in accordance with federal and institutional IRB guidelines, and samples were de-identified to ensure the anonymity of subjects. The study conforms with The Code of Ethics of the World Medical Association. Pathological and clinical analysis was provided by the University of Kentucky Alzheimer’s Disease Center.
Table 1.
Demographics and Clinical Characteristics of AD Subjects
| Subject # | Disease state | Age (y) | Gender | ApoE | PMI (h) | Braak Stage | MMSE | NFT | Diffuse plaques | Neuritic plaques |
|---|---|---|---|---|---|---|---|---|---|---|
| 1180 | AD | 91 | F | 3/3 | 3.00 | 6 | 1 | 15.6 | 50 | 16.4 |
| 1185 | AD | 79 | F | 4/4 | 3.00 | 6 | 10 | 26.2 | 49 | 13.2 |
| 1188 | AD | 75 | F | 3/4 | 2.50 | 6 | 1 | 36.5 | 50 | 15.4 |
| 1196 | AD | 83 | F | 3/4 | 2.25 | 6 | 0 | 3.4 | 50 | 7 |
| 1202 | AD | 80 | F | 3/4 | 3.33 | 6 | 22 | 22.2 | 49.2 | 24.8 |
| 1093 | AD | 82 | F | 3/4 | 2.50 | 6 | 1 | 25.8 | 42.6 | 17.2 |
| 1092 | Norm | 86 | F | 3/3 | 1.75 | 0 | 29 | 0 | 0 | 0 |
| 1131 | Norm | 80 | F | 3/3 | 2.25 | 0 | 28 | 0 | 0 | 0 |
| 1163 | Norm | 84 | F | 3/3 | 3.00 | 0 | 26 | 0 | 0 | 0 |
Pathology reported is for frontal cortex.
PMI = post mortem interval; MMSE = Mini-Mental Status Exam; NFT = neurofibrillary tangles; ApoE = apoE genotype
Plaques per 10X objective field (2.35 mm2); tangles per 20X objective field (0.586 mm2).
Statistical analysis
All values are expressed as means ± standard deviations.
3H-PIB binding assay
3H-PIB binding was assessed in homogenates of crude and fractionated human frontal cortical brain tissue. The amount of each fraction assayed was adjusted to reflect the same initial wet weight of brain tissue. Aliquots containing 167 mg original wet tissue per milliliter of solution were further diluted 1:25 in PBS to a final concentration of 6.7 mg wet tissue/ml, and 20 μl of this solution containing the equivalent of 133.3 μg of wet weight tissue were added to each of triplicate wells of a 96-well polypropylene plate (Costar 3365). Two hundred μl of 1.2 nM 3H-PIB (SA = 70.2 Ci/mmol, custom synthesized by Vitrax (Placentia, CA), or by Quotient Bioresearch, Ltd [formerly Amersham] (Cardiff, UK) in PBS was added to duplicate wells, and 200 μl of 1.2 nM 3H-PIB containing 1 μM nonradioactive competitor in PBS was added to the third well of the triplicate as a control for nonspecific PIB binding. Unless otherwise specified, BTA-1 was used as a commercially available substitute competing ligand for PIB.
Samples were incubated for 3 h at room temperature without shaking, transferred to a 96-well Millipore Multiscreen HTS Hi Flow FB (GF/B) filter plate, and filtered with a multiwell plate vacuum manifold (Millipore Corporation, Bedford, MA). The filters were rapidly washed four times with 200 μl of 10 mM Tris-HCl, 35 mM NaCl, 5% ethanol, pH 7.4, (TBS-5% EtOH) and placed in scintillation vials. Two ml of BudgetSolve scintillation fluid were added, and the vials capped and intensively shaken for 1 min before counting for 3H in a Packard TriCarb 2500 TR scintillation counter. Specific binding of each triplicate (total minus nonspecific binding) was calculated as (mean CPM of the two filters from wells containing only radioactive PIB minus the CPM value from the well containing radioactive PIB + 1 μM nonradioactive BTA-1 competitor).
Immunoprecipitation of Pre-3H-PIB bound PIB Binding site
Four microliters of SDS-extracted PIB binding site prior to the sucrose density gradient floatation step were added to 100 μl of PBS in 1.5 ml polypropylene microfuge tubes and incubated for 10 min. Six hundred microliters of 1.2 nM 3H-PIB in PBS were then added. A separate incubation with 3H-PIB in the presence of 1 μM non-radioactive BTA-1 was also prepared and all tubes were incubated at room temperature for 2 hours. The tubes were centrifuged at 100,000 × g for 1 hour at 4°C and the pellet resuspended in 100 μl of RIPA buffer and incubated for 10 min. Nine microliters (9 μg) of monoclonal IgG antibody to the desired epitope or non-immune mouse IgG were added and the mixture incubated at 4°C overnight. Thirty microliters of Protein G-Dynabeads were added and the suspension rotated for 3 hours at 4°C. The beads were collected on a magnetic stand and washed twice with RIPA buffer, retaining the combined washes. 3H-PIB binding to the (supernatant + washes) was measured by filtration and washing 4 times with TBS-5% EtOH to quantify the PIB binding site not retained on the beads.
Extraction of lipids from low buoyant density sucrose gradient fractions
The low buoyant density of the PIB binding site and co-fractionating proportion of Aβ indicates association with lipid. Treatment of the binding site by the Folch method (Folch et al. 1957) with 2:1 (v/v) chloroform:methanol to extract non-covalently bound lipids produced an organic phase (lipids) and an aqueous phase (proteins). PIB binding was destroyed by the Folch solvent extraction. Neither the protein fraction nor the lipid fraction dried and resuspended in PBS bound 3H-PIB (data not shown). PIB binding to synthetic Aβ(1–40) fibrils was not affected by combining with the same resuspended AD lipid fraction.
Aβ electrophoresis and immunoblotting
The PIB binding site fraction was extremely resistant to solubilization for SDS-PAGE analysis. Samples of 30 μg total protein in 1.5 ml polypropylene Eppendorf tubes were adjusted to 1 ml with water, and precipitated by the addition of 100 μl of 76% w/v trichloroacetic acid (TCA), 0.1% w/v deoxycholate. The pellet after centrifugation was washed twice with 1 ml of acetone and then once with 1 ml of hexane : isopropanol (3:2 v/v) to remove lipids that interfere with immunodetection of Aβ in the PIB binding site. The wash steps included brief sonication in a sonicator bath, vortexing, then centrifugation for 10 min at 15,000 × g at room temperature. The supernatant was aspirated leaving 20–50 μl of liquid to avoid disturbing the pellet.
After the final hexane-isopropanol wash, the pellet was dried in a hood at room temperature overnight. Sixty μl of sample buffer (10% glycerol, 1% SDS, 1% Ficoll-400, 0.2 M triethanolamine-HCl, pH 7.6, 0.025% w/v bromophenol blue, 8 M urea, 0.5 mM EDTA, 0.6 M 2-mercaptoethanol) was added, sonicated for 5 min in a sonicator bath, and allowed to solubilize at room temperature overnight. Before electrophoresis, the samples were sonicated for 5 min in a sonicator bath, incubated for 30 min at 37ºC, and immediately applied to a gel.
