

Abstract
AIM: To investigate the role of TIP30 in apoptotic signal pathway in hepatoblastoma cells and to provide a basis for TIP30 as a gene therapy candidate in the regression of hepatoblastoma cells.
METHODS: Apoptosis of human hepatoblastoma cell lines HepG2 (p53 wild), Hep3B (p53 null) and PLC/RPF/5 (p53 mutant) infected with Ad-TIP30 (bearing a wild type human Tip30 gene) were analyzed and p53, Bax and Bcl-xl expression levels were compared among these cells. MTT assay, DNA fragmentation, in situ 3’ end labeling of DNA, annexin-V FITC staining were used to detect cell death and apoptosis in cells at various time intervals subsequent to infection, and to determine whether TIP30 had an effect on the expression levels of some apoptosis-related gene products such as Bax, p53 and Bcl-xl. A similar time course experiment was performed by Western blotting.
RESULTS: In MTT assay, the viability of HepG2 cells decreased significantly from 99.7% to 10% and displayed more massive cell death within 5-8 d than Hep3B and PLC/RPF/5 cells, with their viability decreased from 97.8% to 44.3% and 98.1% to 50.4%, respectively. In annexin-V FITC assay, the percentage of apoptosis cells in HepG2 cells was two to three-fold higher than that in control cells (infected with Ad-GFP), two-fold higher than that in Hep3B cells and 1.4-fold higher than that in PLC/RPF/5 cells 36 h after infection, respectively. Moreover, in HepG2 cells, the p53 began to increase 6-8 h after infection, reaching a maximum level between 8 and 12 h after infection and then dropped. Bax showed a similar increase in the cells as p53 reached the maximum at 8-12 h and subsequently decreased. Interestingly, Bcl-xl protein levels were down regulated during 24 to 36 h after Ad-TIP30 infection. In contrast, ectopic expression of TIP30 in Hep3B and PLC/RPF/5 cells had no effect on the regulation of Bax expression, but had an effect on Bcl-xl levels. In comparison with HepG2 cells, these data suggested that up-regulation of p53 levels by TIP30 might be a pre-requisite for Bax and Bax/Bcl-xl ratio increase. We hypothesized that TIP30 might regulate Bax gene partly through p53, which sensitizes cells to apoptosis by involving a p53 apoptosis signal transduction pathway.
CONCLUSION: TIP30 plays an important role in predisposing hepatoblastoma cells to apoptosis through regulating expression levels of these genes. Ad-TIP30 carrying exogenous TIP30-anti-tumor genes may be regarded as a potential candidate for the treatment of hepatocellular carcinoma.
Keywords: Hepatoblastoma, TIP30, Apoptosis, Signal transduction
INTRODUCTION
TIP30 is an evolutionarily conserved gene that is expressed ubiquitously in human tissues and some tumor tissues with properties of serine/threonine kinase, coding a polypeptide of 242 amino acids. It is identified as a cofactor to specially enhance human immunodeficiency virus-1 (HIV-1) Tat-activated transcription by phosphorylating heptapeptide repeats of C-terminal domain (CTD) of the largest RNA polymerase II subunit in a Tat-dependent manner[1,2]. Furthermore, Baker et al[2] have shown that TIP30 is an important metastasis suppression factor that accounts for a significant suppression growth retardation in small cell lung carcinoma (SCLC), which could be largely mediated by the ability of TIP30 to promote the apoptosis pathway. Moreover, ectopic expression of TIP30 in SCLC cells induces activation of apoptosis-related genes: Bad and Siva. Nevertheless, the molecular mechanism by which the TIP30 gene mediates apoptosis remains largely unknown.
We employed in vitro analysis to gain an insight into the molecular candidates other than Tat with which TIP30 interacted. In this respect, many pro-apoptotic proteins were investigated to reveal a TIP30 triggering apoptosis pathway. Among them, p53 is considered as one of the best target genes because of its pivotal role in regulating cell cycle progression as well as apoptotic cell death[3,4]. The biochemical activity of p53 closely associated with apoptosis is its function as a sequence-specific DNA-binding protein and a transcriptional factor that controls the expression of a large panel of genes implicated in engagement of apoptosis[5]. Moreover, p53 protein levels have been shown to dramatically rise in response to diverse stresses including ionizing radiation, UV radiation, growth factor deprivation or virus infection[6]. Bax gene is a pro-apoptosis member of Bcl-2 whose promoter is highly p53-responsive. Furthermore, Bcl-xl is another member of Bcl-2 family that has been found to express high levels in more than half of all human tumors[7-9]. Consistent with its anti-apoptotic role by antagonizing the Bax-dependent apoptosis pathway, it prevents all mitochondrial changes including release of cytochrome C and loss of mitochondrial membrane potential as well as caspase activation by sequestering Apaf-1[10,11]. It seems that in some cells, p53 protein-involving apoptosis modification can result in a general up-regulation of protein expression of pro-apoptotic members and down-regulation of anti-apoptotic proteins in Bcl-2 family, shifting the balance in favor of apoptosis[12]. In hepatocellular carcinoma, Bax and Bcl-xl proteins are especially involved in the regulation[13]. Although the central role of p53 as a pro-apoptotic-focus protein has been established, the complicated mechanism by which p53 exerts its interaction with some components and how many regulating factors are involved in p53-dependent pathway are far unknown.
