

Abstract
The 22q11.2 deletion syndrome (22q11.2DS) is a common microdeletion disorder. Most of the patients show the common 3 Mb deletion but proximal 1.5 Mb deletion and unusual deletions located outside the common deleted region, have been detected particularly with the advance of comparative cytogenomic microarray technologies. The individuals reported in the literature with unusual deletions involving the 22q11 region, showed milder facial phenotypes, decreased incidence of cardiac anomalies and intellectual disability. We describe two sibs with an atypical 0.8 Mb microdeletion of chromosome 22q11 who both showed myelomeningocele and mild facial dysmorphisms. The association between neural tube defect and the clinical diagnosis of Di George anomaly/velocardiofacial syndrome is well documented in the literature, but not all cases had molecular studies to determine breakpoint regions. This report helps to narrow a potential critical region for neural tube defects associated with 22q11 deletions.
Keywords: Myelomeningocele, del22q11, FISH, neural tube defects, Kousseff syndrome, velocardiofacial syndrome, DiGeorge anomaly
Introduction
The 22q11.2 deletion syndrome (22q11.2DS) is the most common microdeletion disorder with an estimated prevalence of 1 in 3000-6000 live births [Tezenas et al., 1996; Goodship et al., 1998]. The majority of individuals with 22q11.2DS have the “common” ≈3Mb deletion, and a smaller percentage of individuals exhibit a smaller “proximal” 1.5 Mb deletion (see Fig. 1). Both the “common” and “proximal” deletions result from nonallelic homologous recombination utilizing low copy number repeats (LCRs) sequences as substrates for recombination [Shaikh et al., 2000; McDonald-McGinn et al., 2011], and can be de novo as well as familial [McDonald-McGinn et al., 2001]. A subset of individuals harbors a variety of deletions, not usually detected by classical fluorescent in situ hybridization technology (FISH), that can be identified by comparative cytogenomic microarrays. These include individuals with “distal deletions”, located outside and distally to the common 3Mb deletion region or other “atypical deletions” with unusual breakpoints [Kurahashi et al., 1997; Saitta et al., 1999; Garcia-Miñaur et al., 2002; Rauch et al., 2005; Shaikh et al., 2007; Ben-Sachar et al., 2008; Rødningen et al., 2008; Garavelli et al., 2011;Verhagen et al., 2012].
Fig. 1.
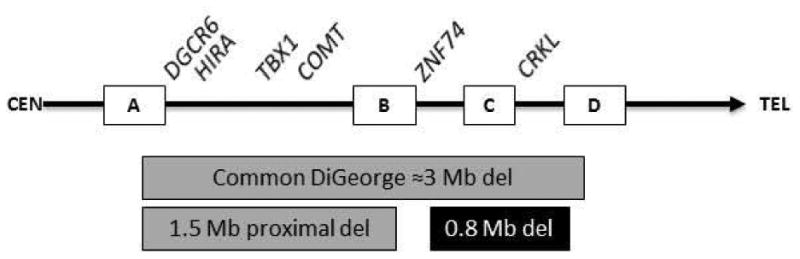
Schematic of the 22q11 chromosome with centromeric (CEN) region on the left and telomeric (TEL) region on the right. Examples of genes in the respective regions are listed at the top of the figure. The squares along the chromosome represent regions of low copy repeats. The typical 3Mb “classic” deletion (del) between low copy repeat regions “A” and “D”, and the smaller “proximal” 1.5Mb deletion between low copy repeat regions “A” and “B” (gray boxes) according to Shaikh et al. [2000]. Examples of genes are listed at the top of the figure. Representative interval of the ≈0.8Mb deletion of the herein reported two sibs with myelomeningoceles (black box). Adapted from Garavelli et al., 2011; Rauch et al., 2005; Shaikh et al., 2000.