Electrophoresis was performed in a MES-bis-Tris NuPAGE buffer system in a 0.75 mm thick 4–12% w/v acrylamide gradient or 12% acrylamide gel. The proteins were transferred to a 0.2 μm nitrocellulose membrane (BioRad), boiled in PBS for 5 min, blocked with 3% w/v BSA (Sigma fraction V) in TBS, and immunoblotted with mixed antibodies (0.5 μg/ml 4G8 and 0.5 μg/ml 6E10). Detection was with an Odyssey fluorescent imager after incubation with a fluorescently-labeled antimouse IgG secondary antibody (1:20,000).
Protein measurement
Brain extract samples containing 1–50 μg of protein were adjusted to 1 ml with water, and precipitated by the addition of 100 μl of 76% w/v TCA, 0.1% w/v deoxycholate. The pellet after centrifugation was washed twice with 1 ml acetone, dried under a gentle air stream, and incubated in 100 μl of 0.5 M NaOH, 1% w/v deoxycholate, 0.1% w/v SDS for at least 12h at room temperature with occasional (2–3 times) sonication in a sonicator bath. This method was required to remove interfering substances and to dissolve the highly insoluble, sedimentable protein fractions. No pellet was observed following this treatment after centrifugation at 100,000 × g for 1h. Protein in samples was determined by bicinchoninic acid (BCA) (Pierce) or by the method of Bradford (Bradford 1976) in a final volume of 200 μl in a 96-well clear polystyrene plate (Costar 3795). Comparable amounts of protein were determined by the two methods. Protein standard curves were constructed with 0.5–10 μg of BSA.
Aβ ELISA measurements
Samples precipitated with TCA/deoxycholate and extracted with acetone as described for the protein measurements were dissolved in 70% formic acid and the Aβ content determined by sandwich ELISA capturing with 6E10 and detecting with biotinyl-4G8/streptavidin-HRP as described (Beckett et al. 2012).
Determination of PIB binding site composition
Protein analysis and identification
Two independently prepared samples of frontal cortex from the 6 pooled Alzheimer’s brains and 3 pooled control brains were used for the mass spectral analysis. Control brain is defined as age-matched brain from a subject with normal cognition (without the characteristic clinical functional deficits of Alzheimer’s disease). See Table 1 for subject demographics and Supporting Information for methods and details.
Results
The scheme for the purification of the PIB binding site from AD brain is shown in Figure 1. Differential centrifugation of AD brain homogenates in isotonic salt-containing solutions resulted in a variable and significant proportion of PIB binding co-sedimenting with the low speed (1,000 × g) nuclei/debris pellet. Brief probe-sonication prior to centrifugation released the PIB binding and allowed separation from the nuclei/debris by differential centrifugation without losing PIB binding. Combining sonication with a 2% w/v SDS detergent extraction used to purify Aβ plaques from AD brain (Roher & Kuo 1999) followed by centrifugation at 15,000 × g produced a two-layered pellet, a loosely compacted (fluffy), light upper layer over a tightly packed dark-colored pellet of debris on the tube bottom. The dark-colored hard pellet contained <5% of the PIB binding, which was readily separated from the upper pellet. The majority of the PIB binding was recovered in the combined 15,000 × g supernatant and light upper pellet layer. Significant Aβ was present in the 15,000 × g hard pellet, but very little PIB binding was present. After ultracentrifugation of the combined 15,000 × g supernatant and the resuspended light upper pellet for 1 h at 100,000 × g at 4°C, most of the protein remained in the 100,000 × g supernatant containing SDS, while all of the specific PIB binding was recovered in the insoluble 100,000 × g pellet (Supporting Table 1).
Figure 1.

Purification scheme for the AD brain PIB binding site.
Buoyant density separation indicates that the purified PIB binding site has properties similar to lipid rafts
The options for fractionating complexes of highly insoluble proteins are limited compared to those available for separating buffer- or detergent-soluble mixtures. Equilibrium density centrifugation in sucrose gradients separates components by their buoyant density and can distinguish unmodified proteins from those associated with carbohydrates, nucleic acids, or lipids, regardless of their size or charge. Washed SDS-extracted PIB binding site was applied in 2.4M sucrose at the bottom of a six-step sucrose gradient, and then centrifuged at 100,000 × g for 18 hours at 16ºC. The majority of the PIB binding and the 6E10/biotinyl-4G8-immunoreactive Aβ in the SDS-extracted sample floated to low-density fractions 2–6 near the top of the gradient, separate from a visible light-scattering lipid layer that floated to the top (fraction 1) (Figure 2). The same separation and distribution of PIB binding was observed when the sample was applied in PBS to the top of the gradient indicating that sedimentation equilibrium had been achieved. The majority of the protein (determined by the Bradford method (Bradford 1976)) accumulated near the top of the gradient (fraction 1), separated from the low buoyant density PIB binding fractions 2–6 and from the higher density fractions 7–11 where non-lipid modified/associated proteins accumulate. Buoyant density profiles of the PIB binding site showed some variability between individuals, but PIB binding was always associated with low buoyant density. Age-matched control human frontal cortex showed a similar profile of protein fractionation, but no specific 3H-PIB binding, and only small amounts of Aβ were detected in control brain in the gradient (Figure 2). The similarity of both the amount and shape of the total protein distribution on the sucrose density gradients between control and AD subjects indicate that, after enrichment of the PIB binding site, the AD pathology does not grossly distort the total protein distribution pattern.
Figure 2.

Equilibrium buoyant density separation indicates that the PIB binding site has a density suggesting associated lipid. Control (pool of 3) and AD brain (pool of 6) showing protein, Aβ, and PIB binding. Error bars show standard deviations across 3 gradients for each pool.
3H-PIB binding activity is highly resistant to dissociation
A variety of conditions were tested for their ability to affect or solubilize the PIB binding site from the SDS-resistant pellet. Attempts to release the PIB binding with high salt (2M NaCl), high pH (1M NaOH) or low pH (30% glacial acetic acid) were unsuccessful, as were treatments with 8M urea, 6M guanidine-HCl, or 2M KSCN. More than 50% of the PIB binding was recovered after washing the pellet with PBS. No binding activity was detected in the supernatants. In complementary experiments, prebound 3H-PIB remained associated with the binding site when treated under conditions that did not decrease post-treatment binding, and dissociated under those that reduced binding, indicating that the bound PIB ligand was not drastically affecting the extractability of the binding site.