Our study was designed to evaluate the expression of TIP30 and its target gene after infection with Ad-TIP30 to enhance our understanding of molecular basis of human hepatoma and to raise the realistic possibility of targeting the p53 pathway in cancer therapy. With the advantages of both maximum effect and minimum cytotoxicity to the target cells in genetic therapeutic approaches, adenovirus vectors are being wildly developed for gene therapy[14].
MATERIALS AND METHODS
Cells, chemicals and antibodies
Human heptoblastoma carcinoma cell lines HepG2, Hep3B and PLC/RPF/5 were grown in DMEM supplemented with 10% FBS (Hyclone). Anti-p53 antibody (Ab-2) was purchased from Sigma (Stlouis, MO, USA). Anti-Bax and anti-Bcl-xl antibodies were obtained from Oncogene Research Products (San Diego, CA).
Recombinant adenovirus infection
Cells were grown in 96-well and 6-well dishes at a density of 5×103 or 5×106 per well. Then the cells were infected with either Ad-TIP30 or Ad-GFP (no TIP30) at MOI of 20-50 MOI by co-culturing with viruses at 37 °C for 1 hour in 200-500 μL of serum free medium. An additional 100-1000 μL of complete medium was then added and cells were cultured in the presence of viruses for the indicated length of time.
Cell viability assays
XTT colorimetric assay (Oncogene Research Products, San Diego, CA) was used to determine cell proliferation and death. Briefly, cells were plated in a 96-well-plate at a concentration of 8000-10000 cells/well and infected with adenoviruses. After cells were incubated for 24 h per day, XTT reaction mixture was added, which contained XTT labeling reagent and the electron-coupling reagent in a 20:1 ratio. Cell/solution mixture was then read using a colormetric plate reader at a wavelength of 500 nm. Each sample was assayed in triplicate. Data were then analyzed with Microsoft Excel software. Cells were continually cultured for 8 d after-infection.
Apoptosis assay
Flow cytometry Cells were infected with Ad-TIP30 and Ad-GFP with mock (no virus) as control. Afterwards, infected and control cells were disassociated with 0.25% trypsin and collected by centrifugation at 8000 g with cells floating in the culture, washed twice with PBS and stained with annexin-V-FITC and propidium iodide. Fluorescence was measured with a minimum of 10000 events for each sample in a FACSCAN according to the method suggested by the manufacturers. Data analysis was performed.
DNA fragmentation analysis (DNA ladder) Infected and control cells were lysed at 0, 12, 24 or 36 h after infection. The lysate buffer contained 10 mmol/L Tris-HCl (pH7.5), 10 mmol/L EDTA, 0.3% Triton X-100, 100 μg/mL RNase and 200 μg/mL proteinase K. After incubated at 50 °C for 4 h or overnight, lysate was extracted twice with phenol/chloroform, then precipitated at -20 °C for 10 min by adding sodium acetate/ethanol. DNA concentration was determined, and an equal amount of DNA (10 μg) was electrophoresed on 2% agarose gel containing 0.1 μg/mL ethidium bromide. DNA bands were examined using UVPC system.
In situ 3’ end labeling of DNA in apoptotic cells 3’ end labeling of DNA in situ was performed using the Apoptag Plus Kit (Oncogene Research Products, San Diego, CA). Cells grown in vitro were spun onto a plastic slide and fixed with buffered 4% formaldehyde at 0, 12, 24 h after infection. The percentage of positive cells was also determined by flow cytometry.
Western blotting
Cells were harvested and lysed in ice-cold lysis buffer containing 10 mmol/L Tris-HCl (pH7.5), 20% SDS, and 1mmol/L PMSF. After 30 min incubation on ice, lysate was stored at -80 °C before use. Protein concentration was determined by the Bradford method[35]. Equal amounts of protein (50 μg) were loaded onto SDS-polyacrylamide gels and the protein was electrophoretically transferred to a PVDF film (Millipore, Bedford, MA). Immunoblots were analyzed using specific primary antibodies (antibodies to Bcl-2, Bcl-xl, Bax, p53, β-actin), and protein was visualized using a chemiluminescence detection kit (Amersham, Ayesbury, UK). β-actin was used as an internal control to confirm whether the amount of protein was equal.
Statistical analysis
The data in this paper were the mean of three replications. Unless otherwise stated. Test of significant differences in significance was performed with Student’s t-test. P<0.05 was considered statistically significant and P<0.01 very statistically significant.