Individuals with 22q11.2DS can show a broad spectrum of phenotypic abnormalities [Lindsay et al., 2001; Vantrappen et l., 2001; Greenhalgh et al., 2003; Oskarsdóttir et al., 2005] ranging from a combination of mild facial dysmorphisms, learning disabilities, cardiovascular malformations (CVM), genitourinary anomalies, immunologic abnormalities, palatal anomalies, and variable degrees of developmental delay. It is suggested that individuals with distal or atypical deletions excluding TBX1 have milder phenotypes with a decreased incidence of CVM and cognitive impairment [Verhagen et al., 2012; Rauch et al., 2005; Ryan et al., 1997, McDonald-McGinn et al. 2013]. Central nervous system (CNS) abnormalities have also been reported in 22q11.2DS and include neural tube defects (NTD) such as myelomeningocele [Botto et al., 2003]. The association between conotruncal heart defects and sacral meningoceles was first described by Kousseff in 1984. Kousseff [1984] reported three of four sibs, all with sacral meningoceles and mild facial dysmorphisms; the first child had transposition of great vessels and the second sib had truncus arteriosus type I, and both died soon after birth due to CVM. All three affected sibs had normal karyotypes [Kousseff 1984]. Subsequently in 2002, the third child previously reported by Kousseff [1984] was found to have a deletion on 22q11 by FISH [Forrester et al., 2002]. A few other reports of myelomeningocele in individuals with 22q11.2DS have been reported in detail in the medical literature [Toriello et al., 1985; Palacios et al., 1993; Nickel et al., 1994; Nickel and Magenis, 1996; Seller et al., 2002; Maclean et al., 2004; Kinoshita et al., 2010; Canda et al., 2012; McDonald-McGinn et al., 2013].
We report on two sibs with myelomeningocele and a maternally inherited “atypical deletion” on chromosome 22q11 (0.8 Mb) identified by oligonucleotide microarrays.
Methods
The proband was initially tested using an Agilent 44K customized oligonucleotide microarray representing a consensus design through an academic consortium organized by Emory Genetics Laboratory (Baldwin et al. Genet Med 10:415-429, 2008). The proband's sib was tested using the Affymetrix CytoScan HD Array® (1,953,246 oligonucleotides and 743,304 SNP probes). The proband, the proband's sib, both parents, and both maternal grandparents were also tested using fluorescence in situ hybridization (FISH) with the RP11-505B16 probe from Empire Genomics (Buffalo, NY). For the FISH experiments, the proband and the proband's sib served as positive controls, and a pooled sample collected from normal males served as the negative control.
Whole exome sequencing was performed on DNA from the sister of the proband using the methods described in Wooderchak-Donahue et al. [2013] with the following alterations: the Agilent SureSelectXT Human All Exon V4 kit (Agilent) was used on the automated library generation Bravo instrument option a (Agilent), and sequencing was performed on the HiSeq™ 2500 instrument (Illumina). Sequence wasaligned to the human reference genome (GRCh37 or Hg19) using Burrows-Wheeler Aligner (0.5.11) and variants from the reference were called with Samtools and Genome Analysis Toolkit (v.1.6). After the first alignment, a second refined alignment was done which removed PCR duplicate reads, identified read bias, and realigned around deletions and insertions. Variant lists were in vcf file format or csv format with Annovar annotation (http://www.openbioinformatics.org/annovar/). Variants with a quality score <10 and >0.1% population frequency (based on 1,000 Genomes data and an internal database of previously exome sequenced samples) were excluded from further analysis for potential dominant conditions, while variants with >5% population frequency were excluded for potential recessive conditions. Variants identified by exome sequencing were evaluated based on their predicted pathogenicity, zygosity, and phenotypic relevance to NTDs. Computational analysis of missense variants was performed using SIFT [Kumar et al. 2009] and Polyphen2 [Adzhubei et al. 2010]. Nomenclature for the AIFM3 variant is based on transcript NM_001018060.
Informed consent was obtained on all participants using an Institutional Review Board approved protocol from the University of Utah.
