

Abstract
Primary sclerosing cholangitis (PSC) is a chronic cholestatic syndrome of unknown origin mostly found in males, and characterized by diffuse inflammation and fibrosis of both intra- and extra-hepatic bile ducts. So far, PSC is considered as an autoimmune hepatobiliary disease. In most cases the progression of PSC towards liver cirrhosis and liver failure is slow but irreversible, and liver transplantation is currently the only definitive treatment. In recent years, PSC has been an area of active research worldwide with great interest in etiology, pathogenesis, diagnosis, and therapeutic options such as hydrophilic ursodeoxycholic acid and immunosuppressive agent tacrolimus. Recent updates on clinical and therapeutic aspects of PSC are discussed in the present review.
Keywords: Sclerosing cholangitis, Diagnosis, Therapy
INTRODUCTION
Primary sclerosing cholangitis (PSC), first described by a French author Delbet in 1924[1], is a chronic cholestatic syndrome characterized by diffuse inflammation and fibrosis of both intra- and extra-hepatic bile ducts[2]. The mean age at diagnosis is 40 years and men are affected about two times more than women[3]. The natural history of the disease is variable from patient to patient although in most cases the progression towards liver failure is slow but irreversible. In the end stages, PSC results in biliary cirrhosis, portal hypertension, and is associated with bile duct carcinoma with a high frequency (8%). Currently, PSC is the fifth most common indication for liver transplantation in the USA, but in the Nordic countries, PSC is the most important indication for orthotopic liver transplantation (OLT). With a still unknown etiology, establishing the correct therapy for PSC is difficult. Unlike primary biliary cirrhosis (the other most common chronic cholestatic disease in the adult), PSC lacks a definitive medical therapy. The ultimate goal of the therapy should be symptom improvement and longer survival. Promising regimens are high doses of ursodeoxycholic acid (UDCA) alone or in combination with other drugs, and tacrolimus (FK506). Presently, liver transplantation is the only definitive treatment.
The present review will address recent aspects of PSC and focus on pathogenesis, diagnosis and treatment.
EPIDEMIOLOGY, ETIOLOGY AND PATHOGENESIS
The prevalence of PSC is currently unknown. About 75% are associated with inflammatory bowel disease (IBD), especially ulcerative colitis (UC) (87% of associations with IBD). Given the prevalence of UC in USA between 40 and 225 per 100000, and knowing that about 2.5-7.5% of patients with this disease suffer from PSC[4,5], the prevalence in USA has been estimated as 1-6 cases per 100000 persons. However, this data is likely to underestimate the true prevalence of PSC, since 20-30% of cases of PSC are not associated with IBD[6]. Males are two times more affected than females, and the average age of clinical onset of PSC is 39-40 years, but the range can be between 1 and 90 years[7]. Although PSC is most likely a multifactorial disease, the exact etiology remains unknown so far. Among the many pathogenic theories formulated, the most important are discussed below.
Genetic predisposition
There is evidence about the familial occurrence of PSC and many studies have focused on the relationship between PSC and the human major histocompatibility complex HLA. Findings suggest a genetic background for PSC predisposition. HLA type II haplotypes B8 or DR3 are most commonly associated with PSC (60% and 56%, respectively)[8-10], suggesting a central role of DR3-β locus. DRw52a is also very frequently associated (52-100% of patients)[11,12]. DR2 is associated with a younger onset of the disease[9] while DR4 seems to be an important marker of more rapid disease progression[13]. For HLA type I haplotypes, the association involves A1 and Cw7 genes.
Immunological causes
This seems to be the most attractive hypothesis for PSC. The strong association of PSC with a series of autoimmune diseases underscores the role of immunological alterations in the pathophysiology of the disease (Table 1). Moreover, specific autoantibodies can be found in patients with PSC, i.e., antineutrophil cytoplasmatic antibodies (p-ANCA)[14], anticolon antibodies[15], antineutrophil nuclear antibodies[16] with a high frequency, while anti-mitochondrial auto-antibodies (AMA), anti-nuclear auto-antibodies (ANA), anti-smooth muscle auto-antibodies (ASMA) with a lower frequency[17]. Circulating immune complexes are found in as many as 80% of patients[18]. Other immunological abnormalities may include hypergammaglobulinemia (30%), high serum IgM (50%)[14], decreased circulating T cells, increased ratio of CD4:CD8[19], decreased C3[20]. At histology, it is possible to find lymphocytic bile duct destruction[21] and an increase of class II major histocompatibility complex (MHC II) on biliary epithelial cells[22]. However, the exact role of immune system alterations (primary or secondary involvement?) in the development, behaviour and progression of the disease is still not completely understood.
Table 1.
Diseases most commonly associated with PSC.
| Celiac disease |
| Rheumatoid arthritis |
| Thyroiditis |
| Sjogren’s syndrome |
| Lupus erythematosus |
| Lupic nephritis |
| Chronic pancreatitis |
| Retroperitoneal fibrosis |
| Systemic sclerosis |
| Peyronie’s disease |
| Autoimmune hemolytic anemia |
| Immune thrombocytopenic purpura |
| Membranous nephropathy |
| Histiocytosis X |
| Cystic fibrosis |
| Angioblastic lymphadenopathy |
| Intra-abdominal adenopathy |
| Vasculitis |
| Pseudotumor of the orbit |
| Gallbladder disease |
Bacterial-toxic damage
This theory is based on the frequent association of PSC with IBD, especially UC[23]. The combined activity of detergent bile acid with bacteria in a diseased colon may result in an increased mucosal permeability. The presence of bacteria[23] and/or their toxins, and the increased concentration of potentially toxic bile acids in the portal vein[6] may cause Kupffer cell activation to produce tumor necrosis factor (TNF)[24]. Overproduction of TNF may ultimately result in bile duct inflammation and hepatobiliary lesions leading to portal fibrosis and PSC. It is a fact, however, that an accurate study employing liver histology in PSC patients found only a mild or absent portal phlebitis, as a marker of portal vein bacteraemia[21]. The development of PSC, moreover, is not related to the severity of IBD. PSC may be diagnosed years before the onset of colitis or years after total colectomy, and this finding suggests that bacteremia alone may not be the sole determinant in the pathogenesis of PSC[25].
Viral infection
Several viruses including CMV and retrovirus type III have been implicated in the pathogenesis of PSC. This theory is less attractive, since investigators have only shown induction of secondary cholangitis and biliary atresia but not PSC[21].
Smoking behaviour
In a controlled study, we found that the frequency of PSC and UC was markedly increased in non-smoking patients[26], suggesting that smoking is associated with a decreased risk of PSC. Nicotine may be the active agent responsible for the negative correlation between smoking and disease risk. Indeed, the addition of transdermal nicotine to conventional maintenance therapy could improve symptoms in patients with ulcerative colitis[27]. In another study, however, we found that transdermal nicotine did not have a clear short-term beneficial effect on PSC[28]. Thus, further studies are needed to clarify this issue.
Biliary arteriolar injury
The rationale for this theory is that all conditions that can alter the peribiliary vascular plexus may cause ischemic damage and biliary tract necrosis and potential evolution to PSC. Such conditions include liver transplantation, chronic rejection, or diseases characterized by a high frequency of thrombosis[29,30]. Vascular injury, however, was absent at histology in the liver of PSC patients undergoing liver transplantation[31]. Although suggestive, this theory has been abandoned so far.
DIAGNOSIS
Diagnosis of PSC may be difficult, especially at early stages, since patients are asymptomatic or poorly symptomatic. Diagnostic steps must include clinical assessment, laboratory tests, imaging, and histology. The ultimate diagnosis of PSC requires that all secondary causes of cholangitis are ruled out, namely bacterial infections (chronic and acute, secondary to surgery or to acquired immunodeficiency syndromes), abnormalities of the biliary tree, ischemic bile duct damage (secondary to floxuridine treatment), and neoplasms[6].
Clinical assessment
At an early stage, PSC is frequently asymptomatic. Symptoms appear with the progression of the disease and include pruritus, jaundice, fatigue, weight loss, and steatorrhoea. Fever, pain in the right upper quadrant of the abdomen, night sweating, and chills are present in 10-15% of patients at the time of the diagnosis[6]. In children the onset may be characterized by anorexia, nausea, fatigue, and weight loss[7]. The physical examination is usually negative in early stages. If positive, it may disclose hepatomegaly (55%), intermittent jaundice (45%), splenomegaly (35%), skin hyper pigmentation (25%), excoriations (21%), other signs such as xanthomas, ascites and edema[32]. Progressive portal hypertension is characterized by abundant ascites, variceal bleeding, and portal systemic encephalopathy[33].
Laboratory tests
A cholestatic biochemical profile for six months or more is frequently found in PSC patients, but findings are not specific[32]. Alkaline phosphatase (AP) can be normal[34] or up to 3 or 4 times normal[2,35]. A mild-to-moderate elevation in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) is usually present. Bilirubin fluctuates but is elevated, albumin can be normal or decreased, partial thromboplastin time (PTT) can be normal or increased. This picture may be different in children: Feldstein et al[36] found an increased AST - ALT level and an increased γGT level respectively in 90% and 94% of cases at the time of the diagnosis of PSC. Although AP was increased in 75% of patients, there was a high variability due to faster bone turnover during growth. These findings suggest that γGT is the most sensitive test for the diagnosis of PSC in children. Eosinophilia can be found in 5% of patients[37]. Some immunological tests may help in the diagnosis of PSC. Hyper-γ-globulinemia is found in 30% of patients, an increase of IgM in 40-50%[37,38], ANA in 6%, ASMA in 11%, and AMA in 5% of patients[37]. In children, hyper-γ-globulinemia was found in 66% of patients, an increase of IgM in 23%, an increase of IgG in 70%, ANA and ASMA in 69%, and ANCA in 72% of patients[36].