RESULTS
Cooperation of TIP30 and p53 predisposed cells to death
The effect of TIP30 and p53 gene products on the in vitro growth characteristics of HepG2, Hep3B and PLC/RPF/5 cells was examined by colorimetric XTT assay. The three kinds of cells infected with Ad-TIP30 and Ad-GFP and mock (no virus) were incubated in DMEM medium and monitored by viability staining for 8 d. As shown in Figure 1, the viability of HepG2 cells decreased significantly from 99.7%, 68.3% to 10% and further displayed more massive cell deaths within 5-8 d than Hep3B and PLC/RPF/5 cells, with the latter decreased from 97.8%, 62.1% to 44.3% and 98.1%, 68.6% to 50.4%, respectively. All the three kinds of cells infected with Ad-GFP were found to have relatively lower cell deaths until 5-8 d. On the contrary, control cells (no virus) displayed a relatively high growth rate and maintained a high percentage of live cells even until 8-10 d after plating. These results demonstrated that expression of exogenous TIP30 in HepG2 cells could cooperate with p53 to reduce cell proliferation and then led them to massive deaths.
Figure 1.
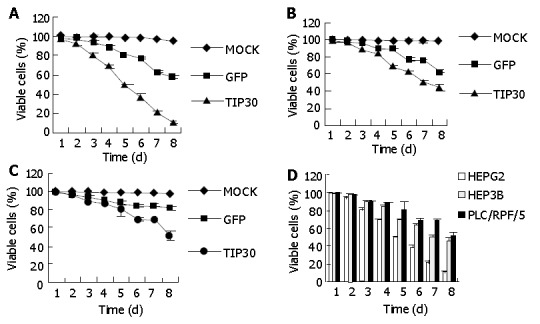
Cell viability of hepatoblastoma cell lines: HepG2, Hep3B and PLC/RPF/5 on indicated days as measured by XTT assay following infection with Ad-TIP30 and GFP at MOI of 20 or treatment of PBS (mock) (A, B, C). Three cell lines were infected with TIP30 at MOI of 20 in 8 continuous days (D). Values in the figures are mean±SD.
TIP30 induced cell death was due to apoptosis
To verify whether the cell death induced by TIP30 was ascribed to the activation of apoptosis in cells, in situ 3’ end labeling of DNA assay was performed to examine 3’-OH groups at the end of DNA fragments in apoptotic cells (Figure 2), which was a hallmark of apoptosis. Percentage of apoptotic was calculated by FACS assay. This quantitative analysis indicated that the proportion of apoptosis cells progressively increased in cells bearing Ad-TIP30. The maximum cell apoptosis was obtained at 24 h in HepG2, Hep3B, and PLC/RPF/5 cells, respectively. Quantitative analysis of apoptosis revealed that the percentage of apoptotic cells in HepG2 cells at 24 h was 1.5-to 2-fold (P<0.01) higher than those in Hep3B (48.1%) and PLC/RPF/5 (46.3%) cells. However, there was almost no apoptotic cell in mock (no virus).
Figure 2.
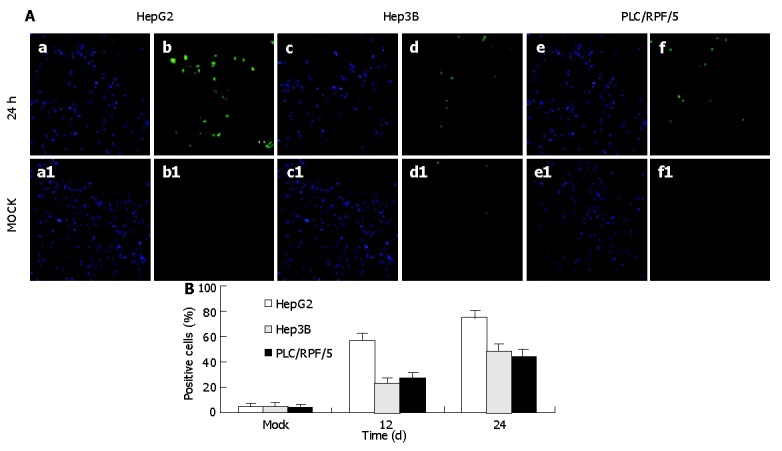
TUNEL (in situ) assay for TIP30 infection induced apoptosis at 24 h after infection and description of cell population with DAPI (a, c, d) and fluorescent (b, d, f) microscopy with mock as control. A: The mean of percentages of positive cells was calculated from the analysis; B: Data in the figure are mean±SD (P<0.01).
Similar results were attained by flow cytometry assay with cells treated as previously. Cells were labeled with annexin-V-FITC followed by propidium staining in order to differentiate the necrosis-cells or apoptotic cells. The apoptotic cell proportions in HepG2 cells were 32.36%, 61.04%, and 81.67% at 12, 24 and 36 h after infection respectively. Meanwhile, in Hep3B and PLC/RPF/5 cells, the apoptotic cell proportions were only 12.38%, 34.08%, 50.71% and 11.26%, 23.17%, 53.52% at the same time after infection respectively, while mock cells did not display significant apoptosis (Figure 3).