Results
Clinical Reports
Proband
The proband was the first child of healthy parents, born at 30 weeks of gestation by cesarean due to fetal distress. The pregnancy was remarkable for oligohydramnios, decreased fetal movements, maternal migraine and tachycardia treated with acetaminophen and a beta blocker. Ultrasonography at 20 weeks gestation showed a myelomeningocele. The mother reported taking prenatal vitamins during the pregnancy. Growth parameters at birth were: weight = 1060 g (10-25th centile), length = 37 cm (10-25th centile), and head circumference = 4.5 cm (3-10th centile). After birth, the infant had transient hypoglycemia treated by intravenous dextrose infusion. A magnetic resonance image (MRI) of the spine confirmed a myelomeningocele at the lumbosacral junction with tethered cord that required surgical intervention the day after birth. An MRI of the brain showed callosal dysgenesis, colpocephaly, partial absence of the septum pellucidum, but no signs of a Chiari malformation. An echocardiogram showed a patent foramen ovale and patent ductus arteriosus, but no other major cardiovascular malformations. During neonatal period she had periodic breathing with apnea of prematurity and gastro-esophageal reflux.
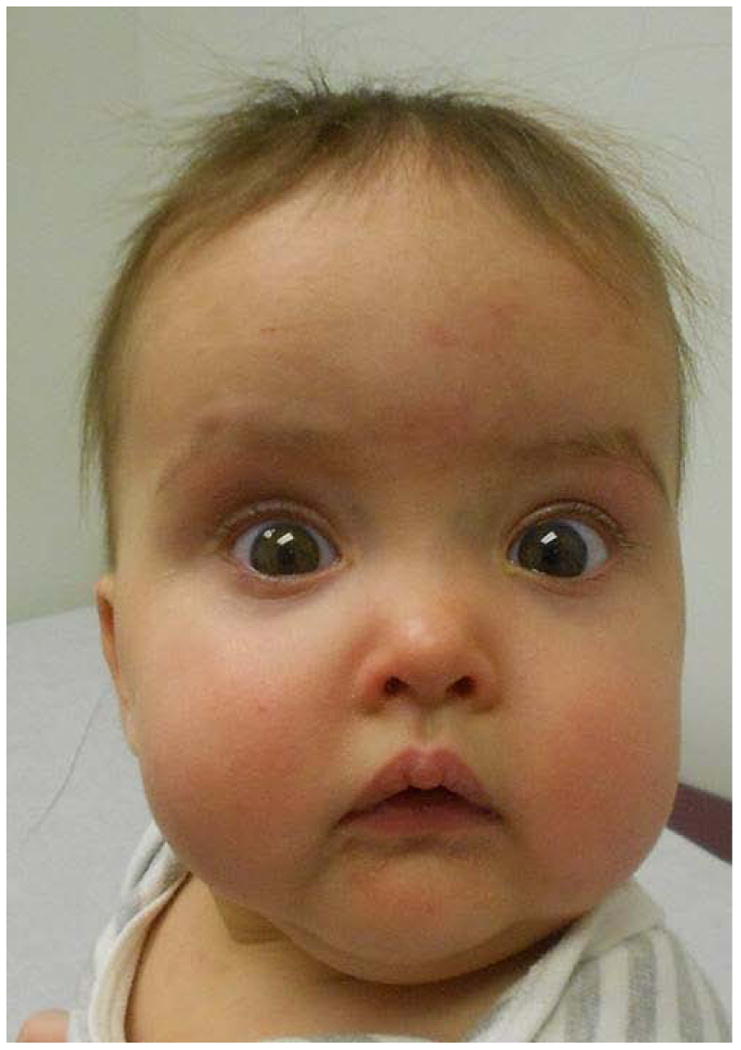
Clinical follow up evaluation at 7 months of age showed the following growth parameter centiles: head circumference at ≈90th centile, weight at ≈5th centile, and length at ≈50th centile. The child had a few nonspecific facial minor anomalies (e.g. prominent forehead, hypertelorism, telecanthus) (Fig. 2), a small cutaneous vascular malformation of the anterior neck, a right single palmar crease, normal development, a neurogenic bladder secondary to the myelomeningocele, and mild caliectasis of collecting system.
Fig. 2.

Face of proband with atypical distal deletion on 22q11.2.
Sib
The sister of the proband was born at 37 weeks gestation by repeat cesarean. The mother received a multivitamin, and folic acid 4 mg/day through the 14th week of pregnancy. Prenatal ultrasonography detected a myelomeningocele at 21 weeks gestation. Growth parameters at birth were small for gestational age: weight = 2071g, length = 44 cm, head circumference = 31.5 cm. MRI of the brain showed a Chiari type II malformation, posterior thinning of the corpus callosum, with mild enlargement of the lateral ventricles. A renal ultrasound was normal. An echocardiogram showed an aneurysm of the septum primum flap that bowed bidirectionally, but was otherwise unremarkable. A lumbosacral myelomeningocele (≈3×5cm) was noted and was repaired surgically after birth. She developed hydrocephalus after repair of the myelomeningocele that decreased after endoscopic third ventriculostomy and choroid plexus coagulation.