Imaging
This is the most important step for the diagnosis of PSC. At the end of the 1970s, ERCP and percutaneous transhepatic cholangiography (PTC) represented the gold standard for the diagnosis of PSC (Figure 1). Nowadays, most reliable techniques are magnetic resonance (MR) and MR-cholangiopancreaticography (MRCP)[39]. Distinctive features are a multifocal stricture and bead involving bile ducts[40,41], which appear as normal or slightly dilated[42], and diffuse strictures[42]. However, in the early stages, fine or deep ulcerations of the common bile duct can be the only findings[6]. Gallbladder and cystic ducts are involved in 15% of patients[43]. In small-duct PSC, a PSC variant, cholangiographic features may be silent, because affected bile ducts are too small to be seen by radiology[6]. The finding of a polipoid mass into dilated ducts may be predictive of cholangiocarcinoma and needs further investigations including biopsy, brushing, needle aspiration and evaluation of serum and bile tumoral markers (CEA and CA19.9)[44,45]. An important role of PSC diagnosis is the emerging of MRCP (Figure 2)[46,47]. Weber et al[47] recently compared MRCP with ERCP in 55 patients with suspected PSC. Morphologic criteria of PSC were documented with ERCP as the gold standard, and sensitivity, specificity and diagnostic accuracy were calculated. Of the 55 patients with PSC at ERCP, 40 were positive for MRCP imaging and 37 for liver biopsy. The authors concluded that MRCP could be a reliable non-invasive imaging method for the diagnosis and follow up of PSC. Nowadays, MR imaging can be a useful tool to establish the diagnosis of advanced PSC leading to cirrhosis, in the presence of large regenerative nodules. In another recent study[39], 52 patients with PSC underwent MR imaging, 87% of PSC patients had classic findings of liver cirrhosis, but with different patterns and there was a high variability among the patients. The common findings were hypertrophy of the caudate lobe (58-63%), large regenerative nodules (54%) localized in the central part of liver in about two-third of the cases, biliary ductal dilatation (80%), peripheral bile duct dilatation due to compression of central ducts by central regenerative nodules (29%), peripheral wedge-shaped areas of parenchymal atrophy (50% of patients with cirrhosis patterns) and fibrosis. The authors, however, did not evaluate the sensivity and specificity of MR imaging in PSC, thus more studies are needed in this field.
Figure 1.
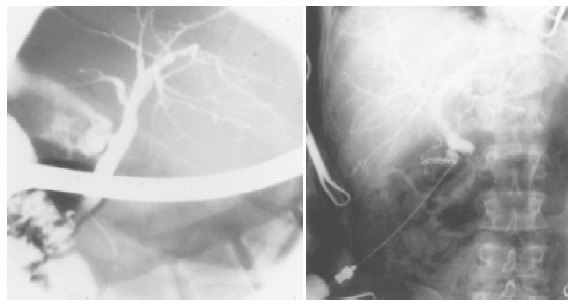
Cholangiographic pictures of enlarged bile ducts in a PSC patient. On the left picture of ERCP, and on the right picture of PTC, multifocal stricturing and slightly dilated bile ducts are visible in both pictures.
Figure 2.
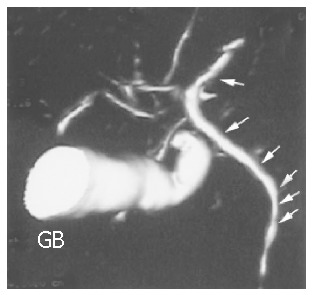
MRCP pictures of a PSC patient. Wall irregularities (see arrows) are visible in undilated bile ducts. The gallbladder (GB) is enlarged.
Ultrasonography
We reported for the first time that fasting gallbladder volume was greatly enlarged in PSC patients. The enlargement could be noteworthy (i.e., >100 mL) and in one case a volume of 324 mL was found without cystic duct obstruction[48]. Nevertheless, postprandial gallbladder contraction was preserved and comparable to normal. Thus, when associated with altered biochemistry, the finding of an increased fasting gallbladder volume at ultrasonography (i.e., >50 mL) could be a useful, non-invasive, and easy to perform screening test in patients suspected of having PSC. However, the sensitivity of this test is low in early stages, and a normal gallbladder volume does not rule out the diagnosis of PSC.
Histology
Histological findings are not specific for PSC and false negatives are frequent (5-10%) because in the early stages the disease is focal[49]. Extra-hepatic and large intra-hepatic bile ducts are characterized by necrosis of epithelial cells, a thickened fibrous wall with inflammatory infiltrates that tend to cluster around biliary glands (Figure 3)[42,50]. Intra-hepatic bile ducts are characterized by necrosis of epithelial cells, bile duct proliferation, ductopenia in some tracts, edema in some others, fibrous cholangitis with features in portal triads of concentric fibrosis around bile ducts (Figure 4)[50,51]. In advanced stages, bile ducts become a solid fibrous cord, which is a distinctive feature of PSC. There is also a typical reactive hyperplasia of intramural glands of the extra hepatic bile ducts while dysplasia is rare[52]. Hepatic parenchyma shows some changes, which are common to primary biliary cirrhosis and not specific but important for staging and prognosis. Histological features can be classified in four stages. In the first stage, inflammation is focal and limited to portal triads. In the second stage, lesions are more widespread, infiltrates and fibrosis are more predominant, and bile ducts are enlarged. In the third stage, portal to portal fibrous septa are commonly found, while stage four is a typical and nonspecific picture of cirrhosis[6].
Figure 3.
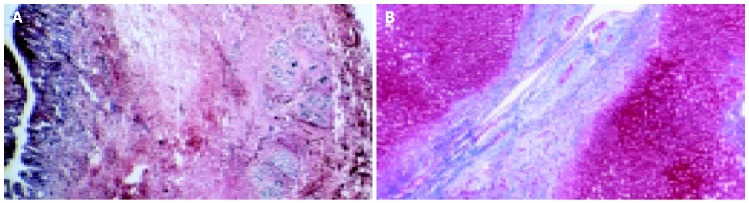
Histological appearance of the common bile duct (A) and a large intralobular bile duct (B) in PSC (Cross section of liver, 4× and 40× magnification, Masson Stain).
Figure 4.
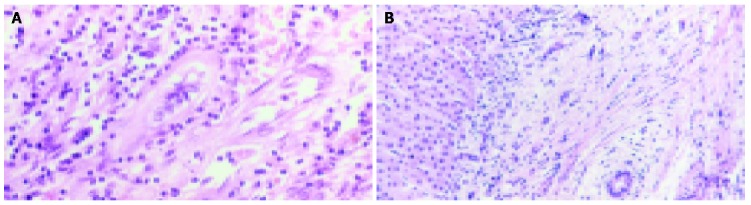
Histological appearance of a small bile duct with inflammatory cells (A) and a small intra-hepatic bile duct with concentric rings of fibrosis (B) in PSC (40× magnification, H&E).
NATURAL HISTORY
Since PSC progression can be silent for years, its detection may result from abnormal liver function tests and histological features[53]. However, an earlier diagnosis means prolonged survival since therapy might interfere with the natural history of the disease. The mean survival from the time of diagnosis has been reported to be 9-11 years[54] and 17 years[55]. In children, the mean survival without therapy is 12.7 years but it is shorter with overlapping autoimmune hepatitis (AIH)[36]. The most common complications in PSC include osteoporosis (related to the osteoblast inhibitors found in serum of patients with cholestasis)[56], portal hypertension and liver failure, cholestasis, cholelitiasis and choledocholithiasis (in 30% of patients, probably related to chronic cholestasis)[57], deficiency in vitamins A, B, C, D (50% vitamin A deficiency), ascites, bleeding from esophageal varices, spontaneous bacterial peritonitis, portal encephalopathy, bleeding from peristomal varices (after proctocolectomy and ileal stoma), bacterial cholangitis (spontaneous or secondary to ERCP or biliary surgery). The presence of dominant strictures of the biliary tract (15-20% of patients) may result in jaundice, pruritus, fever[58-60], and cholangiocarcinoma (from 6% to 30%, specially in patients with cirrhosis or with UC associated)[61]. All above-mentioned complications may reduce survival.