Figure 3.
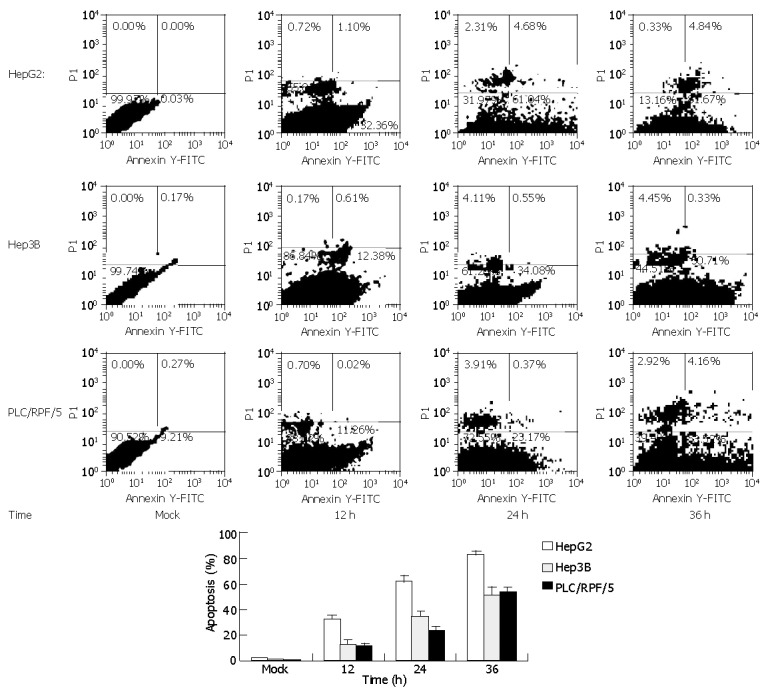
FACS analyses of annexin-V-FITC and PI double-stained cells. Cells were infected with Ad-TIP30 and mock as control (no virus). After treatment for 12, 24, 36 h, cells were double-stained at various indicated time points with annexin-V-FITC and PI and analyzed by flow cytometry. Cells appearing in the lower right quadrant showed positive annexin-V-FITC staining, which indicated phosphatidylserine translocation to the cell surface and no DNA staining by PI. Data in the figures are mean±SD. The percentage of apoptosis cells of three cell lines treated as previously (P<0.01).
To further confirm the kinetics of this programmed cell death, DNA fragmentation assay was performed with the same treatment in cells as previously. As indicated in Figure 4, DNA fragmentation became detectable at 24 h in HepG2 cells infected with Ad-TIP30, whereas it was detectable at 36 h in Hep3B and PLC/RPF/5 cells. Nevertheless, DNA ladder never emerged during the whole incubation process (72 h and more) of control cells (infected with Ad-GFP and mock, data not shown).
Figure 4.
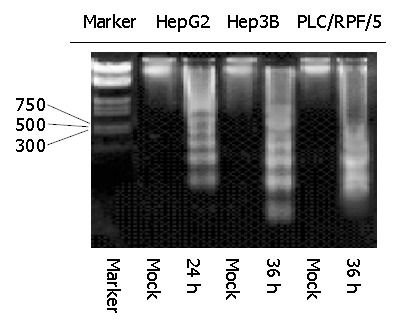
Kinetics of apoptosis in HepG2 and Hep3B and PLC/RPF/5 cells infected with Ad-TIP30. Twelve, twenty-four or thirty-six hours after infection, analysis of DNA laddering was performed by gel electrophoresis. An overexposed photograph was provided to show the beginning of DNA laddering at 24 h in HepG2 cells and at 36 h in Hep3B and PLC/RPF/5 cells.
Infection with Ad-TIP30 induced profound elevation of p53 protein
In response to DNA damage, the tumor suppressor protein, p53, was stabilized, accumulated inside the cells and was detectable by Western blotting. It was found to be a reliable initiator of apoptosis. We used HepG2 to study the expression level of p53 after infection with Ad-TIP30. Levels of p53 protein in HepG2 cells were examined at 6, 8, 12, 24 and 36 h after infection with 50 MOI of Ad-TIP30. In Figure 5, at 6-8 h after infection, p53 protein levels were significantly higher than those at other time points and drastically reduced at 12-24 h (Figure 5). Although the level of p53 after infection with Ad-GFP was a little higher than that of mock cells, no changes were observed at different time points (data not shown). According to the data quantified by densitometry, levels of p53 protein in Ad-TIP30 infected HepG2 cells were elevated by a range of 2-to 3- fold compared with cells infected with Ad-GFP. A comparable reduction in p53 protein was observed at 36 h, when a large percentage of apoptotic HepG2 cells was found. Expression of exogenously introduced wild type TIP30 gene was confirmed in Ad-TIP30 infected cells by Western blotting (data not shown).