At 8 months of age she had normal weight (10th centile) and head circumference (50th centile), but length was <2nd centile. On examination, nonspecific facial minor anomalies (Fig. 3) (eg. depressed nasal root, anteverted nares, telecanthus) were noted. She had delayed gross motor development with weakness in the lower extremities. In addition, examination of the lower extremities showed dorsal lymphedema of the feet, dimpling at the knees, a vertical crease on left anterior lower leg, and the appearance of decreased muscle mass in lower limbs compared to the rest of body. As a consequence of the myelomeningocele, she had a neurogenic bowel and bladder with need for intermittent catheterization. She had a left supranumerary nipple, and a large irregular capillary malformation surrounding the region of the repaired myelomeningocele (Fig. 4).
Fig. 3.
Face of affected sib with atypical distal deletion on 22q11.2.
Fig. 4.
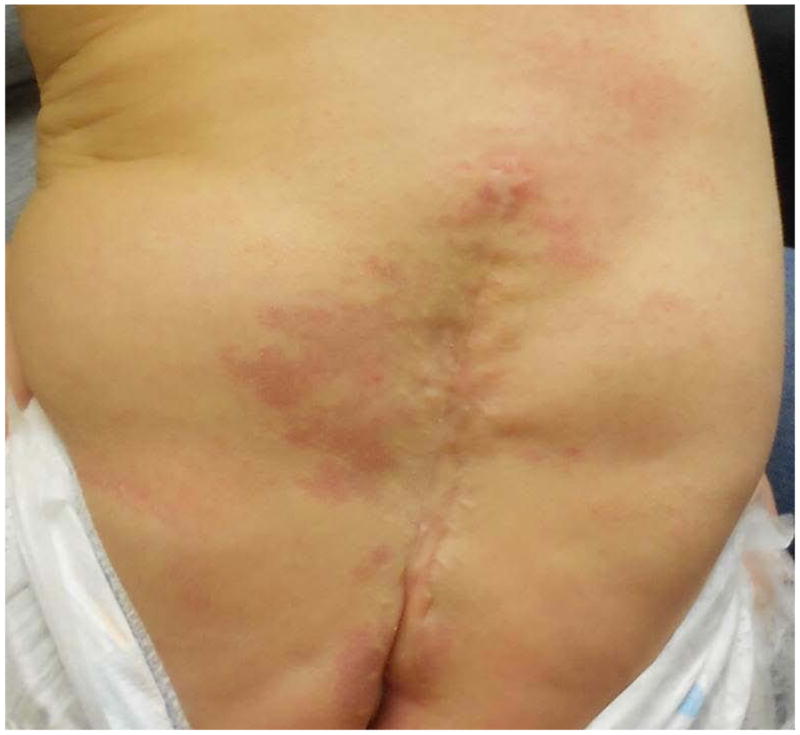
Photograph of the back of the affected sib of the proband, showing repaired myelomeningocele and surrounding capillary malformation.
Molecular/Cytogenetic results
Karyotype of the proband at the 550 band level was normal (46,XX). The cytogenomic microarray analysis of the proband from peripheral blood showed an atypical microdeletion on chromosome 22q11 (≈751 kb in size), distal to TBX1 between LCR B and LCR D (Fig. 1) [arr 22q11.21 (20,754,222-21,505,558×1)(hg19)]. This deletion involved 12 genes listed in the Online Mendelian Inheritance in Man (OMIM) (www.omim.org) (ZNF74, SCARF2, MED15, PI4KA, SERPIND1, SNAP29, CRKL, LZTR1, THAP7, P2RX6, SLC7A4, BCRP2), and 8 other genes (KLHL22, POM12L4P, TMEM191A, AIFM3, FLJ39582, MCG16703, P2RX6P, LOC400891). The proband's sib had the atypical deletion on 22q11 (≈748 kb in size) using the Affymetrix CytoScan array [ISCN: arr 22q11.21 (20,716,876-21,465,661×1)(hg19)]. The variation in deletion sizes in the sibs likely differed due to differences in probe coverage between the two comparative microarray platforms. Molecular FISH on interphase and metaphase cells from peripheral blood showed a deletion of one copy of the RP11-505B16 probe locus in the proband, the proband's sib, and in the proband's mother; neither the father nor the mother's parents showed a deletion by FISH. Targeted array analysis showed that the mother carried the same atypical 22q11 deletion. The mother denied any significant medical problems. Maternal grandparents did not have the same atypical 22q11 deletion and hence the deletion was de novo in the mother.