Predicting survival on the basis of clinical, biochemical, and histological features is of great importance to monitoring therapy and timing liver transplantation. Thus, many prognostic models and risk score models have been constructed, including the Child-Pugh score[62], the Mayo Clinic survival model[63] and the Kaplan-Meier survival curve, which have been corrected and integrated with ERCP findings[64]. Results, however, are not always related to the true evolution of the disease. PSC is most commonly associated with IBD. The prevalence of IBD in PSC patients is 54-100% (90% UC, 10% Crohn’s disease) and in most of the cases PSC follows IBD (94% of patients have IBD at the time of diagnosis), but the correlation is lacking between liver and colon damage[35]. In the adult population AIH appears to coexist with PSC as an overlap syndrome[65,66] in 7.1-10.6% of cases, the prevalence in children averages 35%[67]. Usually patients with mixed findings of the two diseases have predominant manifestations of AIH and their histological assessment may show only features of periportal hepatitis. The prognosis of this association is unknown, but since there is no gain with corticosteroids, it is likely that the PSC component dictates the clinical course of the illness. PSC has been found to be associated with a large number of other syndromes. As previously mentioned, the high frequency of association with autoimmune diseases indeed supports the autoimmune pathogenesis theory (Table 1).
THERAPY
Since the etiology and pathogenesis of PSC are still unknown, therapy is difficult and remains mostly endoscopic. Although several medications have been evaluated alone or in combination, liver transplantation stands as the definitive therapy for PSC.
Ursodeoxycholic acid (UDCA)
UDCA is the dihydroxy bile acid produced in a small amount by colon microflora from dehydroxylation of the primary bile salt chenodeoxycholic acid. UDCA is found in human bile as 4-5% of the total bile acid pool. Because of its chemical structure, UDCA is more hydrophilic (i.e., less detergent and less cytotoxic) than other primary and secondary bile acids. Orally, the absorption of UDCA is between 30% and 60%, mainly in the small intestine (80%) and less in the colon[68]. Advanced cholestasis may diminish the oral bioavailability of UDCA[69]. Hepatocytes are able to pick up UDCA from the portal vein via specific transporters (NTCP and OATP)[70] and after that, UDCA is conjugated to glycine and taurine[71]. From the liver, UDCA is secreted in bile ducts via another transporter protein, the bile salt export pump (BSEP)[70]. The first pass hepatic metabolism is 70%, so its blood level in systemic circulation is very low[72] and peak levels in bile are found 1-3 h after administration. The half-life of UDCA is 3.5-5.8 d[73], and UDCA is mainly eliminated by faeces. In cholestatic diseases, however, renal secretion of UDCA may increase. UDCA is responsible for a number of effects in the body (Table 2). These effects include decreased serum and biliary cholesterol levels, increased conversion of cholesterol to bile acids, decreased ileal absorption of endogenous bile acids[74-76], increased total serum bile acid pool[77,78], improvement of bile acid hepatic excretory rates and transit time[79]. In experimental animals, UDCA induces hypercholeresis, i.e., a greater than expected choleresis[80] via the so-called “cholehepatic shunt” process[81]. When protonated, in fact, UDCA is more lipophilic and can be rapidly reabsorbed from the bile ductules into the peribiliary plexuses. In this way, it comes back directly to the liver and can be re-secreted. Additional effects of UDCA include reduction of T-cells that mediate hepatocellular damage[82,83], cell damage induced by decreased hydrophobic bile acid[84-86], and inhibition of neoplasm proliferation[87-89]. Regimens of UDCA used in PSC are depicted in Table 3 and include UDCA alone (at low or high doses) or in combination with other medications. Though UDCA is still widely used in PSC patients, there is no definitive data regarding the impact of this drug on survival or time to OLT.
Table 2.
Targets, mechanisms and effects of UDCA therapy.
| Target | Mechanisms | Effects | References |
| Cholesterol | Intestinal absorption ↓ | Biliary cholesterol decreased by 40-60% | [118] |
| Conversion to bile acids ↑ | Serum LDL and HDL cholesterol decreased | ||
| Bile acid pool | Ileal absorption of endogenous hydrophobic bile acids ↓ | Serum UDCA increased by 10-64% | |
| Total bile acids ↑ Hydrophobic bile acids ↓ | [74-77,119,120] | ||
| Unchanged hydrophilic bile acid pool | [121,122] | ||
| Bile flow | Exocytocis and canalicular transport ↑(due to ↑ cytoplasmatic free Ca2+) | ||
| Modulation of membrane transport proteins | Excretory rates and bile acids transit time ↑ | [123-125] | |
| Hypercholeresis | [80] | ||
| Gallbladder | Modulation of smooth muscle contractility (CCK receptor + cholinergic nerves) | Fasting gallbladder volume↑ | [126-128] |
| Postprandial gallbladder emptying ν | |||
| Gallbladder bile | Biliary total proteins ↓ | Crystallization-promoting activity ↓ | [129,130] |
| Concanavalin A-binding fraction ↓ | Inhibition of cholesterol crystallization | ||
| Immune system | Expression of MHC class I and II ↓ | Immunomodulatory effect T-cell hepatocellular damage ↓ | [82,83] |
| Cells | Hydrophobic bile acid induced cell damage↓ | Cytoprotection (e.g., liver damage ↓) | [85,86] |
| Apoptosis or necrosis ↓ | |||
| Neoplasms | Unknown (decreased fecal hydrophobic deoxycholate, lithocholate) | Chemo protection (neoplasm proliferation ↓) | [87,89,131] |
↓, decreased; ↑, increased; ν, unchanged; MHC, major histocompatibility complex.
Table 3.
Regimens and effects of UDCA for PSC therapy.
| Regimen | Assessment | Outcome | References | |
| Low doses (single administration) | 8-13 mg/(kg·d) | Liver biochemistry | Improved | [92] |
| Histology, symptoms, survival | Ineffective | |||
| 13-15 mg/(kg·d) | Liver biochemistry | Improved | [90] | |
| Histology, symptoms, survival | Ineffective | |||
| Low doses (multiple administration)1 | 10-12 mg/(kg·d) t.i.d. | Liver biochemistry | Improved | [93] |
| Histology, symptoms | No progression | |||
| 20 mg/(kg·d) | Liver biochemistry | Improved | [94] | |
| Histology | Improved | |||
| High doses | 25-30 mg/(kg·d) | ERCP | No progression | |
| Liver biochemistry | Improved | |||
| Mayo risk score and survival at 4 yr | Improved | [95] | ||
| Combination | UDCA 650 mg/d + azathioprine 1-1.5 mg/(kg·d)+ prednisolone 1-10 mg/(kg·d) | Liver biochemistry | Improved | |
| Histology | Improved | [96] | ||
| ERCP | Improved |
1Comparable effects for multiple vs single administration.
UDCA alone
Several trials used UDCA at low doses (8-15 mg/kg b.w. daily) and showed a relevant improvement in liver biochemistry but not in histology, symptoms and survival[90-92]. One Dutch multicenter randomized study[93] compared a single dose with multiple doses (t.i.d. at meal time) for 2 years in 48 PSC patients. For both groups the total administered doses were 10-12 mg/kg b.w. daily. During the 2-year observation period, symptom and AP, γGT and AST decreased significantly while bilirubin and histology did not deteriorate in both groups. No difference existed between single and multiple doses of UDCA. As biliary enrichment of UDCA is expected to be lower in cholestasis, use of high doses of UDCA in PSC has a rationale. Mitchell et al[94] compared UDCA (20 mg/kg·d) (n = 13) with placebo (n = 13), and found that UDCA in total bile acid pool increased from 3% to more than 70% in the UDCA group. Although there was no difference between the two groups with respect to symptoms like malaise and fatigue, pruritus and jaundice were more frequent in the control group. The UDCA group had improvement in serum levels of AP and γGT (no effect on bilirubin and albumin levels), while there was a minor decrease of the scores of portal inflammation. ERCP showed no progression of the disease. The authors concluded that high dose regime of UDCA might be effective in the therapy of PSC but the heterogeneous stages of patients at the starting point of the study did not allow drawing definitive conclusions. Another study[95] employed UDCA 25-30 mg/(kg·d) in 23 patients (77% with UC), 38% of the patients showed more than 50% improvement of AP compared to baseline, bilirubin was improved by 44% in the 11 patients with prior hyperbilirubinemia, and AST and albumin were improved in 59% of the patients. The Mayo risk score also improved together with the 4-year survival. Taken together, these studies have shown that high doses of UDCA have a positive outcome not only in liver biochemistry, but also in survival of PSC patients. The results of a controlled trial with a high dose of UDCA for PSC are awaited from the Mayo Clinic group.
UDCA in combination
UDCA has been employed in combination with prednisolone and azathioprine[96]. The triple regimen comprised a daily dose of UDCA 650 mg plus prednisolone (from a starting dose of 1 mg/kg b.w. to a final dose of 5-10 mg/kg b.w.) and azathioprine 1-1.5 mg/kg b.w. In the 15 patients followed up for 41 mo, there was a rapid and relevant decrease of liver enzyme levels and also AP and AST (56% decrease), ALT (65% decrease), and bilirubin (27% decrease). ERCP and liver histology were also improved and only 1 patient developed dominant strictures as a complication of the disease. These promising results need to be confirmed by larger and controlled studies.
D-penicillamine
Because of increased copper deposits in PSC liver, the Mayo Clinic group evaluated the effect of D-penicillamine on 70 patients for 36 mo. There was no beneficial effect on disease progression[97]. The onset of important side effects (e.g., proteinuria) was a reason to abandon this treatment.