Figure 5.
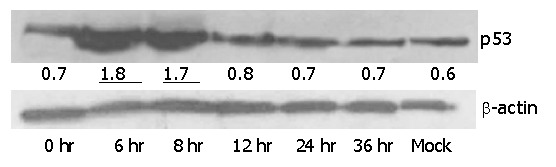
Western blotting analysis of p53 protein in HepG2 cells infected with Ad-TIP30. HepG2 cells were infected with Ad-TIP30. Harvested protein (50 ug/lane) was applied to 12% SDS-polyacrylamide electrophoresis. Proteins were transferred to PVDF film and probed with anti-serum specific for p53. p53 levels were quantified by densitometric analysis and normalized to β-actin. The data shown here were representative of three independent experiments with a similar result.
Expression of Bcl-2 family members after Ad-TIP30 infection
Expression of Bcl-2 family members was examined in three cell lines. Levels of Bax at 8-10 h after infection with Ad-TIP30 were 2-fold higher than those of Ad-GFP infected cells and mock in HepG2 cells, while there was no change in Hep3B and PLC/RPF/5 cells (Figure 6).
Figure 6.
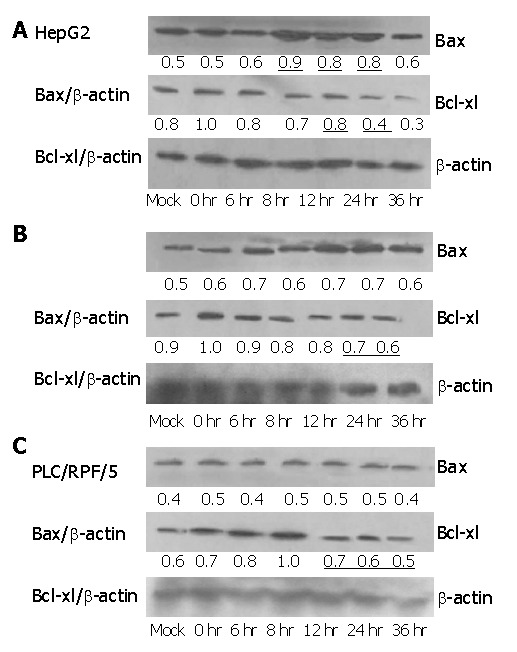
Expression of Bax and Bcl-xl after infection with Ad-TIP30 and mock as control in the three cell lines. Lysates were collected at 0, 6, 8, 12, 24, 36 h after infection. Ad-TIP30 infection resulted in increased Bax protein expression which was at its peak at 12-24 h after infection in HepG2 cells (Figure 6A), and decreased Bcl-xl protein levels in three cell lines (Figures 6A-C).
We examined Bcl-xl level in the three cell lines treated as previously. Its levels in HepG2, Hep3B and PLC/RPF/5 cells infected with AD-TIP30 were 2-fold to 1.5-fold lower than those infected with Ad-GFP (data not shown). Furthermore, in HepG2 cells, at 24-36 h after infection, Bax/Bcl-xl ratio reached the maximum of 2-fold higher than at other time points, with a requisite increase in p53 levels (Figure 7). In the mean time, a little decrease was found in Hep3B and PLC/RPF/5 cells at 36 h after infection.
Figure 7.
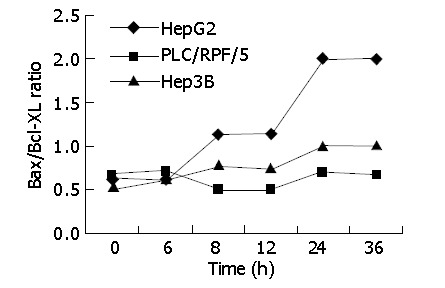
Infection of Ad-TIP30 with immunoblot analysis: densitometric analysis of the bands. The cells were infected with Ad-TIP30 and mock (no virus) as control in HepG2, Hep3B and PLC/RF/5 cells. The variation of Bax/Bcl-xl ratio in the three cell lines with time course was shown.
DISCUSSION
TIP30 is a novel Tat-cooperation protein that enhances human immunodeficiency virus-1 (HIV-1) by phosphorylating RNA polymerase II subunit. Recently, Xiao et al[1] and Baller et al[2] have shown that the 242- amino-acid-long- protein TIP30 plays an important role in suppression of metastasis of small cell lung carcinoma (SCLC) leading it to apoptosis.
We are in favor of connecting TIP30 with some genes as a preferential representative in apoptosis pathway. p53 gene and its related genes have been regarded as a model for the following reasons. P53 is a key factor in regulating apoptosis and shares one common down-regulator with TIP30: pig12[15]. The manner of p53 activation appears to be primarily controlled through phosphorylating its N-terminal for its stability. Sherr et al[16] and several other studies suggest that the major manner of evaluating its levels is at post-transcription. Xiao et al[17] demonstrated that TIP30 had a characteristic of phosphorylase kinase activity to trigger apoptosis.