Treatment (10 min) with a variety of detergents (Tween 20, Triton X-100, digitonin, sarkosyl, CHAPS, cholate, deoxycholate, octyl glucoside, RIPA buffer, cetyl trimethylammonium bromide, SDS), proteases (trypsin, bacterial collagenase), nucleases (RNase, DNase), or glycosidases (heparinase III, chitinase) failed to affect or solubilize the PIB binding activity. This resistance is characteristic of amyloid fibrils and may also be explained by chemically modified, racemized, isomerized proteins and covalently associated (cross-linked) complexes, which are a prominent feature of the AD brain (Roher et al. 1993a, Roher et al. 1993b). Steric effects of SDS-resistant crosslinked proteins or tightly associated lipids or glycoconjugates may also play a stabilizing or screening role.
Since denaturants such as SDS, urea, and guanidine were ineffective at solubilizing or inactivating the PIB binding site, we resorted to more stringent methods used to extract insoluble Aβ amyloid from tissue. Treatment with high concentrations of formic acid (>50%), which are typically used to extract insoluble forms of Aβ from AD brain, solubilized the particulate fraction including the Aβ and destroyed 3H-PIB binding. Formic acid treatment of tissue sections also has been shown to abolish PIB binding (Ikonomovic et al. 2008). Importantly, similar concentrations of acetic acid had no effect on the solubility or amount of PIB binding recovered after washing the pellet with PBS. Pre-bound 3H-PIB was released by the concentrated formic acid. After 100,000 × g centrifugation for 1 hour, neutralization of the concentrated formic acid extract or dialysis against deionized water (1.5 kD cutoff tubing) produced protein aggregates that did not bind PIB. No PIB binding was detectable in the formic acid-insoluble pellet (primarily lipofuscin).
Components of the low buoyant density PIB binding site fraction of AD and normal human brain
The properties of the low buoyant density PIB-binding fraction resemble, but are not identical to, those previously reported for non-ionic detergent Triton X-100-extracted lipid raft-like material containing GM1 and cholesterol associated with a fraction of the total Aβ peptide (Yanagisawa et al. 1995, Yanagisawa & Ihara 1998, Lee et al. 1998). The ganglioside:Aβ complex (GAβ) of Yanagisawa specifically reacts with their monoclonal antibody 4396 raised against the GM1:Aβ protein:lipid complex (Yanagisawa et al. 1997). Aβ sequence-dependent epitopes in this complex are obscured by the lipid. Thin layer chromatography of the lipids extracted from the low buoyant density PIB binding fraction is consistent with a lipoprotein complex containing gangliosides, sphingomyelins, and cholesterol (Supporting Figure 3). Western blotting of our delipidated gradient fractions containing PIB binding fractions reveals Aβ but no caveolin or flotillin (data not shown).
Proteins associated with specialized regions of cellular membranes can experience a lipid environment distinct from the bilayer average. Many phospholipids, but not all lipid classes such as cholesterol and gangliosides, can be readily extracted by detergent treatment. Lipid domains and rafts defined by differential detergent solubility can be separated from each other and from non-lipidated proteins. The non-standard detergent extraction (anionic SDS vs. non-ionic TX-100) complicates direct comparison to established protein patterns found in rafts.
Structural requirements for ligand binding of the PIB binding site are preserved during purification
Of concern in isolating a ligand binding structure is that the molecular composition and/or configuration of the ligand binding site had been altered by the purification procedure. Displacement of 3H-BTA-1 binding to synthetic Aβ(1–40) fibrils by a series of substituted benzothiazole anilines (BTAs) was reported to produce parallel displacement curves of the radioligand in AD brain (Klunk et al. 2003b). We find (Supporting Figures 1 and 2, and Supporting Table 2) that the EC50’s of displacement of 3H-PIB by a series of BTA analogs at different stages of purification (AD brain frontal cortex 100,000 × g pellet, SDS-extracted PIB binding site, and the low buoyant density PIB binding site from the sucrose density gradient) are indistinguishable from one another and nearly coincident with the 1:1 reference line for their EC50’s. The ligand displacement curves are parallel and have a Hill plot slope of ~1.
High-affinity 3H-PIB binding to synthetic Aβ peptide fibrils is strikingly lower on a per mole peptide basis than to AD brain or the purified AD brain PIB binding site. One hundred ng of synthetic fibrils bind the same amount of 1.2 nM 3H-PIB as 0.15 ng Aβ in the AD brain-derived material. In addition, synthetic Aβ(1–40) and Aβ(1–42) fibrils showed an overall decrease in apparent affinity for the BTA analogs compared to the AD brain material, although the relative potencies of the analogs were maintained (Supporting Figure 2 and Supporting Table 2). The EC50 reference lines for the BTA analogs against synthetic peptides were displaced, but parallel to those from AD brain and the purified PIB binding site.
Immunoprecipitation of the purified PIB binding site with Aβ-specific antibodies indicates that Aβ peptide is a likely component
Figure 3 shows that a significant fraction of the PIB binding can be immunoprecipitated with the monoclonal Aβ antibody 4G8 (Aβ17–24) using magnetic Protein G-Dynabeads to capture the antibody-bound particulate PIB binding site. Interestingly, the monoclonal Aβ antibody 6E10 (Aβ3–8) is unable to immunoprecipitate the PIB binding site or the pre-formed 3H-PIB-binding site complex. Both Aβ epitopes are present in a 4.5 kDa m.w. band when the PIB binding site is subjected to SDS-PAGE and western blotting separately with the respective antibodies. No high m.w. bands corresponding to antibody cross-reactivity with full-length APP are present (Figure 4). The major band in lane 3 is likely an SDS-stable Aβ dimer (~9 kDa), although it could also be the C-terminal C83 fragment of APP. C99 (~10.7 kDa) was not observed. These observations suggest that either the N-terminal 6E10 (aa 3–8) epitope of Aβ in the PIB binding site is relatively inaccessible to antibodies as seen in the GAβ ganglioside:Aβ complex (Yanagisawa et al. 1995, Yanagisawa et al. 1997), even after extraction of lipids with hexane : isopropanol, or the epitope is modified/missing.
Figure 3.

PIB binding site is immunoprecipitated by anti-Aβ monoclonal antibody 4G8. 3H-PIB prebound to purified PIB binding site is depleted by incubation with anti-Aβ 4G8 (but not by pre-immune mouse IgG or anti-Aβ 6E10) followed by isolation of the immune complexes on Protein G-Dynabead magnetic beads. See Methods for details.
Figure 4.