Corticosteroids and other immunosuppressants
Based on the hypothesis that PSC has an immunologic cause, corticosteroids and other immunosuppressants were used for PSC. Oral corticosteroids yielded an initial improvement in the biochemical profile. However, lack of evidence for the long term benefit as well as bone demineralization, is an argument against the use of this regimen[98]. Whereas tacrolimus (FK506) resulted in a significant improvement of liver biochemistry in 10 PSC patients after 1 year of treatment[99]. In another study, methotrexate was ineffective[6]. Other medications such as azathioprine, cyclosporine, tested in association with corticosteroids and UDCA, have never been evaluated alone in the therapy of PSC[61].
Other drugs for chronic cholestasis
Pruritus in PSC can be common and often disabling. As far as bile flow is preserved, a suitable approach is sequestering luminal bile salts. Cholestyramine, the chloride salt of a non-absorbed basic anion-exchange resin is effective at an oral dose of 4 g t.i.d.[100]. In patients who do not tolerate cholestyramine, an alternative is the ammonium resin cholestipol hydrochloride. Due to their affinity to di-hydroxy bile salts, these resins must be taken apart from UDCA. In patients not responding to resins, rifampine 150 mg b.i.d. can be effective as well as phenobarbital (60-100 mg at bedtime), anti-histamines, naloxone and naltrexone[61]. There is no proven therapy for osteoporosis in PSC, options might include drugs such as 25-hydroxyvitamin D plus calcium[100], calcitonin, and biphosphonates. Studies performed with biphosphonates like etidronate in PBC[101,102] suggested that these drugs could be valuable in PSC, too. When chronic jaundice develops, it is necessary to monitor fat-soluble vitamin levels in order to treat deficiencies with supplements. Antibiotics usually manage bacterial cholangitis with a high penetration rate in biliary tract like cyprofloxacine. Alternative drugs are amoxycillin and trimethoprim-sulfametoxazole[61].
Endoscopic treatment
Therapeutic ERCP may be effective in PSC patients with symptomatic dominant strictures (i.e., discrete areas of narrowing within the extrahepatic biliary tree), gallstones or debris[103-106]. Other studies found that PSC patients undergoing endoscopic treatment had an increased survival, which was much higher than that predicted from survival models[103,107]. Endoscopic treatment may prevent biliary obstruction, which seems to be the main cause of cirrhosis in these patients. Methods include catheter or balloon dilatation (Figure 5), temporary stent placement, and nasobiliary drainage with or without lavage. Endoscopic treatment is considered to be a valuable option in addition to medical treatment[2,106].
Figure 5.
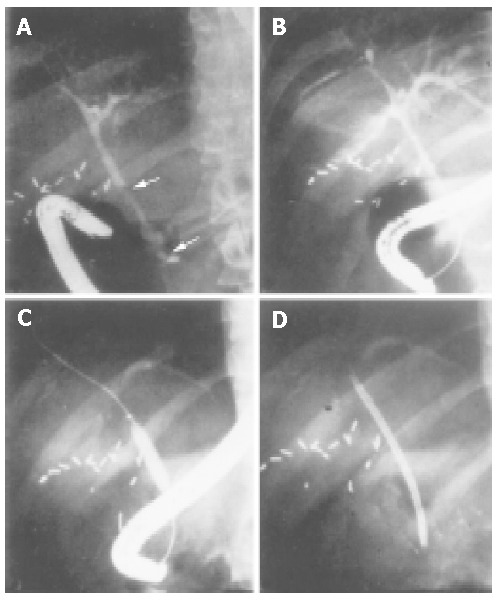
Sequence of balloon dilatation during ERCP treatment in a PSC patient with prior multiple bile duct strictures.
Liver transplantation
Orthotopic liver transplantation (OLT) is an effective therapy for PSC and the only life-saving option for the end-stage disease (>85% survival at 3 years)[108-110]. In patients with PSC and UC undergoing OLT, intestinal symptoms subside or remain quiescent in the post transplantation period[111]. Following OLT, however, PSC tends to recur in 15-30% of patients, and there is also a high recurrence rate of biliary strictures, chronic rejection, and reflux cholangitis[112]. Unfortunately, use of immunosuppr-essants such as orthoclone or corticosteroids could not improve survival and recurrence of the disease[112]. Indications for OLT are well accepted and have been recently reviewed. Each patient should be assessed individually keeping in mind that important factors for OLT are both difficult prediction of disease course and the overall increased risk of hepatobiliary malignancies (i.e., cholangiocarcinoma and hepatocellular carcinoma). Indications related to the end-stage disease include jaundice, which cannot be alleviated endoscopically or with medical therapy, cirrhosis with reduced liver function, variceal bleeding, portal gastropathy, intractable ascites, hepatic encephalopathy, severe recurrent bacterial cholangitis, progressive muscle wasting, disabling fatigue, and suspected hepatocellular carcinoma or cholangiocarcinoma[7,61,113,114].
Proctocolectomy
In theory this procedure could improve the natural history of PSC. Two studies, however, found no effect on symptoms, biochemical, radiological, histological features of PSC and survival after proctocolectomy[54,115]. This surgical approach, however, should be always performed in case of intractable IBD, colonic dysplasia, and colonic cancer.
Biliary surgery
This approach should be avoided because of the risk of complicating cholangitis[116] and because previous surgery is a contraindication for liver transplantation[117].
CONCLUSIONS
PSC is a disease of unknown cause implying progressive fibrosis and ultimately disappearance of intra- and/or extra hepatic ducts. Although PSC is not a common disease, it represents a diagnostic and therapeutic challenge for the physicians and ultimately involves several body regions. The disease is poorly symptomatic in most cases and cholestatic profile appears only at a later stage, in particular when a dominant stenosis develops. Moreover, signs and symptoms are not specific and overlap with other biliary diseases, while laboratory findings are poorly diagnostic since all liver enzymes can be normal or only slightly increased. Indeed, AP levels in adults and γGT levels in children are the most sensitive tests when PSC is suspected. Immunological tests, on the other hand, can be misleading since hyper-γ-globulinemia and increased IgM levels are found only in less than half of the patients with different types of autoantibodies and a low frequency of occurrence. Whereas both ERCP and PTC are the only useful tools for diagnosing PSC, they become diagnostic only in advanced PSC. In the future, as the sensitivity and specificity raise, less invasive tools such as MRCP and MR will need to be included in the diagnostic workup for PSC. Lastly, liver histology is useful for PSC diagnosis but a high number of false negatives are possible at earlier stages, due to the focal distribution of lesions. There is no established therapy for PSC but some drugs may relieve symptoms and prolong survival. Such drugs include high doses of UDCA, alone or standard doses of UDCA in combination with azathioprine and prednisolone. Tacrolimus shows promising results, although longer trials are needed to show an ultimate effect on the progression of the disease. Waiting for more effective medical treatments, liver transplant is the only definitive therapy for PSC, although 15-30% of transplanted patients would have PSC recurrence.