Additionally, the pro-apoptotic activity of TIP30 by stresses such as chemical oxidants, growth factor deprivation, leads to the expression of a number of genes such as Bad and Siva, which are responsible for apoptosis[2]. Despite the fact that TIP30 is defined unambiguously as a tumor suppressor and a pro-apoptotic factor, the molecular mechanism by which TIP30 gene mediates apoptosis and which component is involved in TIP30 pathway remain largely unknown. Therefore, we employed in vitro analysis to reveal the apoptosis pathway in which TIP30 takes part.
p53 protein is a transcription factor that can inhibit cell cycle progression or induce apoptosis in response to stress or DNA damage, with a short life and a low expression level in normal cells[5]. Normally, the half-life of p53 is attributable to degradation mediated by Mdm2, which targets p53 for ubiquitin-dependent proteolysis[18]. Several stress-responsive kinases have been shown to phosphorylate p53 in a manner that could potentially inhibit Mdm2-mediated degradation. Avantaggiati et al[19] showed that DNA damage triggered accumulation and functional activation of p53 by converting the protein from a latent to an active form. Two proteins have been recently identified as potential regulators of p53 stability: one is kinase CHK2, which could inhibit Mdm2-mediated ubiquitination and proteolysis of p53[18]. Hemmatip et al[20] have shown that introduction of Ad-P14ARF into U-20s osteosarcoma cells could efficiently induce apoptosis in p53 pathway, in turn upregulation of P14ARF could mediate accumulation of p53 via sequestration of the p53-antagonist Mdm2 through the ubiquitin /proteasome pathway. Thus, P14ARF relieves the Mdm2-mediated degradation of p53, thereby prolonging the half-life of p53 protein[21]. In human breast cancer cells-MCF-7, Ad-P14ARF has cytotoxicity to MCF-7 cells (p53 wild type), resulting in a considerable increase of p53. Jiang et al[13] demonstrated that staurosprine, quinacrine and ultraviolet irradiation were all capable of triggering apoptosis of HepG2 cells by affecting the expression levels of p53. A cytotoxic agent of rodent liver, 7H-dibenzocc carbazole (DBC) could increase p53 protein levels followed by apoptotic cell death[22]. In our studies, when HepG2 cells were infected with Ad-TIP30, to our expectation, levels of wide p53 were increased in a time-dependent manner with an asynchronous apoptosis, indicating that apoptosis induced by TIP30 is attributable to the accumulation of p53, which supports the hypothesis that TIP30 is involved in apoptosis pathway partly by cooperating with p53 protein.
It is well known that one mechanism by which p53 is stabilized within infected cells is that p53 is substantially modified post-translationally via serine targeted phosphorlation at both the N-terminus and C-terminus as well as acetylation at lysine residues in C-terminus[23], implicating that p53 appears to be optimal in post-translation level under a stress response. In addition, the primary consequence of this induction is cell cycle arrest or apoptotic cell death. Xiao et al[17] found that TIP30 had an intrinsic kinase activity in phosphorylating the heptapeptide repeats of the C-terminal domain (CTD) of the largest subunit of RNA polymerase II in concurrence with the ability to trigger SCLC cells to apoptosis. We also speculate that TIP30 might phosphorylate the p53 C- or N- terminus in advance to enhance the p53-dependent pathway. Nevertheless, verification of the hypothesis requires further experiment. Interestingly, in Hep3B and PLC/RPF/5 cells where p53 protein is absent or mutational, introduction of TIP30 also predispose cells to apoptosis at a less extent than in HepG2 cells, implicating that TIP30 might trigger apoptosis through other pathways that occur independently of the p53/Bax pathway. Further investigation might be essential and helpful.
In p53/mitochondrial pathway, members of Bcl-2 family are critical regulators of the apoptotic pathway[24]. Bcl-xl, an anti-apoptotic member of Bcl-2 family, inhibits the release of cytochrome C from mitochondria whereas Bax, a pro-apoptotic member of Bcl-2 family, has been shown to stimulate release of cytochrome C from mitochondria thereby enhancing apoptotic response[25].
In the present study, we evaluated the role of Bax and Bcl-xl in Ad-TIP30 mediated apoptosis. Early studies have shown that Bax gene plays an important role in the course of carcinogenesis in stomach, colorectum and endometrium in humans[26]. Furthermore, the Bax gene promoter is highly p53-responsive and its expression is upregulated by p53[27].