In order to exclude the possibility that a mutation within the deleted region, or a second genetic condition may account for the NTDs, whole exome sequencing was performed on the sib of the proband. Genes residing within the previously identified chromosomal deletion were specifically scrutinized for rare coding variants or variants located +/- 10 base pairs into flanking introns. Whole exome sequencing did not identify any genetic variations that are likely to cause NTDs. A single rare hemizygous variant, c.1021G>A;p.Gly341Ser, was identified in the apoptosis-inducing factor, mitochondrion-associated 3 (AIFM3) gene. AIFM3 is a poorly studied gene that has been shown to induce apoptosis in response to cellular stress [Yang et al. 2012]. At this time AIFM3 has not previously been linked to NTDs in humans, and we are unaware of any published animal model. The c.1021G>A variant has a minor allele frequency of 0.25% in European Americans [Exome Variant Server, accessed 5/21/2014] and the computational prediction programs SIFT and PolyPhen-2 predict this variant to be tolerated. The Gly341 residue is not well evolutionarily conserved, as this position is Ala in opossum and Ser in chicken, Xenopus and zebrafish. In light of the evidence available, the variant identified in AIFM3 is unlikely to be pathogenic.
Discussion
The herein reported sibs had non-distinctive facial dysmorphisms and myelomeningocele with the absence of major cardiovascular malformations. Both sibs harbored an “atypical” deletion within 22q11.21. The phenotypic presentation of 22q11.2DS comprises a broad spectrum of clinical findings and the involvement of multiple organ systems including the central nervous system. NTDs have been rarely associated with 22q11 microdeletions [Nickel and Magenis, 1996], but these sibs provide further evidence of a link between deletions on 22q11 and NTDs.
NTDs were initially reported in association with conotruncal anomalies by Kousseff in 1984. Subsequently, Toriello et al. [1985] described one individual with similar clinical findings and named the condition “Kousseff syndrome”. The individuals reported by Kousseff [1984] and Toriello et al. [1985] had normal karyotypes but no FISH studies or additional molecular studies were performed. Palacios et al. [1993] reported an association between DiGeorge anomaly and lumbar meningocele based on one individual, although no FISH studies for deletions on 22q11 were performed. In 1994, Nickel et al. confirmed the presence of a 22q11.2 microdeletion, in three individuals, two with a clinical diagnosis of velocardiofacial syndrome and one with DiGeorge sequence, all having NTDs. Moreover, Forrester et al. [2002], reported the FISH analysis of the third sib and his father, originally described by Kousseff in 1984 showing deletions of the DiGeorge critical region on 22q11.2. Subsequently, the sacral meningocele was attributed as part of the phenotypic spectrum of 22q11.2DS rather than the patient having Kousseff syndrome as a specific separate entity. Interestingly, Maclean et al. [2004] reported two individuals with the association of conotruncal anomaly and myelomenigocele, one of which was negative on FISH analysis for a 22q11.2 microdeletion (N25 locus probe) suggesting that despite the identification of a deletion on 22q11 in the original family, Kousseff syndrome may be causally heterogeneous. In 2013, McDonald-McGinn et al. performed exome, targeted exome and/or Sanger sequencing on 17 individuals with 22q11.2DS associated with atypical clinical findings such as polymicrogyria, skin anomalies, myelomeningocele, hypospadia, and laryngo-tracheal abnormalities. They reported four unrelated patients with three novel mutations in SNAP29, a gene mapping to 22q11.2 potentially associated with CEDNIK syndrome (cerebral dysgenesis, neuropathy, ichthyosis and keratoderma) [Sprecher et al., 2005; Fuchs-Telem et al., 2011] and genitourinary anomalies [Zhang et al., 2009]. The authors [McDonald-McGinn et al., 2013] also hypothesized that the phenotypic variability observed in some patients with 22q11.2DS could be due to additional mutations on the non-deleted chromosome, leading to the unmasking of autosomal recessive conditions such as CEDNIK syndrome. Within the group of individuals evaluated by McDonald-McGinn [2013], two individuals had myelomeningocele along with tetralogy of Fallot, but only one had a mutation in SNAP29 and neither showed any clinical features suggesting CEDNIK syndrome. We did not observe any mutations in SNAP29 by exome sequencing. It is possible that the myelomeningocele phenotype and other lower penetrance manifestations of 22q11DS could be due to somatic events or additional germline mutations on the non-deleted chromosome not identified on exome sequencing.