Footnotes
Assistant Editor Guo SY Edited by Wang XL and Ma JY
References
- 1.Delbet P. Retrecissement du choledoque: cholecystoduo-denostomie. Bull Mem Soc Nat Chir. 1924;50:1144–1146. [Google Scholar]
- 2.Sherlock S, Dooley J. Diseases of the liver and biliary system. Oxford: Blackwell Science; 2002. [Google Scholar]
- 3.Olsson R, Danielsson A, Järnerot G, Lindström E, Lööf L, Rolny P, Rydén BO, Tysk C, Wallerstedt S. Prevalence of primary sclerosing cholangitis in patients with ulcerative colitis. Gastroenterology. 1991;100:1319–1323. [PubMed] [Google Scholar]
- 4.Schrumpf E, Fausa O, Elgjo K, Kolmannskog F. Hepatobiliary complications of inflammatory bowel disease. Semin Liver Dis. 1988;8:201–209. doi: 10.1055/s-2008-1040541. [DOI] [PubMed] [Google Scholar]
- 5.Shepherd HA, Selby WS, Chapman RW, Nolan D, Barbatis C, McGee JO, Jewell DP. Ulcerative colitis and persistent liver dysfunction. Q J Med. 1983;52:503–513. [PubMed] [Google Scholar]
- 6.Lee YM, Kaplan MM. Primary sclerosing cholangitis. N Engl J Med. 1995;332:924–933. doi: 10.1056/NEJM199504063321406. [DOI] [PubMed] [Google Scholar]
- 7.Angulo P, Lindor KD. Primary sclerosing cholangitis. Hepatology. 1999;30:325–332. doi: 10.1002/hep.510300101. [DOI] [PubMed] [Google Scholar]
- 8.Schrumpf E, Fausa O, Førre O, Dobloug JH, Ritland S, Thorsby E. HLA antigens and immunoregulatory T cells in ulcerative colitis associated with hepatobiliary disease. Scand J Gastroenterol. 1982;17:187–191. doi: 10.3109/00365528209182038. [DOI] [PubMed] [Google Scholar]
- 9.Donaldson PT, Farrant JM, Wilkinson ML, Hayllar K, Portmann BC, Williams R. Dual association of HLA DR2 and DR3 with primary sclerosing cholangitis. Hepatology. 1991;13:129–133. [PubMed] [Google Scholar]
- 10.Chapman RW, Varghese Z, Gaul R, Patel G, Kokinon N, Sherlock S. Association of primary sclerosing cholangitis with HLA-B8. Gut. 1983;24:38–41. doi: 10.1136/gut.24.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zetterquist H, Broomé U, Einarsson K, Olerup O. HLA class II genes in primary sclerosing cholangitis and chronic inflammatory bowel disease: no HLA-DRw52a association in Swedish patients with sclerosing cholangitis. Gut. 1992;33:942–946. doi: 10.1136/gut.33.7.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prochazka EJ, Terasaki PI, Park MS, Goldstein LI, Busuttil RW. Association of primary sclerosing cholangitis with HLA-DRw52a. N Engl J Med. 1990;322:1842–1844. doi: 10.1056/NEJM199006283222603. [DOI] [PubMed] [Google Scholar]
- 13.Mehal WZ, Lo YM, Wordsworth BP, Neuberger JM, Hubscher SC, Fleming KA, Chapman RW. HLA DR4 is a marker for rapid disease progression in primary sclerosing cholangitis. Gastroenterology. 1994;106:160–167. doi: 10.1016/s0016-5085(94)95085-7. [DOI] [PubMed] [Google Scholar]
- 14.Duerr RH, Targan SR, Landers CJ, LaRusso NF, Lindsay KL, Wiesner RH, Shanahan F. Neutrophil cytoplasmic antibodies: a link between primary sclerosing cholangitis and ulcerative colitis. Gastroenterology. 1991;100:1385–1391. [PubMed] [Google Scholar]
- 15.Zauli D, Schrumpf E, Crespi C, Cassani F, Fausa O, Aadland E. An autoantibody profile in primary sclerosing cholangitis. J Hepatol. 1987;5:14–18. doi: 10.1016/s0168-8278(87)80055-7. [DOI] [PubMed] [Google Scholar]
- 16.Snook JA, Chapman RW, Fleming K, Jewell DP. Anti-neutrophil nuclear antibody in ulcerative colitis, Crohn's disease and primary sclerosing cholangitis. Clin Exp Immunol. 1989;76:30–33. [PMC free article] [PubMed] [Google Scholar]
- 17.Wiesner RH, LaRusso NF, Ludwig J, Dickson ER. Comparison of the clinicopathologic features of primary sclerosing cholangitis and primary biliary cirrhosis. Gastroenterology. 1985;88:108–114. doi: 10.1016/s0016-5085(85)80141-4. [DOI] [PubMed] [Google Scholar]
- 18.Bodenheimer HC, LaRusso NF, Thayer WR, Charland C, Staples PJ, Ludwig J. Elevated circulating immune complexes in primary sclerosing cholangitis. Hepatology. 1983;3:150–154. doi: 10.1002/hep.1840030203. [DOI] [PubMed] [Google Scholar]
- 19.Lindor KD, Wiesner RH, Katzmann JA, LaRusso NF, Beaver SJ. Lymphocyte subsets in primary sclerosing cholangitis. Dig Dis Sci. 1987;32:720–725. doi: 10.1007/BF01296138. [DOI] [PubMed] [Google Scholar]
- 20.Brinch L, Teisberg P, Schrumpf E, Akesson I. The in vivo metabolism of C3 in hepatobiliary disease associated with ulcerative colitis. Scand J Gastroenterol. 1982;17:523–527. doi: 10.3109/00365528209182243. [DOI] [PubMed] [Google Scholar]
- 21.Ludwig J, Barham SS, LaRusso NF, Elveback LR, Wiesner RH, McCall JT. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hepatology. 1981;1:632–640. doi: 10.1002/hep.1840010612. [DOI] [PubMed] [Google Scholar]
- 22.Chapman RW, Kelly PM, Heryet A, Jewell DP, Fleming KA. Expression of HLA-DR antigens on bile duct epithelium in primary sclerosing cholangitis. Gut. 1988;29:422–427. doi: 10.1136/gut.29.4.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eade MN, Brooke BN. Portal bacteraemia in cases of ulcerative colitis submitted to colectomy. Lancet. 1969;1:1008–1009. doi: 10.1016/s0140-6736(69)91802-9. [DOI] [PubMed] [Google Scholar]
- 24.Warren KW, Athanassiades S, Monge JI. Primary sclerosing cholangitis. A study of forty-two cases. Am J Surg. 1966;111:23–38. doi: 10.1016/0002-9610(66)90339-4. [DOI] [PubMed] [Google Scholar]
- 25.Steckman M, Drossman DA, Lesesne HR. Hepatobiliary disease that precedes ulcerative colitis. J Clin Gastroenterol. 1984;6:425–428. doi: 10.1097/00004836-198410000-00006. [DOI] [PubMed] [Google Scholar]
- 26.van Erpecum KJ, Smits SJ, van de Meeberg PC, Linn FH, Wolfhagen FH, vanBerge-Henegouwen GP, Algra A. Risk of primary sclerosing cholangitis is associated with nonsmoking behavior. Gastroenterology. 1996;110:1503–1506. doi: 10.1053/gast.1996.v110.pm8613056. [DOI] [PubMed] [Google Scholar]
- 27.Pullan RD, Rhodes J, Ganesh S, Mani V, Morris JS, Williams GT, Newcombe RG, Russell MA, Feyerabend C, Thomas GA. Transdermal nicotine for active ulcerative colitis. N Engl J Med. 1994;330:811–815. doi: 10.1056/NEJM199403243301202. [DOI] [PubMed] [Google Scholar]
- 28.Vleggaar FP, van Buuren HR, van Berge Henegouwen GP, Hop WC, van Erpecum KJ. No beneficial effects of transdermal nicotine in patients with primary sclerosing cholangitis: results of a randomized double-blind placebo-controlled cross-over study. Eur J Gastroenterol Hepatol. 2001;13:171–175. doi: 10.1097/00042737-200102000-00013. [DOI] [PubMed] [Google Scholar]
- 29.Northover JM, Terblanche J. A new look at the arterial supply of the bile duct in man and its surgical implications. Br J Surg. 1979;66:379–384. doi: 10.1002/bjs.1800660603. [DOI] [PubMed] [Google Scholar]
- 30.Ludwig J, Kim CH, Wiesner RH, Krom RA. Floxuridine-induced sclerosing cholangitis: an ischemic cholangiopathy? Hepatology. 1989;9:215–218. doi: 10.1002/hep.1840090209. [DOI] [PubMed] [Google Scholar]
- 31.Ludwig J, LaRusso NF, Wiesner RH. The syndrome of primary sclerosing cholangitis. Prog Liver Dis. 1990;9:555–566. [PubMed] [Google Scholar]
- 32.Wiesner RH, Porayko MK, La Russo NF, Ludwig J. Primary Sclerosing Cholangitis. Schift Schift JBLCP, editor, 7th [Disease of the liver]. 1991. In: Schift Schift JBLCP, editor. Ref Type: Serial (Book,Monograph); [Google Scholar]
- 33.Chapman RW, Arborgh BA, Rhodes JM, Summerfield JA, Dick R, Scheuer PJ, Sherlock S. Primary sclerosing cholangitis: a review of its clinical features, cholangiography, and hepatic histology. Gut. 1980;21:870–877. doi: 10.1136/gut.21.10.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balasubramaniam K, Wiesner RH, LaRusso NF. Primary sclerosing cholangitis with normal serum alkaline phosphatase activity. Gastroenterology. 1988;95:1395–1398. doi: 10.1016/0016-5085(88)90378-2. [DOI] [PubMed] [Google Scholar]
- 35.Wiesner RH, Ludwig J, LaRusso NF, MacCarty RL. Diagnosis and treatment of primary sclerosing cholangitis. Semin Liver Dis. 1985;5:241–253. doi: 10.1055/s-2008-1040621. [DOI] [PubMed] [Google Scholar]