Pearson and Francis[28] have shown that adenovirus-mediated over-expression of p53 in lung cancer cell lines H1299, H358 and HK322 simultaneously upregulates Bax protein levels 18 to 36 h after introduction. In Tu-138 and Tu-167 cells, after infection with Ad5CMV-p53[29], Bax levels were elevated 2-to 6-fold higher than controls, demonstrating that influence upon Bax is caused by asynchronous activation of p53. Mohiuddin et al[30] reported that in hepablastoma cells multiple stimuli such as ultraviolet and carcumin could not change Bax levels. On the contrary, in this case, exogenous TIP30 and endogenous p53 expression in HepG2 cells led to elevation of Bax level at 18-24 h after infection whereas no change was found in Hep3B cells with absence of p53, indicating that it is alternatively possible that the Bax levels are not influenced directly by TIP30, but rather through p53 involvement when apoptotic cell death is initiated. In support of this model, more observations have shown that in many other cancer cell lines, a high level of Bax follows the activation and stabilization of p53[31].
Previous reports have shown that alternation in the ratio of proapoptotic to anti-apoptotic members of the Bcl-2 family, rather than the absolute expression level of any single Bcl-2 family member, determines apoptotic sensitivity[32]. As to hepatoblastoma cells[33], the ratio of Bax to Bcl-xl protein plays a major role in sensitivity to apoptotic stimuli, because the expression of Bcl-2 is uncommon in hepatoblastoma cells according to several researches. Ectopic expression of transcriptionally active p53 alone is not sufficient for the activation of apoptosis in p53-null Hep3B cells, except when endogenous Bcl-xl is simultaneously inhibited by anti-sense oligonucleotide in these cells[34-36]. In our studies, immunoblot analysis revealed decreased Bcl-xl levels and increased Bax levels in HepG2 cells after Ad-TIP30 infection. In addition, in Hep3B and PLC/RPF/5 cells, Bcl-xl decrease was observed at a less extent than in HepG2 cells, while there was no significant change in Bax levels, indicating that TIP30 might regulate the Bax and Bcl-xl ratio in a p53 dependent and independent manner. Therefore, our studies clearly suggest that shift in the ratio of proapoptotic factor (Bax) to anti-apoptotic factor (Bcl-xl) in favor of proapoptotic factor (Bax) is an important factor for determining the response toward Ad-TIP30 triggering apoptosis.
What are the mechanisms by which TIP30 causes apoptosis in hepotablastoma cells? We suggest that wild type p53 upregulates Bax expression with concomitant down-regualtion of Bcl-xl is the consequence of p53 accumulation due to exogenous TIP30 expression. Thus, TIP30 gene may serve as a good sample for heptoblastoma gene therapy, not only because it may kill cancer cells directly, but also because it may be regarded as a useful mediator in elevating apoptosis-related gene expression in its apoptotic pathway.
ACKNOWLEDGEMENTS
We thank Dr. Hua Xiao for generously providing a cDNA encoding full-length human TIP30.
Footnotes
Assistant Editor Guo SY Edited by Zhang JZ and Wang XL
References
- 1.Xiao H, Tao Y, Greenblatt J, Roeder RG. A cofactor, TIP30, specifically enhances HIV-1 Tat-activated transcription. Proc Natl Acad Sci USA. 1998;95:2146–2151. doi: 10.1073/pnas.95.5.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker ME, Yan L, Pear MR. Three-dimensional model of human TIP30, a coactivator for HIV-1 Tat-activated transcription, and CC3, a protein associated with metastasis suppression. Cell Mol Life Sci. 2000;57:851–858. doi: 10.1007/s000180050047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chène P. Inhibition of the p53-MDM2 interaction: targeting a protein-protein interface. Mol Cancer Res. 2004;2:20–28. [PubMed] [Google Scholar]
- 4.Batinac T, Gruber F, Lipozencić J, Zamolo-Koncar G, Stasić A, Brajac I. Protein p53--structure, function, and possible therapeutic implications. Acta Dermatovenerol Croat. 2003;11:225–230. [PubMed] [Google Scholar]
- 5.Mirza A, Wu Q, Wang L, McClanahan T, Bishop WR, Gheyas F, Ding W, Hutchins B, Hockenberry T, Kirschmeier P, et al. Global transcriptional program of p53 target genes during the process of apoptosis and cell cycle progression. Oncogene. 2003;22:3645–3654. doi: 10.1038/sj.onc.1206477. [DOI] [PubMed] [Google Scholar]