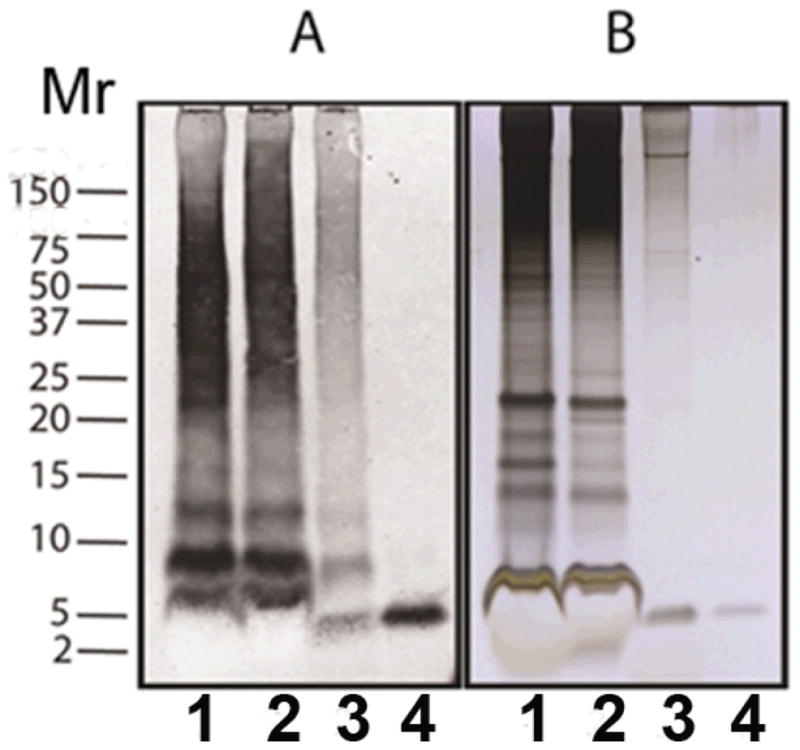
Silver stain and western blot analysis of PIB binding site purification. Duplicate samples, each representing 1.33 mg wet weight of AD brain tissue, were separated on a Novex 12% acrylamide gel in Mes buffer. The gel was cut, and one set of samples was transferred to a nitrocellulose membrane. Aβ was detected with a mixture of the monoclonal antibodies 6E10 and 4G8 (panel A); the other set of samples was stained with silver (panel B). See Methods for details. Lane (1) SDS-extracted brain homogenate 100 kG pellet; lane (2) SDS-extracted 15–100 kG pellet; lane (3) equilibrium buoyant density-purified PIB binding site; lane (4) rPeptide Aβ(1–40) (20 ng western blot, 60 ng silver stain).
Proteomic analysis of the AD brain PIB binding site
Since the PIB binding complex is relatively refractory to separation on SDS-PAGE gels, and epitope accessibility is potentially an issue, we pursued a mass spectrometric proteomics approach combining chemical cleavage under strongly dissociating conditions of 70% formic acid followed by proteolytic digestion to identify proteins in the complex. Equilibrium buoyant density sedimentation of the SDS-insoluble fractions of age-matched normal and AD brain reveals a similar distribution of total protein, including the low buoyant density protein fraction, but the normal brain lacks detectable Aβ or PIB binding (Figure 2). The aggregation state of the particulate floated PIB binding site hampers analysis due to its insolubility in SDS and chaotropes, protease resistance, and poor behavior in a variety of SDS-PAGE systems. Solubility of its components requires the continuous presence of concentrated formic acid, hampering further purification. We found that after CHCl3:CH3OH (C:M) extraction to remove lipids, combining cleavage of the delipidated proteins at methionine by CNBr in 70% formic acid provided soluble peptides after neutralization of the formic acid. Trypsinization of the solution gave peptides suitable for protein identification by mass spectroscopy. For details see Supporting Material.
Three separate floatation purifications of pooled AD and normal brain low buoyant density PIB binding sites were subjected to proteomic analysis. The results are summarized in Table 2. Detailed analysis of the mass spectral data is presented in Supporting Information (Supporting Tables 3 and 4, and Supporting Figures 4–21). Because of the potential for biologically processed or ragged ends of the isolated peptides, a no enzyme (no protease selected) search was conducted on all four samples and the results yielded no new relevant results (data not included). The AD brain fraction is distinguished by the presence of Aβ peptides and three known Aβ-binding and previously demonstrated plaque-associated proteins: ApoE, collagen XXVα1 (CLAC = collagen-like Alzheimer amyloid-associated plaque component), and the microtubule-associated protein tau as well as ubiquitin. The rest of the proteins detected were either common to both normal and AD brain samples or present only in the normal brain samples. The floatation purification reduced the number of detectable proteins identified in the AD brain PIB binding site from >110 in the PIB binding site before the density gradient to 17 in the floated PIB binding site. Only four of these 17 were found in the AD brain PIB binding site. No tubulin was found in the floated PIB binding site, suggesting that the microtubule cytoskeleton is not an integral component of the PIB binding site.
Table 2.
Proteins identified by mass spectrometry in the sucrose density gradient-purified PIB binding site.
| Protein | Sequences from pooled sample 1 | Sequences from pooled sample 2 |
|---|---|---|
| Aβ | LVFFAEDVGSNK DAEFRHDSGYEVHHQK* DAEFRHDSGYEVHHQKLVFFAEDVGSNK* |
LVFFAEDVGSNK* DAEFRHDSGYEVHHQK* |
| Collagen XXVα | INHGFLSADQQLIK | INHGFLSADQQLIK |
| Ubiquitin protein | TITLEVEPSDTIENVK | TITLEVEPSDTIENVK |
| Microtubule Associated protein-tau | IGSLDNITHVPGGGNK IGSLDNITHVPGGGNKK |
IGSLDNITHVPGGGNK IGSLDNITHVPGGGNKK |
| ApolipoproteinE | AKLEEQAQQIRLQAEAFQAR | GEVQAmLGQSTEELRδ |
Proteins found in 2 independently prepared samples of 6 pooled AD brains but not in 2 independently prepared samples of 3 pooled control brains (with the exception of Aβ, which was found in all samples). The ubiquitin sequence found in both sample preparations corresponds to 4 different ubiquitin proteins, and without further analysis it is uncertain which one or ones may be present in the AD samples.
Peptides appeared multiple times in the same sample. This was due to the same peptide having multiple precursor ions and fragment ions.
m designates an oxidized methionine
Discussion
We describe here the isolation and initial characterization of a highly purified proteolipid complex from human AD frontal cortex that is responsible for the high-affinity binding of the amyloid imaging ligand PIB in AD brain. This high-affinity 3H-PIB binding site is a distinct population of highly insoluble Aβ assemblies with low buoyant density, separable from other Aβ assemblies. This complex is undetectable in normal age-matched human frontal cortex.