- 36.Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary sclerosing cholangitis in children: a long-term follow-up study. Hepatology. 2003;38:210–217. doi: 10.1053/jhep.2003.50289. [DOI] [PubMed] [Google Scholar]
- 37.Wiesner RH, LaRusso NF. Clinicopathologic features of the syndrome of primary sclerosing cholangitis. Gastroenterology. 1980;79:200–206. [PubMed] [Google Scholar]
- 38.van Milligen de Wit AW, van Deventer SJ, Tytgat GN. Immunogenetic aspects of primary sclerosing cholangitis: implications for therapeutic strategies. Am J Gastroenterol. 1995;90:893–900. [PubMed] [Google Scholar]
- 39.Bader TR, Beavers KL, Semelka RC. MR imaging features of primary sclerosing cholangitis: patterns of cirrhosis in relationship to clinical severity of disease. Radiology. 2003;226:675–685. doi: 10.1148/radiol.2263011623. [DOI] [PubMed] [Google Scholar]
- 40.MacCarty RL, LaRusso NF, Wiesner RH, Ludwig J. Primary sclerosing cholangitis: findings on cholangiography and pancreatography. Radiology. 1983;149:39–44. doi: 10.1148/radiology.149.1.6412283. [DOI] [PubMed] [Google Scholar]
- 41.Chen LY, Goldberg HI. Sclerosing cholangitis: broad spectrum of radiographic features. Gastrointest Radiol. 1984;9:39–47. doi: 10.1007/BF01887799. [DOI] [PubMed] [Google Scholar]
- 42.Ludwig J, MacCarty RL, LaRusso NF, Krom RA, Wiesner RH. Intrahepatic cholangiectases and large-duct obliteration in primary sclerosing cholangitis. Hepatology. 1986;6:560–568. doi: 10.1002/hep.1840060403. [DOI] [PubMed] [Google Scholar]
- 43.Jeffrey GP, Reed WD, Carrello S, Shilkin KB. Histological and immunohistochemical study of the gall bladder lesion in primary sclerosing cholangitis. Gut. 1991;32:424–429. doi: 10.1136/gut.32.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gluskin LE, Payne JA. Cystic dilatation as a radiographic sign of cholangiocarcinoma complicating sclerosing cholangitis. Am J Gastroenterol. 1983;78:661–664. [PubMed] [Google Scholar]
- 45.MacCarty RL, LaRusso NF, May GR, Bender CE, Wiesner RH, King JE, Coffey RJ. Cholangiocarcinoma complicating primary sclerosing cholangitis: cholangiographic appearances. Radiology. 1985;156:43–46. doi: 10.1148/radiology.156.1.2988012. [DOI] [PubMed] [Google Scholar]
- 46.Textor HJ, Flacke S, Pauleit D, Keller E, Neubrand M, Terjung B, Gieseke J, Scheurlen C, Sauerbruch T, Schild HH. Three-dimensional magnetic resonance cholangiopancreatography with respiratory triggering in the diagnosis of primary sclerosing cholangitis: comparison with endoscopic retrograde cholangiography. Endoscopy. 2002;34:984–990. doi: 10.1055/s-2002-35830. [DOI] [PubMed] [Google Scholar]
- 47.Weber C, Krupski G, Lorenzen J, Grotelüschen R, Seitz U, Rogiers X, Adam G. MRCP in primary sclerosing cholangitis. Rofo. 2003;175:203–210. doi: 10.1055/s-2003-37228. [DOI] [PubMed] [Google Scholar]
- 48.van de Meeberg PC, Portincasa P, Wolfhagen FH, van Erpecum KJ, VanBerge-Henegouwen GP. Increased gall bladder volume in primary sclerosing cholangitis. Gut. 1996;39:594–599. doi: 10.1136/gut.39.4.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scheuer PJ. Ludwig Symposium on biliary disorders--part II. Pathologic features and evolution of primary biliary cirrhosis and primary sclerosing cholangitis. Mayo Clin Proc. 1998;73:179–183. doi: 10.4065/73.2.179. [DOI] [PubMed] [Google Scholar]
- 50.LaRusso NF, Ludwig J. Primary sclerosing cholangitis. Dtsch Med Wochenschr. 1986;111:1263. [PubMed] [Google Scholar]
- 51.Wee A, Ludwig J. Pericholangitis in chronic ulcerative colitis: primary sclerosing cholangitis of the small bile ducts? Ann Intern Med. 1985;102:581–587. doi: 10.7326/0003-4819-102-5-581. [DOI] [PubMed] [Google Scholar]
- 52.Katabi N, Albores-Saavedra J. The extrahepatic bile duct lesions in end-stage primary sclerosing cholangitis. Am J Surg Pathol. 2003;27:349–355. doi: 10.1097/00000478-200303000-00008. [DOI] [PubMed] [Google Scholar]
- 53.Wiesner RH, Grambsch P, LaRusso NF, Dickson ER. Is primary sclerosing cholangitis a progressive disease or not? Hepatology. 1988;8:970–972. doi: 10.1002/hep.1840080444. [DOI] [PubMed] [Google Scholar]
- 54.Martin FM, Rossi RL, Nugent FW, Scholz FJ, Jenkins RL, Lewis WD, Gagner M, Foley E, Braasch JW. Surgical aspects of sclerosing cholangitis. Results in 178 patients. Ann Surg. 1990;212:551–556; discussion 556-558. doi: 10.1097/00000658-199010000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aadland E, Schrumpf E, Fausa O, Elgjo K, Heilo A, Aakhus T, Gjone E. Primary sclerosing cholangitis: a long-term follow-up study. Scand J Gastroenterol. 1987;22:655–664. doi: 10.3109/00365528709011139. [DOI] [PubMed] [Google Scholar]
- 56.Janes CH, Dickson ER, Okazaki R, Bonde S, McDonagh AF, Riggs BL. Role of hyperbilirubinemia in the impairment of osteoblast proliferation associated with cholestatic jaundice. J Clin Invest. 1995;95:2581–2586. doi: 10.1172/JCI117959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brandt DJ, MacCarty RL, Charboneau JW, LaRusso NF, Wiesner RH, Ludwig J. Gallbladder disease in patients with primary sclerosing cholangitis. AJR Am J Roentgenol. 1988;150:571–574. doi: 10.2214/ajr.150.3.571. [DOI] [PubMed] [Google Scholar]
- 58.Martin EC, Fankuchen EI, Schultz RW, Casarella WJ. Percutaneous dilatation in primary sclerosing cholangitis: two experiences. AJR Am J Roentgenol. 1981;137:603–605. doi: 10.2214/ajr.137.3.603. [DOI] [PubMed] [Google Scholar]
- 59.May GR, Bender CE, LaRusso NF, Wiesner RH. Nonoperative dilatation of dominant strictures in primary sclerosing cholangitis. AJR Am J Roentgenol. 1985;145:1061–1064. doi: 10.2214/ajr.145.5.1061. [DOI] [PubMed] [Google Scholar]
- 60.Johnson GK, Geenen JE, Venu RP, Hogan WJ. Endoscopic treatment of biliary duct strictures in sclerosing cholangitis: follow-up assessment of a new therapeutic approach. Gastrointest Endosc. 1987;33:9–12. doi: 10.1016/s0016-5107(87)71475-8. [DOI] [PubMed] [Google Scholar]
- 61.Lee YM, Kaplan MM. Management of primary sclerosing cholangitis. Am J Gastroenterol. 2002;97:528–534. doi: 10.1111/j.1572-0241.2002.05585.x. [DOI] [PubMed] [Google Scholar]
- 62.Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–649. doi: 10.1002/bjs.1800600817. [DOI] [PubMed] [Google Scholar]
- 63.Ponsioen CI, Tytgat GN. Primary sclerosing cholangitis: a clinical review. Am J Gastroenterol. 1998;93:515–523. doi: 10.1111/j.1572-0241.1998.157_b.x. [DOI] [PubMed] [Google Scholar]
- 64.Olsson RG, Asztély MS. Prognostic value of cholangiography in primary sclerosing cholangitis. Eur J Gastroenterol Hepatol. 1995;7:251–254. [PubMed] [Google Scholar]
- 65.Kaya M, Angulo P, Lindor KD. Overlap of autoimmune hepatitis and primary sclerosing cholangitis: an evaluation of a modified scoring system. J Hepatol. 2000;33:537–542. doi: 10.1034/j.1600-0641.2000.033004537.x. [DOI] [PubMed] [Google Scholar]
- 66.van Buuren HR, van Hoogstraten HJE T, Schalm SW, Vleggaar FP. High prevalence of autoimmune hepatitis among patients with primary sclerosing cholangitis. J Hepatol. 2000;33:543–548. doi: 10.1034/j.1600-0641.2000.033004543.x. [DOI] [PubMed] [Google Scholar]
- 67.Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary sclerosing cholangitis in children: a long-term follow-up study. Hepatology. 2003;38:210–217. doi: 10.1053/jhep.2003.50289. [DOI] [PubMed] [Google Scholar]
- 68.Rubin RA, Kowalski TE, Khandelwal M, Malet PF. Ursodiol for hepatobiliary disorders. Ann Intern Med. 1994;121:207–218. doi: 10.7326/0003-4819-121-3-199408010-00009. [DOI] [PubMed] [Google Scholar]
- 69.Sauer P, Benz C, Rudolph G, Klöters-Plachky P, Stremmel W, Stiehl A. Influence of cholestasis on absorption of ursodeoxycholic acid. Dig Dis Sci. 1999;44:817–822. doi: 10.1023/a:1026686530785. [DOI] [PubMed] [Google Scholar]
- 70.Kullak-Ublick GA, Stieger B, Hagenbuch B, Meier PJ. Hepatic transport of bile salts. Semin Liver Dis. 2000;20:273–292. doi: 10.1055/s-2000-9426. [DOI] [PubMed] [Google Scholar]