The previous reports of NTDs with 22q11DS suggest a role for gene(s) within the 22q11 region in NTDs, and the herein reported sibs provide additional support for this hypothesis helping to narrow a potential critical region for NTDs associated with 22q11 deletions. However, even if there are causative genes for NTD on 22q11, non-penetrance/clinical expressivity is likely, given that the mother is clinically unaffected but harbors the same deletion. It is possible that the NTD phenotype is multifactorial and/or polygenic. Additional studies investigating the deletion size and location on 22q11 in other individuals with NTDs will be important to help identify causative genes for NTD.
Acknowledgments
We thank the family for their participation. We thank Heather Hanson for enrollment and help with sample collection. This project was supported by the University of Utah Clinical Genetics Research Program: Phenotyping Core (CGRP), and the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1RR025764. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors have no conflict of interest to declare.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shachar S, Ou Z, Shaw CA, Belmont JW, Patel MS, Hummel M, Amato S, Tartaglia N, Berg J, Sutton VR, Lalani SR, Chinault AC, Cheung SW, Lupski JR, Patel A. 22q11.2 distal deletion: a recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome. Am J Hum Genet. 2008;82:214–221. doi: 10.1016/j.ajhg.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O'Leary LA, Wong LY, Elixson EM, Mahle WT, Campbell RM. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101–107. doi: 10.1542/peds.112.1.101. [DOI] [PubMed] [Google Scholar]
- Canda MT, Demir N, Bal FU, Doganay L, Sezer O. Prenatal diagnosis of a 22q11 deletion in a second-trimester fetus with conotruncal anomaly, absent thymus and meningomyelocele: Kousseff syndrome. J Obstet Gynaecol Res. 2012;38:737–740. doi: 10.1111/j.1447-0756.2011.01770.x. [DOI] [PubMed] [Google Scholar]
- Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP) Seattle, WA: May 21, 2014. http://evs.gs.washington.edu/EVS/ [Google Scholar]
- Forrester S, Kovach MJ, Smith RE, Rimer L, Wesson M, Kimonis VE. Kousseff syndrome caused by deletion of chromosome 22q11-13. Am J Med Genet. 2002;112:338–342. doi: 10.1002/ajmg.10625. [DOI] [PubMed] [Google Scholar]
- Fuchs-Telem D, Stewart H, Rapaport D, Nousbeck J, Gat A, Gini M, Lugassy Y, Emmert S, Eckl K, Hennies HC, Sarig O, Goldsher D, Meilik B, Ishida-Yamamoto A, Horowitz M, Sprecher E. CEDNIK syndrome results from loss-of-function mutations in SNAP29. Br J Dermatol. 2011;164:610–616. doi: 10.1111/j.1365-2133.2010.10133.x. [DOI] [PubMed] [Google Scholar]
- Garavelli L, Rosato S, Wischmeijer A, Gelmini C, Esposito A, Mazzanti L, Franchi F, De Crescenzo A, Palumbo O, Carella M, Riccio A. 22q11.2 Distal Deletion Syndrome: Description of a New Case with Truncus Arteriosus Type 2 and Review. Mol Syndromol. 2011;2:35–44. doi: 10.1159/000334262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Miñaur S, Fantes J, Murray RS, Porteous ME, Strain L, Burns JE, Stephen J, Warner JP. A novel atypical 22q11.2 distal deletion in father and son. J Med Genet. 2002;39:E62. doi: 10.1136/jmg.39.10.e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodship J, Cross I, LiLing J, Wren C. A population study of chromosome 22q11 deletions in infancy. Arch Dis Child. 1998;79:348–351. doi: 10.1136/adc.79.