Protein components of the low buoyant density purified PIB binding site
Proteomic analysis of the low buoyant density isolated PIB binding site identified a limited set of proteins (Table 2) that are among those known to increase AD genetic risk and to be associated with Aβ pathology in the AD brain. A recent genome-wide association study of 555 non-Hispanic Caucasian participants following a global cortical measure of Aβ deposition by 18F-florbetapir PET imaging identified the ApoE locus as a major contributor to the variance in PET signal (Ramanan et al. 2013). Since florbetapir and PIB compete for the same binding site in the AD brain (Ni et al. 2013), this observation is consistent with our finding that the fraction of Aβ that binds PIB with high affinity is associated with ApoE. Aβ peptide sequences encompassing residues 1 – 28 (1–16, 17–28, 1–28) were detected in the high-affinity PIB-binding fraction. CNBr cleavage in formic acid is required to produce and recover tryptic peptides from the insoluble complex. CNBr cleaves at methionine 671 in APP (770 aa isoform numbering) …SEVKMDAEF…. which is at the N-terminus of the Aβ peptide. Therefore we cannot distinguish between APP fragments and Aβ peptide produced by BACE cleavage of APP. However, we detect a distinct 4.5 kDa band on SDS-PAGE immunoreactive with both 6E10 and 4G8 (Figure 4, panel A), consistent with full- or near full-length Aβ peptide and no full-length APP. Peptides with the ApoE, Aβ consensus binding sequence (QQIRLQAEA) (Cho et al. 2001, Liu et al. 2011, Wisniewski et al. 1995, Chan et al. 1996) were recovered (Table 2) from the purified PIB binding site, consistent with finding the consensus Aβ(12–28) sequence that binds Apo E. Direct association of the apoE peptides with Aβ has not been determined. The C-terminal residues IGSLDNITHVPGGGNKK (354–370) of the human tau sequence of the 4th microtubule repeat domain (337–369) were recovered in the PIB binding site, which includes S356, a PKA/CAMKII consensus phosphorylation site in tau.
Collagen XXVα1 (CLAC = collagen-like Alzheimer amyloid-associated plaque component) is a type II membrane-bound and neuronal enriched protein precursor processed by furin to a 50 kDa monomer of triple-helical collagen. It is found in tight protease-resistant complex with Aβ(1–42), rarely Aβ(1–40), in a Thioflavin-S negative plaque population distinct from diffuse plaques in AD and older Down Syndrome brain (Kowa et al. 2004). Interestingly, immunoreactive CLAC is not found in complex with cerebrovascular amyloid, which is primarily Aβ(1–40), or in aged monkeys with abundant Aβ deposition, or in aged PDAPP transgenic mice with human-sequence Aβ and 90% amino acid identity of human to murine CLAC (Hashimoto et al. 2002, Lemere et al. 1999). A weak but reproducible genetic association between the COL25A1 gene and increased risk for AD on chromosome 4q25 has been identified in a Swedish population (Forsell et al. 2010). The MS sequence we identified is adjacent to the known cationic binding sequence (for heparin) within collagen XXVa1 in the COL1 region (Soderberg et al. 2004, Osada et al. 2005). Fibrillar Aβ, but not monomeric Aβ, is bound through anionic and aromatic residues in the N-terminal half of the peptide (Kakuyama et al. 2005). Collagen XXVα1 is believed to be deposited following fibrillization of Aβ(1–40) peptide (Soderberg et al. 2005). Transgenic overexpression of human collagen XXV in non-AD mice promotes AD-like pathology, including increased endogenous murine Aβ production and deposition as well as increased BACE1 and p35/p25 levels, and synaptic and behavioral consequences similar to AD (Tong et al. 2010).
Ubiquitin-C is also present in the isolated PIB binding site, although it is unclear whether it is free or covalently attached to other proteins. The presence and type of ubiquitin linkage are differentially coupled to cellular processes such as degradation (lysosome, proteasome, ERAD), cell cycle, DNA repair, protein kinase regulation, and endocytosis (Kulathu & Komander 2012), which could be useful in tracing the biology of the PIB binding site. While not surprising, the presence in the functional PIB binding site of these plaque-associated proteins suggests that the minimal complex for PIB binding contains Aβ and/or APP Aβ-region peptides and some or all of these additional proteins. High-affinity PIB binding is concentrated in this lipid-associated, low buoyant density subpopulation of insoluble Aβ assemblies.
Proteins of possible interest found in both the AD samples and the controls were brain soluble acidic protein, myristoylated alanine-containing C-Kinase substrate (MARCKS) and ferritin heavy chain. Interestingly, two proteins, S100-A9, an inflammatory signaling molecule, and synaptosomal-associated protein 25 (SNAP-25), a key element in rapid endocytosis/exocytosis processes, were found in 2 independently prepared samples of 3 pooled control brains that were missing from 2 independently prepared samples of 6 pooled AD brains. It is possible that these components are depleted as part of the pathological process in AD.
3H-PIB-Binding Aβ is only a fraction of total AD brain Aβ
Svedberg et al. (Svedberg et al. 2009) found that the amount of insoluble Aβ measured by immunoassay and by histology exceeded the 11C-PIB binding to AD brain homogenate. We find that around 65% of frontal cortical AD brain Aβ is SDS-insoluble, sediments at low centrifugal force, and accounts for only about 3.5% of the PIB binding. The remaining 96% of 3H-PIB binding is SDS-insoluble and resides in a low buoyant density lipid-containing fraction. This compartmentalization may reflect differential localization of aggregated Aβ in the tissue. Importantly, by quantifying in situ binding to tissue sections of several brain areas by autoradiography, Svedberg et al. noted that 11C-PIB binding is concentrated in certain brain laminae, and not in others. Similar results were found for 3H-PIB in AD brain (Marutle et al. 2013). PIB binding is not detected in the inner laminar layers of the cerebral cortex, while immunoreactive Aβ is clearly present in outer and inner layers. The authors comment that it might be of great importance to find out why PIB failed to recognize the Aβ accumulation in the inner cortical layers. Our ability to isolate a discrete population of Aβ that binds a high stoichiometry of PIB, approaching 1:1, is a reminder that not all insoluble Aβ in the brain is the same, which may account in part for the discrepancy in correlation of amyloid accumulation and disease progression. Possibly the Aβ pathology in the brain regions that have deposits but little PIB binding could be similar to the deposits in the natural and transgenic animal models, which are generally not associated with the profound neuronal death that characterizes AD (Jucker 2010, Rosen et al. 2011).
The purified PIB binding fraction in other brain regions of both male and female subjects with AD is concentrated in a low buoyant density of Aβ fraction. There is, however, variation among individuals, with some showing some higher buoyant density Aβ PIB binding. The detailed composition and whether this is reflective of the extent of pathology progression remain to be determined.
Some humans can also have less PIB binding and accumulate a larger fraction of Aβ pathology with low PIB-binding stoichiometry. We reported on a subject with clinical AD who had ten-fold higher Aβ concentrations than the normal Aβ load in the AD brain and no mutations in the APP, PS1, or PS2 genes (Rosen et al. 2010). We were unable to detect postmortem 3H-PIB binding in a filtration assay of temporal and occipital cortical tissue homogenates from this subject’s brain. One possible interpretation of this finding is that the form of Aβ deposits in this subject’s brain was relatively non-toxic and that it required the observed massive accumulation of Aβ to elicit the clinical symptoms that are observed with a lesser amount of a more pathogenic, high PIB-binding stoichiometry form of Aβ. A small number of low PIB binding AD cases with significant Aβ accumulation have also been reported by others (Cairns et al. 2009, Ikonomovic et al. 2012). These apparent exceptions could furnish useful clues to the factors that govern the pathogenicity of aggregated Aβ in AD. While Aβ is clearly associated with the PIB binding complex, there is also the possibility that uncharacterized components of the complex may account for pathogenicity.