- 71.Hofmann AF. Pharmacology of ursodeoxycholic acid, an enterohepatic drug. Scand J Gastroenterol Suppl. 1994;204:1–15. doi: 10.3109/00365529409103618. [DOI] [PubMed] [Google Scholar]
- 72.Saksena S, Tandon RK. Ursodeoxycholic acid in the treatment of liver diseases. Postgrad Med J. 1997;73:75–80. doi: 10.1136/pgmj.73.856.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ward A, Brogden RN, Heel RC, Speight TM, Avery GS. Ursodeoxycholic acid: a review of its pharmacological properties and therapeutic efficacy. Drugs. 1984;27:95–131. doi: 10.2165/00003495-198427020-00001. [DOI] [PubMed] [Google Scholar]
- 74.Stiehl A, Benz C, Sauer P. Mechanism of hepatoprotective action of bile salts in liver disease. Gastroenterol Clin North Am. 1999;28:195–209, viii. doi: 10.1016/s0889-8553(05)70050-9. [DOI] [PubMed] [Google Scholar]
- 75.Stiehl A, Raedsch R, Rudolph G. Acute effects of ursodeoxycholic and chenodeoxycholic acid on the small intestinal absorption of bile acids. Gastroenterology. 1990;98:424–428. doi: 10.1016/0016-5085(90)90834-n. [DOI] [PubMed] [Google Scholar]
- 76.Stiehl A, Czygan P, Kommerell B, Weis HJ, Holtermüller KH. Ursodeoxycholic acid versus chenodeoxycholic acid. Comparison of their effects on bile acid and bile lipid composition in patients with cholesterol gallstones. Gastroenterology. 1978;75:1016–1020. [PubMed] [Google Scholar]
- 77.Crosignani A, Podda M, Battezzati PM, Bertolini E, Zuin M, Watson D, Setchell KD. Changes in bile acid composition in patients with primary biliary cirrhosis induced by ursodeoxycholic acid administration. Hepatology. 1991;14:1000–1007. [PubMed] [Google Scholar]
- 78.Trauner M, Graziadei IW. Review article: mechanisms of action and therapeutic applications of ursodeoxycholic acid in chronic liver diseases. Aliment Pharmacol Ther. 1999;13:979–996. doi: 10.1046/j.1365-2036.1999.00596.x. [DOI] [PubMed] [Google Scholar]
- 79.Jazrawi RP, de Caestecker JS, Goggin PM, Britten AJ, Joseph AE, Maxwell JD, Northfield TC. Kinetics of hepatic bile acid handling in cholestatic liver disease: effect of ursodeoxycholic acid. Gastroenterology. 1994;106:134–142. doi: 10.1016/s0016-5085(94)94899-2. [DOI] [PubMed] [Google Scholar]
- 80.Dumont M, Erlinger S, Uchman S. Hypercholeresis induced by ursodeoxycholic acid and 7-ketolithocholic acid in the rat: possible role of bicarbonate transport. Gastroenterology. 1980;79:82–89. [PubMed] [Google Scholar]
- 81.Yoon YB, Hagey LR, Hofmann AF, Gurantz D, Michelotti EL, Steinbach JH. Effect of side-chain shortening on the physiologic properties of bile acids: hepatic transport and effect on biliary secretion of 23-nor-ursodeoxycholate in rodents. Gastroenterology. 1986;90:837–852. doi: 10.1016/0016-5085(86)90859-0. [DOI] [PubMed] [Google Scholar]
- 82.Terasaki S, Nakanuma Y, Ogino H, Unoura M, Kobayashi K. Hepatocellular and biliary expression of HLA antigens in primary biliary cirrhosis before and after ursodeoxycholic acid therapy. Am J Gastroenterol. 1991;86:1194–1199. [PubMed] [Google Scholar]
- 83.Calmus Y, Gane P, Rouger P, Poupon R. Hepatic expression of class I and class II major histocompatibility complex molecules in primary biliary cirrhosis: effect of ursodeoxycholic acid. Hepatology. 1990;11:12–15. doi: 10.1002/hep.1840110104. [DOI] [PubMed] [Google Scholar]
- 84.Rodrigues CM, Fan G, Ma X, Kren BT, Steer CJ. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest. 1998;101:2790–2799. doi: 10.1172/JCI1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lazaridis KN, Gores GJ, Lindor KD. Ursodeoxycholic acid 'mechanisms of action and clinical use in hepatobiliary disorders'. J Hepatol. 2001;35:134–146. doi: 10.1016/s0168-8278(01)00092-7. [DOI] [PubMed] [Google Scholar]
- 86.Moschetta A, vanBerge-Henegouwen GP, Portincasa P, Renooij WL, Groen AK, van Erpecum KJ. Hydrophilic bile salts enhance differential distribution of sphingomyelin and phosphatidylcholine between micellar and vesicular phases: potential implications for their effects in vivo. J Hepatol. 2001;34:492–499. doi: 10.1016/s0168-8278(00)00046-5. [DOI] [PubMed] [Google Scholar]
- 87.Bayerdörffer E, Mannes GA, Richter WO, Ochsenkühn T, Wiebecke B, Köpcke W, Paumgartner G. Increased serum deoxycholic acid levels in men with colorectal adenomas. Gastroenterology. 1993;104:145–151. doi: 10.1016/0016-5085(93)90846-5. [DOI] [PubMed] [Google Scholar]
- 88.Pardi DS, Loftus EV, Kremers WK, Keach J, Lindor KD. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003;124:889–893. doi: 10.1053/gast.2003.50156. [DOI] [PubMed] [Google Scholar]
- 89.Hill MJ, Melville DM, Lennard-Jones JE, Neale K, Ritchie JK. Faecal bile acids, dysplasia, and carcinoma in ulcerative colitis. Lancet. 1987;2:185–186. doi: 10.1016/s0140-6736(87)90766-5. [DOI] [PubMed] [Google Scholar]
- 90.Beuers U, Spengler U, Kruis W, Aydemir U, Wiebecke B, Heldwein W, Weinzierl M, Pape GR, Sauerbruch T, Paumgartner G. Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: a placebo-controlled trial. Hepatology. 1992;16:707–714. doi: 10.1002/hep.1840160315. [DOI] [PubMed] [Google Scholar]
- 91.Stiehl A, Walker S, Stiehl L, Rudolph G, Hofmann WJ, Theilmann L. Effect of ursodeoxycholic acid on liver and bile duct disease in primary sclerosing cholangitis. A 3-year pilot study with a placebo-controlled study period. J Hepatol. 1994;20:57–64. doi: 10.1016/s0168-8278(05)80467-2. [DOI] [PubMed] [Google Scholar]
- 92.Okolicsanyi L, Groppo M, Floreani A, Morselli-Labate AM, Rusticali AG, Battocchia A, Colombo M, Galatola G, Gasbarrini G, Podda M, et al. Treatment of primary sclerosing cholangitis with low-dose ursodeoxycholic acid: results of a retrospective Italian multicentre survey. Dig Liver Dis. 2003;35:325–331. doi: 10.1016/s1590-8658(03)00076-8. [DOI] [PubMed] [Google Scholar]
- 93.van Hoogstraten HJ, Wolfhagen FH, van de Meeberg PC, Kuiper H, Nix GA, Becx MC, Hoek AC, van Houte DP, Rijk MC, Salemans JM, et al. Ursodeoxycholic acid therapy for primary sclerosing cholangitis: results of a 2-year randomized controlled trial to evaluate single versus multiple daily doses. J Hepatol. 1998;29:417–423. doi: 10.1016/s0168-8278(98)80059-7. [DOI] [PubMed] [Google Scholar]
- 94.Mitchell SA, Bansi DS, Hunt N, Von Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology. 2001;121:900–907. doi: 10.1053/gast.2001.27965. [DOI] [PubMed] [Google Scholar]
- 95.Harnois DM, Angulo P, Jorgensen RA, Larusso NF, Lindor KD. High-dose ursodeoxycholic acid as a therapy for patients with primary sclerosing cholangitis. Am J Gastroenterol. 2001;96:1558–1562. doi: 10.1111/j.1572-0241.2001.03777.x. [DOI] [PubMed] [Google Scholar]
- 96.Schramm C, Schirmacher P, Helmreich-Becker I, Gerken G, zum Büschenfelde KH, Lohse AW. Combined therapy with azathioprine, prednisolone, and ursodiol in patients with primary sclerosing cholangitis. A case series. Ann Intern Med. 1999;131:943–946. doi: 10.7326/0003-4819-131-12-199912210-00006. [DOI] [PubMed] [Google Scholar]
- 97.LaRusso NF, Wiesner RH, Ludwig J, MacCarty RL, Beaver SJ, Zinsmeister AR. Prospective trial of penicillamine in primary sclerosing cholangitis. Gastroenterology. 1988;95:1036–1042. doi: 10.1016/0016-5085(88)90180-1. [DOI] [PubMed] [Google Scholar]
- 98.Lindor KD, Wiesner RH, Colwell LJ, Steiner B, Beaver S, LaRusso NF. The combination of prednisone and colchicine in patients with primary sclerosing cholangitis. Am J Gastroenterol. 1991;86:57–61. [PubMed] [Google Scholar]
- 99.Van Thiel DH, Carroll P, Abu-Elmagd K, Rodriguez-Rilo H, Irish W, McMichael J, Starzl TE. Tacrolimus (FK 506), a treatment for primary sclerosing cholangitis: results of an open-label preliminary trial. Am J Gastroenterol. 1995;90:455–459. [PMC free article] [PubMed] [Google Scholar]
- 100.Levy C, Lindor KD. Treatment Options for Primary Biliary Cirrhosis and Primary Sclerosing Cholangitis. Curr Treat Options Gastroenterol. 2003;6:93–103. doi: 10.1007/s11938-003-0010-0. [DOI] [PubMed] [Google Scholar]