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhalgh KL, Aligianis IA, Bromilow G, Cox H, Hill C, Stait Y, Leech BJ, Lunt PW, Ellis M. 22q11 deletion: a multisystem disorder requiring multidisciplinary input. Arch Dis Child. 2003;88:523–524. doi: 10.1136/adc.88.6.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita H, Kokudo T, Ide T, Kondo Y, Mori T, Homma Y, Yasuda M, Tomiyama J, Yakushiji F. A patient with DiGeorge syndrome with spina bifida and sacral myelomeningocele, who developed both hypocalcemia-induced seizure and epilepsy. Seizure. 2010;19:303–305. doi: 10.1016/j.seizure.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Kousseff BG. Sacral meningocele with conotruncal heart defects: a possible autosomal recessive trait. Pediatrics. 1984;74:395–398. [PubMed] [Google Scholar]
- Kurahashi H, Tsuda E, Kohama R, Nakayama T, Masuno M, Imaizumi K, Kamiya T, Sano T, Okada S, Nishisho I. Another critical region for deletion of 22q11: a study of 100 patients. Am J Med Genet. 1997;72:180–185. doi: 10.1002/(sici)1096-8628(19971017)72:2<180::aid-ajmg10>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Lindsay EA. Chromosomal microdeletions: dissecting del22q11 syndrome. Nat Rev Genet. 2001;2:858–868. doi: 10.1038/35098574. [DOI] [PubMed] [Google Scholar]
- Maclean K, Field MJ, Colley AS, Mowat DR, Sparrow DB, Dunwoodie SL, Kirk EP. Kousseff syndrome: a causally heterogeneous disorder. Am J Med Genet A. 2004;124:307–312. doi: 10.1002/ajmg.a.20418. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, Finucane B, Driscoll DA, Emanuel BS, Zackai EH. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet Med. 2001;3:23–29. doi: 10.1097/00125817-200101000-00006. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome) Medicine (Baltimore) 2011;90:1–18. doi: 10.1097/MD.0b013e3182060469. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Fahiminiya S, Revil T, Nowakowska BA, Suhl J, Bailey A, Mlynarski E, Lynch DR, Yan AC, Bilaniuk LT, Sullivan KE, Warren ST, Emanuel BS, Vermeesch JR, Zackai EH, Jerome-Majewska LA. Hemizygous mutations in SNAP29 unmask autosomal recessive conditions and contribute to atypical findings in patients with 22q11.2DS. J Med Genet. 2013;50:80–90. doi: 10.1136/jmedgenet-2012-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel RE, Pillers DA, Merkens M, Magenis RE, Driscoll DA, Emanuel BS, Zonana J. Velo-cardio-facial syndrome and DiGeorge sequence with meningomyelocele and deletions of the 22q11 region. Am J Med Genet. 1994;52:445–449. doi: 10.1002/ajmg.1320520410. [DOI] [PubMed] [Google Scholar]
- Nickel RE, Magenis RE. Neural tube defects and deletions of 22q11. Am J Med Genet. 1996;66:25–27. doi: 10.1002/(SICI)1096-8628(19961202)66:1<25::AID-AJMG6>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Oskarsdóttir S, Persson C, Eriksson BO, Fasth A. Presenting phenotype in 100 children with the 22q11 deletion syndrome. Eur J Pediatr. 2005;164:146–150. doi: 10.1007/s00431-004-1577-8. [DOI] [PubMed] [Google Scholar]
- Palacios J, Gamallo C, García M, Rodríguez JI. Decrease in thyrocalcitonin-containing cells and analysis of other congenital anomalies in 11 patients with DiGeorge anomaly. Am J Med Genet. 1993;46:641–646. doi: 10.1002/ajmg.1320460608. [DOI] [PubMed] [Google Scholar]