Although the floated PIB binding site has similarities to the GAβ isolated from rodent brain (Yanagisawa et al. 1995, Yanagisawa & Ihara 1998, Lee et al. 1998), the neurodegenerative disease scenario in mouse models is clearly different. The differences between GAβ and the PIB binding site are likely more complex than simple lipid-protein interaction, otherwise mice that develop human-sequence Aβ pathology would have high PIB binding. The amount of Aβ and GAβ complex in the human brain is reported to increase slightly with normal aging, but dramatically so with AD progression. A nonionic detergent-resistant membrane fraction with similar properties containing insoluble Aβ has also been observed in Down Syndrome brain and PDAPP mouse brain with Aβ pathology (Oshima et al. 2001), as well as in aged nonhuman primates with Aβ pathology (Hayashi et al. 2004). Only in human brain with AD or Down Syndrome is significant high-affinity PIB binding observed (Handen et al. 2012, Landt et al. 2011). PIB binding of the immune-isolated published GAβ complexes has not been reported. We find that the low buoyant density raft-like fraction isolated by the PIB binding site method from hamster brain does not bind PIB (not shown), nor does the low buoyant density raft-like fraction from normal age-matched human brain (this study). Our prediction would be that any detectable PIB binding of antibody 4396 immuno-isolated murine GAβ complexes would have low PIB/Aβ stoichiometry. We have shown low PIB binding for nonhuman primates (Rosen et al. 2011). Fast et al. found low PIB/Aβ stoichiometry for canines (Fast et al. 2013), as we (data not shown) and the Klunk lab (Klunk et al. 2005) have reported for APP transgenic mice.
The conformation of amyloid fibrils spontaneously formed from synthetic Aβ peptide can be influenced by fibril assembly conditions (Petkova et al. 2005), and also can be specified by the nature of a seed fibril that nucleates fibril growth from monomeric peptide (Paravastu et al. 2008, Paravastu et al. 2009, Kodali et al. 2010, Lu et al. 2013). A different complement of proteins, peptides, and/or lipids in the mouse models is a potential explanation for the human:mouse differences in Aβ PIB binding. In support of this possibility, published comparative lipidomics reveal striking differences in the changes in the lipid profiles comparing control and AD pathology conditions of three transgenic AD mouse models and human AD brain (Chan et al. 2012).
The mechanism inducing PIB binding in Aβ in the AD brain likely involves more than simple molecular composition. Addition of the lipid components extracted from the AD brain PIB binding site to pre-formed synthetic fibrils failed to produce high stoichiometry PIB binding, leaving open the interpretation that there is some additional factor or environmental constraint such as scaffolding associated with the human disease process. Raft lipid component association with human extracellular amyloid fibrils of different proteins other than Aβ is selective and is not recapitulated by mixing preformed fibrils with lipids (Gellermann et al. 2005). Intracellular neurofibrillary tangles of microtubule-associated tau protein are also associated with a similar pattern of specific lipids (Gellermann et al. 2006). Human-specific biology in the immediate environment of the high stoichiometry PIB binding sites may lead to an Aβ fibril form associated with AD but is not present or only in limited amount in animal models. Association of PIB binding with the presence of Aβ and the parallel but displaced affinity of PIB analogs for human AD and synthetic Aβ fibrils supports an Aβ conformational subset with high affinity for PIB. This could potentially result from details in the initial association of Aβ peptide with certain lipid components or the presence of a specific protein component or modification(s) of the Aβ peptide that enables high-stoichiometry, high-affinity PIB binding. Teasing out the details could be complex because the immediate initiating factor may no longer be present after decades in the AD brain. Answering these questions may lead to new diagnostics and therapeutic approaches that address disease progression.
Imaging with the 11C-PIB and other PET amyloid ligands is now proposed as a criterion for a presymptomatic stage of Alzheimer’s disease in which Aβ pathology is detectable by PIB long before the clinical signs of cognitive deficits are detectable (Dubois et al. 2010, Sperling et al. 2011). A hypothesis suggesting the staging of biomarkers (Jack et al. 2010) was supported by a similar analysis based on familial AD cases, in which the age of symptom onset is more predictable than in idiopathic AD (Bateman et al. 2012). Further support from a prospective cohort study (Villemagne et al. 2013) on amyloid accumulation, and revamping of the hypothesis to better account for tau pathology (Jack et al. 2013), provides a time- rather than symptom-based sequential ordering of biomarker changes. This revision of the more than twenty year-old criteria for clinical evidence of memory dysfunction to permit a diagnosis of possible or probable AD has occasioned much debate among clinicians and scientists over whether AD should be defined as a disease of clinical dysfunction or of the presence of pathology. Reliance on an amyloid imaging measurement for early disease detection makes it imperative to understand the formation and relationship of different polymorphic forms of Aβ in the AD brain to different stages of the disease process, as well as to the particular Aβ imaging ligand employed.
Supplementary Material
Acknowledgments
This work was made possible by a generous grant from the Coins for Alzheimer’s Research Trust (C.A.R.T.) to H. L. and L. C. W. by the Rotary Clubs of North Carolina, South Carolina, and Georgia and by an unrestricted Grants4Targets grant from Bayer Healthcare to H. L. A University of Kentucky Research Support Grant funded custom synthesis of 3H-PIB in the initial stages of this work. Additional 3H-PIB from Vitrax was a kind gift of Dr. Brian Ciliax, Emory University. Thanks to Dr. William Klunk for the list of substituted benzothiazole anilines (BTAs). AD brain tissue was provided by the Alzheimer’s Disease Center at the University of Kentucky. Research reported in this publication was supported by the National Institute on Aging of the National Institutes of Health under awards P30AG028383 and P50AG025688, the National Institute of Neurological Disorders and Stroke R21 NS080576 to H.L. and H.P.S., National Institute of Aging R21AG040589 to L.C.W., and the National Center for Research Resources P20RR020171, P51RR165, and P51OD11132.
We acknowledge the assistance of Tina Beckett for Aβ determinations and Drs. Jeffrey Rush and Charles Waechter for expertise and equipment for lipid analysis. We also thank Linda Van Eldik, Ph.D. Sanders-Brown Director, Peter Nelson, M.D., Ph.D., Neuropathology Core Director, and Sonya Anderson, Brain Bank Coordinator for human brain tissue and their contributions on behalf of the ADC. We are forever in debt to the patients whose brain donations made this investigation possible.
ABBREVIATIONS
- Aβ
amyloid β-peptide
- PIB
Pittsburgh Compound B
Footnotes
The authors declare no conflicts of interest.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Contributor Information
Sergey V. Matveev, Email: sergey.matveev@uky.edu.
H. Peter Spielmann, Email: hps@uky.edu.
Brittney M. Metts, Email: brittney.metts@uky.edu.