- 101.Wolfhagen FH, van Buuren HR, Vleggaar FP, Schalm SW. Management of osteoporosis in primary biliary cirrhosis. Baillieres Best Pract Res Clin Gastroenterol. 2000;14:629–641. doi: 10.1053/bega.2000.0108. [DOI] [PubMed] [Google Scholar]
- 102.Vleggaar FP, van Buuren HR, Wolfhagen FH, Schalm SW, Pols HA. Prevention and treatment of osteoporosis in primary biliary cirrhosis. Eur J Gastroenterol Hepatol. 1999;11:617–621. doi: 10.1097/00042737-199906000-00006. [DOI] [PubMed] [Google Scholar]
- 103.Baluyut AR, Sherman S, Lehman GA, Hoen H, Chalasani N. Impact of endoscopic therapy on the survival of patients with primary sclerosing cholangitis. Gastrointest Endosc. 2001;53:308–312. doi: 10.1016/s0016-5107(01)70403-8. [DOI] [PubMed] [Google Scholar]
- 104.Cotton PB, Nickl N. Endoscopic and radiologic approaches to therapy in primary sclerosing cholangitis. Semin Liver Dis. 1991;11:40–48. doi: 10.1055/s-2008-1040421. [DOI] [PubMed] [Google Scholar]
- 105.Johnson GK, Geenen JE, Venu RP, Schmalz MJ, Hogan WJ. Endoscopic treatment of biliary tract strictures in sclerosing cholangitis: a larger series and recommendations for treatment. Gastrointest Endosc. 1991;37:38–43. doi: 10.1016/s0016-5107(91)70618-4. [DOI] [PubMed] [Google Scholar]
- 106.Stiehl A, Rudolph G, Klöters-Plachky P, Sauer P, Walker S. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: outcome after endoscopic treatment. J Hepatol. 2002;36:151–156. doi: 10.1016/s0168-8278(01)00251-3. [DOI] [PubMed] [Google Scholar]
- 107.Fogel EL, Sherman S, Park SH, McHenry L, Lehman GA. Therapeutic biliary endoscopy. Endoscopy. 2003;35:156–163. doi: 10.1055/s-2003-37011. [DOI] [PubMed] [Google Scholar]
- 108.Langnas AN, Grazi GL, Stratta RJ, Wood RP, Marujo W, Markin RS, Donovan J, Burnett D, Zetterman R, Sorrell M. Primary sclerosing cholangitis: the emerging role for liver transplantation. Am J Gastroenterol. 1990;85:1136–1141. [PubMed] [Google Scholar]
- 109.McEntee G, Wiesner RH, Rosen C, Cooper J, Wahlstrom E. A comparative study of patients undergoing liver transplantation for primary sclerosing cholangitis and primary biliary cirrhosis. Transplant Proc. 1991;23:1563–1564. [PubMed] [Google Scholar]
- 110.Scharschmidt BF. Human liver transplantation: analysis of data on 540 patients from four centers. Hepatology. 1984;4:95S–101S. doi: 10.1002/hep.1840040723. [DOI] [PubMed] [Google Scholar]
- 111.Gavaler JS, Delemos B, Belle SH, Heyl AE, Tarter RE, Starzl TE, Gavaler C, van Thiel DH. Ulcerative colitis disease activity as subjectively assessed by patient-completed questionnaires following orthotopic liver transplantation for sclerosing cholangitis. Dig Dis Sci. 1991;36:321–328. doi: 10.1007/BF01318204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kugelmas M, Spiegelman P, Osgood MJ, Young DA, Trotter JF, Steinberg T, Wachs ME, Bak T, Kam I, Everson GT. Different immunosuppressive regimens and recurrence of primary sclerosing cholangitis after liver transplantation. Liver Transpl. 2003;9:727–732. doi: 10.1053/jlts.2003.50143. [DOI] [PubMed] [Google Scholar]
- 113.Bjøro K, Schrumpf E. Liver transplantation for primary sclerosing cholangitis. J Hepatol. 2004;40:570–577. doi: 10.1016/j.jhep.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 114.Brandsaeter B, Isoniemi H, Broomé U, Olausson M, Bäckman L, Hansen B, Schrumpf E, Oksanen A, Ericzon BG, Höckerstedt K, et al. Liver transplantation for primary sclerosing cholangitis; predictors and consequences of hepatobiliary malignancy. J Hepatol. 2004;40:815–822. doi: 10.1016/j.jhep.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 115.Cangemi JR, Wiesner RH, Beaver SJ, Ludwig J, MacCarty RL, Dozois RR, Zinsmeister AR, LaRusso NF. Effect of proctocolectomy for chronic ulcerative colitis on the natural history of primary sclerosing cholangitis. Gastroenterology. 1989;96:790–794. [PubMed] [Google Scholar]
- 116.Farges O, Malassagne B, Sebagh M, Bismuth H. Primary sclerosing cholangitis: liver transplantation or biliary surgery. Surgery. 1995;117:146–155. doi: 10.1016/s0039-6060(05)80078-9. [DOI] [PubMed] [Google Scholar]
- 117.Ahrendt SA, Pitt HA. Surgical treatment for primary sclerosing cholangitis. J Hepatobiliary Pancreat Surg. 1999;6:366–372. doi: 10.1007/s005340050132. [DOI] [PubMed] [Google Scholar]
- 118.Poupon RE, Ouguerram K, Chrétien Y, Verneau C, Eschwège E, Magot T, Poupon R. Cholesterol-lowering effect of ursodeoxycholic acid in patients with primary biliary cirrhosis. Hepatology. 1993;17:577–582. doi: 10.1002/hep.1840170408. [DOI] [PubMed] [Google Scholar]
- 119.Bachrach WH, Hofmann AF. Ursodeoxycholic acid in the treatment of cholesterol cholelithiasis. Part II. Dig Dis Sci. 1982;27:833–856. doi: 10.1007/BF01391378. [DOI] [PubMed] [Google Scholar]
- 120.Bachrach WH, Hofmann AF. Ursodeoxycholic acid in the treatment of cholesterol cholelithiasis. part I. Dig Dis Sci. 1982;27:737–761. doi: 10.1007/BF01393771. [DOI] [PubMed] [Google Scholar]
- 121.Beuers U, Spengler U, Zwiebel FM, Pauletzki J, Fischer S, Paumgartner G. Effect of ursodeoxycholic acid on the kinetics of the major hydrophobic bile acids in health and in chronic cholestatic liver disease. Hepatology. 1992;15:603–608. doi: 10.1002/hep.1840150409. [DOI] [PubMed] [Google Scholar]
- 122.Rudolph G, Endele R, Senn M, Stiehl A. Effect of ursodeoxycholic acid on the kinetics of cholic acid and chenodeoxycholic acid in patients with primary sclerosing cholangitis. Hepatology. 1993;17:1028–1032. [PubMed] [Google Scholar]
- 123.Beuers U, Nathanson MH, Boyer JL. Effects of tauroursodeoxycholic acid on cytosolic Ca2+ signals in isolated rat hepatocytes. Gastroenterology. 1993;104:604–612. doi: 10.1016/0016-5085(93)90433-d. [DOI] [PubMed] [Google Scholar]
- 124.Jazrawi RP, de Caestecker JS, Goggin PM, Britten AJ, Joseph AE, Maxwell JD, Northfield TC. Kinetics of hepatic bile acid handling in cholestatic liver disease: effect of ursodeoxycholic acid. Gastroenterology. 1994;106:134–142. doi: 10.1016/s0016-5085(94)94899-2. [DOI] [PubMed] [Google Scholar]
- 125.Shimokura GH, McGill JM, Schlenker T, Fitz JG. Ursodeoxycholate increases cytosolic calcium concentration and activates Cl- currents in a biliary cell line. Gastroenterology. 1995;109:965–972. doi: 10.1016/0016-5085(95)90407-7. [DOI] [PubMed] [Google Scholar]
- 126.van Erpecum KJ, van Berge Henegouwen GP, Stolk MF, Hopman WP, Jansen JB, Lamers CB. Fasting gallbladder volume, postprandial emptying and cholecystokinin release in gallstone patients and normal subjects. J Hepatol. 1992;14:194–202. doi: 10.1016/0168-8278(92)90158-l. [DOI] [PubMed] [Google Scholar]
- 127.Dopico AM, Walsh JV, Singer JJ. Natural bile acids and synthetic analogues modulate large conductance Ca2+-activated K+ (BKCa) channel activity in smooth muscle cells. J Gen Physiol. 2002;119:251–273. doi: 10.1085/jgp.20028537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Portincasa P, DiCiaula A, Palmieri V, Velardi A, Van Berge-Henegouwen GP, Palasciano G. Tauroursodeoxycholic acid, ursodeoxycholic acid and gallbladder motility in gallstone patients and healthy subjects. Ital J Gastroenterol. 1996;28:111–113. [PubMed] [Google Scholar]
- 129.Van Erpecum KJ, Portincasa P, Eckhardt E, Go PM, VanBerge-Henegouwen GP, Groen AK. Ursodeoxycholic acid reduces protein levels and nucleation-promoting activity in human gallbladder bile. Gastroenterology. 1996;110:1225–1237. doi: 10.1053/gast.1996.v110.pm8613013. [DOI] [PubMed] [Google Scholar]
- 130.Portincasa P, van Erpecum KJ, Jansen A, Renooij W, Gadellaa M, vanBerge-Henegouwen GP. Behavior of various cholesterol crystals in bile from patients with gallstones. Hepatology. 1996;23:738–748. doi: 10.1002/hep.510230414. [DOI] [PubMed] [Google Scholar]
- 131.Pardi DS, Loftus EV, Kremers WK, Keach J, Lindor KD. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003;124:889–893. doi: 10.1053/gast.2003.50156. [DOI] [PubMed] [Google Scholar]