- Rauch A, Zink S, Zweier C, Thiel CT, Koch A, Rauch R, Lascorz J, Hüffmeier U, Weyand M, Singer H, Hofbeck M. Systematic assessment of atypical deletions reveals genotype-phenotype correlation in 22q11.2. J Med Genet. 2005;42:871–876. doi: 10.1136/jmg.2004.030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rødningen OK, Prescott T, Eriksson AS, Røsby O. 1.4Mb recurrent 22q11.2 distal deletion syndrome, two new cases expand the phenotype. Eur J Med Genet. 2008;51:646–650. doi: 10.1016/j.ejmg.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brøndum-Nielsen K, Scambler PJ. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitta SC, McGrath JM, Mensch H, Shaikh TH, Zackai EH, Emanuel BS. A 22q11.2 deletion that excludes UFD1L and CDC45L in a patient with conotruncal and craniofacial defects. Am J Hum Gen. 1999;65:562–566. doi: 10.1086/302514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seller MJ, Mohammed S, Russell J, Ogilvie C. Microdeletion 22q11.2, Kousseff syndrome and spina bifida. Clin Dysmorphol. 2002;11:113–115. doi: 10.1097/00019605-200204000-00007. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Saitta SC, O'Hare AM, Hu P, Roe BA, Driscoll DA, McDonald-McGinn DM, Zackai EH, Budarf ML, Emanuel BS. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet. 2000;9:489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, O'Connor RJ, Pierpont ME, McGrath J, Hacker AM, Nimmakayalu M, Geiger E, Emanuel BS, Saitta SC. Low copy repeats mediate distal chromosome 22q11.2 deletions: sequence analysis predicts breakpoint mechanisms. Genome Res. 2007;17:482–491. doi: 10.1101/gr.5986507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprecher E, Ishida-Yamamoto A, Mizrahi-Koren M, Rapaport D, Goldsher D, Indelman M, Topaz O, Chefetz I, Keren H, O'brien TJ, Bercovich D, Shalev S, Geiger D, Bergman R, Horowitz M, Mandel H. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am J Hum Genet. 2005;77:242–251. doi: 10.1086/432556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tézenas Du Montcel S, Mendizabai H, Aymé S, Lévy A, Philip N. Prevalence of 22q11 microdeletion. J Med Genet. 1996;33:719. doi: 10.1136/jmg.33.8.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriello HV, Sharda JK, Beaumont EJ. Autosomal recessive syndrome of sacral and conotruncal developmental field defects (Kousseff syndrome) Am J Med Genet. 1985;22:357–360. doi: 10.1002/ajmg.1320220220. [DOI] [PubMed] [Google Scholar]
- Vantrappen G, Rommel N, Devriendt K, Cremers CW, Feenstra L, Fryns JP. Clinical features in 130 patients with the velo-cardio-facial syndrome. The Leuven experience. Acta Otorhinolaryngol Belg. 2001;55:43–48. [PubMed] [Google Scholar]
- Verhagen JM, Diderich KE, Oudesluijs G, Mancini GM, Eggink AJ, Verkleij-Hagoort AC, Groenenberg IA, Willems PJ, du Plessis FA, de Man SA, Srebniak MI, van Opstal D, Hulsman LO, van Zutven LJ, Wessels MW. Phenotypic variability of atypical 22q11.2 deletions not including TBX1. Am J Med Genet A. 2012;158:2412–2420. doi: 10.1002/ajmg.a.35517. [DOI] [PubMed] [Google Scholar]
- Wooderchak-Donahue WL, McDonald J, O'Fallon B, Upton PD, Li W, Roman BL, Young S, Plant P, Fulop GT, Langa C, Morrell NW, Botella LM, Bernabeu C, Stevenson DA, Runo JR, Bayrak-Toydemir P. Bmp9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am J Hum Genet. 2013;93:530–537. doi: 10.1016/j.ajhg.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JS, Fu Y, Zhao YH, Li F, Qian AL, Wu B, Li-Ling J. Genetic analysis of genitourinary malformations. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2009;26:134–138. doi: 10.3760/cma.j.issn.1003-9406.2009.02.003. [DOI] [PubMed] [Google Scholar]