Jing Chen, Email: jchen4@email.uky.edu.
Fredrick Onono, Email: fred.onono@uky.edu.
Haining Zhu, Email: haining@uky.edu.
Stephen W. Scheff, Email: sscheff@email.uky.edu.
Lary C. Walker, Email: lcwalke@emory.edu.
Harry LeVine, III, Email: hlevine@email.uky.edu.
References
- Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett TL, Webb RL, Niedowicz DM, Holler CJ, Matveev S, Baig I, Levine H, III, Keller JN, Murphy MP. Postmortem Pittsburgh Compound B (PiB) Binding Increases with Alzheimer’s Disease Progression. J Alzheimers Dis. 2012;32:128–137. doi: 10.3233/JAD-2012-120655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cairns NJ, Ikonomovic MD, Benzinger T, et al. Absence of PIttsburgh Compound B Detection of CerebralAmyloid Beta in a Patient With Clinical, Cognitive, and Cerebrospinal FluidMarkers of Alzheimer Disease. Arch Neurol. 2009;66:1557–1562. doi: 10.1001/archneurol.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, Wenk MR, Shui G, Di Paolo G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem. 2012;287:2678–2688. doi: 10.1074/jbc.M111.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan W, Fornwald J, Brawner M, Wetzel R. Native complex formation between apolipoprotein E isoforms and the Alzheimer’s disease peptide A beta. Biochemistry. 1996;35:7123–7130. doi: 10.1021/bi952852v. [DOI] [PubMed] [Google Scholar]
- Cho HS, Hyman BT, Greenberg SM, Rebeck GW. Quantitation of apoE domains in Alzheimer disease brain suggests a role for apoE in Abeta aggregation. J Neuropathol Exp Neurol. 2001;60:342–349. doi: 10.1093/jnen/60.4.342. [DOI] [PubMed] [Google Scholar]
- Cohen AD, Rabinovici GD, Mathis CA, Jagust WJ, Klunk WE, Ikonomovic MD. Using Pittsburgh Compound B for in vivo PET imaging of fibrillar amyloid-beta. Adv Pharmacol. 2012;64:27–81. doi: 10.1016/B978-0-12-394816-8.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9:1118–1127. doi: 10.1016/S1474-4422(10)70223-4. [DOI] [PubMed] [Google Scholar]
- Fast R, Rodell A, Gjedde A, Mouridsen K, Alstrup AK, Bjarkam CR, West MJ, Berendt M, Moller A. PiB Fails to Map Amyloid Deposits in Cerebral Cortex of Aged Dogs with Canine Cognitive Dysfunction. Front Aging Neurosci. 2013;5:99. doi: 10.3389/fnagi.2013.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Forsell C, Bjork BF, Lilius L, Axelman K, Fabre SF, Fratiglioni L, Winblad B, Graff C. Genetic association to the amyloid plaque associated protein gene COL25A1 in Alzheimer’s disease. Neurobiol Aging. 2010;31:409–415. doi: 10.1016/j.neurobiolaging.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Gellermann GP, Appel TR, Davies P, Diekmann S. Paired helical filaments contain small amounts of cholesterol, phosphatidylcholine and sphingolipids. Biol Chem. 2006;387:1267–1274. doi: 10.1515/BC.2006.157. [DOI] [PubMed] [Google Scholar]
- Gellermann GP, Appel TR, Tannert A, et al. Raft lipids as common components of human extracellular amyloid fibrils. Proc Natl Acad Sci U S A. 2005;102:6297–6302. doi: 10.1073/pnas.0407035102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handen BL, Cohen AD, Channamalappa U, Bulova P, Cannon SA, Cohen WI, Mathis CA, Price JC, Klunk WE. Imaging brain amyloid in nondemented young adults with Down syndrome using Pittsburgh compound B. Alzheimers Dement. 2012;8:496–501. doi: 10.1016/j.jalz.2011.09.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Wakabayashi T, Watanabe A, et al. CLAC: a novel Alzheimer amyloid plaque component derived from a transmembrane precursor, CLAC-P/collagen type XXV. EMBO J. 2002;21:1524–1534. doi: 10.1093/emboj/21.7.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Kimura N, Yamaguchi H, et al. A seed for Alzheimer amyloid in the brain. J Neurosci. 2004;24:4894–4902. doi: 10.1523/JNEUROSCI.0861-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Abrahamson EE, Price JC, et al. Early AD pathology in a [C-11]PiB-negative case: a PiB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathol. 2012;123:433–447. doi: 10.1007/s00401-012-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PIB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M. The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med. 2010;16:1210–1214. doi: 10.1038/nm.2224. [DOI] [PubMed] [Google Scholar]
- Kakuyama H, Soderberg L, Horigome K, Winblad B, Dahlqvist C, Naslund J, Tjernberg LO. CLAC Binds to Aggregated Abeta and Abeta Fragments, and Attenuates Fibril Elongation. Biochemistry. 2005;44:15602–15609. doi: 10.1021/bi051263e. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, et al. Imaging the pathology of Alzheimer’s disease: amyloid-imaging with positron emission tomography. Neuroimaging Clin N Am. 2003a;13:781–789. ix. doi: 10.1016/s1052-5149(03)00092-3. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Lopresti BJ, Ikonomovic MD, et al. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. J Neurosci. 2005;25:10598–10606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Wang Y, Huang GF, et al. The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci. 2003b;23:2086–2092. doi: 10.1523/JNEUROSCI.23-06-02086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodali R, Williams AD, Chemuru S, Wetzel R. Abeta(1–40) forms five distinct amyloid structures whose beta-sheet contents and fibril stabilities are correlated. J Mol Biol. 2010;401:503–517. doi: 10.1016/j.jmb.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowa H, Sakakura T, Matsuura Y, Wakabayashi T, Mann DM, Duff K, Tsuji S, Hashimoto T, Iwatsubo T. Mostly Separate Distributions of CLAC- versus Abeta40- or Thioflavin S-Reactivities in Senile Plaques Reveal Two Distinct Subpopulations of beta-Amyloid Deposits. Am J Pathol. 2004;165:273–281. doi: 10.1016/s0002-9440(10)63295-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulathu Y, Komander D. Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13:508–523. doi: 10.1038/nrm3394. [DOI] [PubMed] [Google Scholar]
- Landt J, D’Abrera JC, Holland AJ, et al. Using Positron Emission Tomography and Carbon 11-Labeled Pittsburgh Compound B to Image Brain Fibrillar {beta}-Amyloid in Adults With Down Syndrome: Safety, Acceptability, and Feasibility. Arch Neurol. 2011;68:890–896. doi: 10.1001/archneurol.2011.36. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT, Jr, Kosik KS. A detergent-insoluble membrane compartment contains A beta in vivo. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Grenfell TJ, Selkoe DJ. The AMY antigen co-occurs with abeta and follows its deposition in the amyloid plaques of Alzheimer’s disease and down syndrome. Am J Pathol. 1999;155:29–37. doi: 10.1016/s0002-9440(10)65095-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wu WH, Fang CL, et al. Mapping ApoE/Abeta binding regions to guide inhibitor discovery. Mol Biosyst. 2011;7:1693–1700. doi: 10.1039/c1mb05019b. [DOI] [PubMed] [Google Scholar]
- Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Molecular Structure of beta-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marutle A, Gillberg PG, Bergfors A, Yu W, Ni R, Nennesmo I, Voytenko L, Nordberg A. 3H-Deprenyl and 3H-PIB autoradiography show different laminar distributions of astroglia and fibrillar beta-amyloid in Alzheimer brain. J Neuroinflammation. 2013;10:90. doi: 10.1186/1742-2094-10-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni R, Gillberg PG, Bergfors A, Marutle A, Nordberg A. Amyloid tracers detect multiple binding sites in Alzheimer’s disease brain tissue. Brain. 2013;136:2217–2227. doi: 10.1093/brain/awt142. [DOI] [PubMed] [Google Scholar]
- O’Nuallain B, Williams AD, Westermark P, Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem. 2004;279:17490–17499. doi: 10.1074/jbc.M311300200. [DOI] [PubMed] [Google Scholar]
- Osada Y, Hashimoto T, Nishimura A, Matsuo Y, Wakabayashi T, Iwatsubo T. CLAC binds to amyloid beta peptides through the positively charged amino acid cluster within the collagenous domain 1 and inhibits formation of amyloid fibrils. J Biol Chem. 2005;280:8596–8605. doi: 10.1074/jbc.M413340200. [DOI] [PubMed] [Google Scholar]
- Oshima N, Morishima Kawashima M, Yamaguchi H, Yoshimura M, Sugihara S, Khan K, Games D, Schenk D, Ihara Y. Accumulation of amyloid beta-protein in the low-density membrane domain accurately reflects the extent of beta-amyloid deposition in the brain. Am J Pathol. 2001;158:2209–2218. doi: 10.1016/s0002-9440(10)64693-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci U S A. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R. Seeded growth of beta-amyloid fibrils from Alzheimer’s brain-derived fibrils produces a distinct fibril structure. Proc Natl Acad Sci U S A. 2009;106:7443–7448. doi: 10.1073/pnas.0812033106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- Ramanan VK, Risacher SL, Nho K, et al. APOE and BCHE as modulators of cerebral amyloid deposition: a florbetapir PET genome-wide association study. Mol Psychiatry. 2013 doi: 10.1038/mp.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roher AE, Kuo YM. Isolation of amyloid deposits from brain. Methods Enzymol. 1999;309:58–67. doi: 10.1016/s0076-6879(99)09006-0. [DOI] [PubMed] [Google Scholar]
- Roher AE, Lowenson JD, Clarke S, et al. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J Biol Chem. 1993a;268:3072–3083. [PubMed] [Google Scholar]
- Roher AE, Palmer KC, Yurewicz EC, Ball MJ, Greenberg BD. Morphological and biochemical analyses of amyloid plaque core proteins purified from Alzheimer disease brain tissue. J Neurochem. 1993b;61:1916–1926. doi: 10.1111/j.1471-4159.1993.tb09834.x. [DOI] [PubMed] [Google Scholar]
- Rosen RF, Ciliax BJ, Wingo TS, Gearing M, Dooyema J, Lah JJ, Ghiso JA, LeVine H, 3rd, Walker LC. Deficient high-affinity binding of Pittsburgh compound B in a case of Alzheimer’s disease. Acta Neuropathol. 2010;119:221–233. doi: 10.1007/s00401-009-0583-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen RF, Walker LC, LeVine H., 3rd PIB binding in aged primate brain: Enrichment of high-affinity sites in humans with Alzheimer’s disease. Neurobiol Aging. 2011;32:223–234. doi: 10.1016/j.neurobiolaging.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snellman A, Lopez-Picon FR, Rokka J, Salmona M, Forloni G, Scheinin M, Solin O, Rinne JO, Haaparanta-Solin M. Longitudinal Amyloid Imaging in Mouse Brain with 11C-PIB: Comparison of APP23, Tg2576, and APPswe-PS1dE9 Mouse Models of Alzheimer Disease. J Nucl Med. 2013;54:1434–1441. doi: 10.2967/jnumed.112.110163. [DOI] [PubMed] [Google Scholar]
- Soderberg L, Dahlqvist C, Kakuyama H, Thyberg J, Ito A, Winblad B, Naslund J, Tjernberg LO. Collagenous Alzheimer amyloid plaque component assembles amyloid fibrils into protease resistant aggregates. Febs J. 2005;272:2231–2236. doi: 10.1111/j.1742-4658.2005.04647.x. [DOI] [PubMed] [Google Scholar]
- Soderberg L, Kakuyama H, Moller A, Ito A, Winblad B, Tjernberg L, Naslund J. Characterization of the Alzheimer’s disease-associated CLAC protein and identification of an Abeta binding site. J Biol Chem. 2004;280:1007–1015. doi: 10.1074/jbc.M403628200. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedberg MM, Hall H, Hellstrom-Lindahl E, Estrada S, Guan Z, Nordberg A, Langstrom B. [(11)C]PIB-amyloid binding and levels of Abeta40 and Abeta42 in postmortem brain tissue from Alzheimer patients. Neurochem Int. 2009;54:347–357. doi: 10.1016/j.neuint.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Tong Y, Xu Y, Scearce-Levie K, Ptacek LJ, Fu YH. COL25A1 triggers and promotes Alzheimer’s disease-like pathology in vivo. Neurogenetics. 2010;11:41–52. doi: 10.1007/s10048-009-0201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama H, Ye D, Ichise M, et al. PET imaging of brain with the beta-amyloid probe, [(11)C]6-OH-BTA-1, in a transgenic mouse model of Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2005;32:593–600. doi: 10.1007/s00259-005-1780-5. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12:357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Lalowski M, Golabek A, Vogel T, Frangione B. Is Alzheimer’s disease an apolipoprotein E amyloidosis? Lancet. 1995;345:956–958. doi: 10.1016/s0140-6736(95)90701-7. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K, Ihara Y. GM1 ganglioside-bound amyloid beta-protein in Alzheimer’s disease brain. Neurobiol Aging. 1998;19:S65–67. doi: 10.1016/s0197-4580(98)00032-3. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K, McLaurin J, Michikawa M, Chakrabartty A, Ihara Y. Amyloid beta-protein (A beta) associated with lipid molecules: immunoreactivity distinct from that of soluble A beta. FEBS Lett. 1997;420:43–46. doi: 10.1016/s0014-5793(97)01484-1. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K, Odaka A, Suzuki N, Ihara Y. GM1 ganglioside-bound amyloid beta-protein (A beta): a possible form of preamyloid in Alzheimer’s disease. Nat Med. 1995;1:1062–1066. doi: 10.1038/nm1095-1062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.