- 6.Courageot MP, Catteau A, Desprès P. Mechanisms of dengue virus-induced cell death. Adv Virus Res. 2003;60:157–186. doi: 10.1016/s0065-3527(03)60005-9. [DOI] [PubMed] [Google Scholar]
- 7.Hayward RL, Macpherson JS, Cummings J, Monia BP, Smyth JF, Jodrell DI. Enhanced oxaliplatin-induced apoptosis following antisense Bcl-xl down-regulation is p53 and Bax dependent: Genetic evidence for specificity of the antisense effect. Mol Cancer Ther. 2004;3:169–178. [PubMed] [Google Scholar]
- 8.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 9.Groeger AM, Esposito V, De Luca A, Cassandro R, Tonini G, Ambrogi V, Baldi F, Goldfarb R, Mineo TC, Baldi A, et al. Prognostic value of immunohistochemical expression of p53, bax, Bcl-2 and Bcl-xL in resected non-small-cell lung cancers. Histopathology. 2004;44:54–63. doi: 10.1111/j.1365-2559.2004.01750.x. [DOI] [PubMed] [Google Scholar]
- 10.Feng WY, Liu FT, Patwari Y, Agrawal SG, Newland AC, Jia L. BH3-domain mimetic compound BH3I-2' induces rapid damage to the inner mitochondrial membrane prior to the cytochrome c release from mitochondria. Br J Haematol. 2003;121:332–340. doi: 10.1046/j.1365-2141.2003.04268.x. [DOI] [PubMed] [Google Scholar]
- 11.Moreau C, Cartron PF, Hunt A, Meflah K, Green DR, Evan G, Vallette FM, Juin P. Minimal BH3 peptides promote cell death by antagonizing anti-apoptotic proteins. J Biol Chem. 2003;278:19426–19435. doi: 10.1074/jbc.M209472200. [DOI] [PubMed] [Google Scholar]
- 12.Dimitrakakis C, Konstadoulakis M, Messaris E, Kymionis G, Karayannis M, Panoussopoulos D, Michalas S, Androulakis G. Molecular markers in breast cancer: can we use c-erbB-2, p53, bcl-2 and bax gene expression as prognostic factors? Breast. 2002;11:279–285. doi: 10.1054/brst.2002.0445. [DOI] [PubMed] [Google Scholar]
- 13.Jiang MC, Yen HF. Differential regulation of p53, c-myc,Bcl-2 and Bax protein expression during apoptosis induced by wildly divergent stimuli in human hepatoblastoma cells. Oncogene. 1996;22:609. [PubMed] [Google Scholar]
- 14.Shi M, Wang FS, Wu ZZ. Synergetic anticancer effect of combined quercetin and recombinant adenoviral vector expressing human wild-type p53, GM-CSF and B7-1 genes on hepatocellular carcinoma cells in vitro. World J Gastroenterol. 2003;9:73–78. doi: 10.3748/wjg.v9.i1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SH, DeJong J. Microsomal GST-I: genomic organization, expression, and alternative splicing of the human gene. Biochim Biophys Acta. 1999;1446:389–396. doi: 10.1016/s0167-4781(99)00112-8. [DOI] [PubMed] [Google Scholar]
- 16.Sherr CJ. Tumor surveillance via the ARF-p53 pathway. Genes Dev. 1998;12:2984–2991. doi: 10.1101/gad.12.19.2984. [DOI] [PubMed] [Google Scholar]
- 17.Xiao H, Palhan V, Yang Y, Roeder RG. TIP30 has an intrinsic kinase activity required for up-regulation of a subset of apoptotic genes. EMBO J. 2000;19:956–963. doi: 10.1093/emboj/19.5.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mocanu MM, Yellon DM. p53 down-regulation: a new molecular mechanism involved in ischaemic preconditioning. FEBS Lett. 2003;555:302–306. doi: 10.1016/s0014-5793(03)01260-2. [DOI] [PubMed] [Google Scholar]
- 19.Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89:1175–1184. doi: 10.1016/s0092-8674(00)80304-9. [DOI] [PubMed] [Google Scholar]
- 20.Hemmati PG, Gillissen B, von Haefen C, Wendt J, Stärck L, Güner D, Dörken B, Daniel PT. Adenovirus-mediated overexpression of p14(ARF) induces p53 and Bax-independent apoptosis. Oncogene. 2002;21:3149–3161. doi: 10.1038/sj.onc.1205458. [DOI] [PubMed] [Google Scholar]
- 21.Stürzenhofecker B, Schlott T, Quentin T, Kube D, Jung W, Trümper L. Abundant expression of spliced HDM2 in Hodgkin lymphoma cells does not interfere with p14(ARF) and p53 binding. Leuk Lymphoma. 2003;44:1587–1596. doi: 10.3109/10428190309178783. [DOI] [PubMed] [Google Scholar]
- 22.Chen YN, Chen JC, Yin SC, Wang GS, Tsauer W, Hsu SF, Hsu SL. Effector mechanisms of norcantharidin-induced mitotic arrest and apoptosis in human hepatoma cells. Int J Cancer. 2002;100:158–165. doi: 10.1002/ijc.10479. [DOI] [PubMed] [Google Scholar]
- 23.Deng X, Kim M, Vandier D, Jung YJ, Rikiyama T, Sgagias MK, Goldsmith M, Cowan KH. Recombinant adenovirus-mediated p14(ARF) overexpression sensitizes human breast cancer cells to cisplatin. Biochem Biophys Res Commun. 2002;296:792–798. doi: 10.1016/s0006-291x(02)00948-8. [DOI] [PubMed] [Google Scholar]
- 24.Kasai H, Yamamoto K, Koseki T, Yokota M, Nishihara T. Involvement of caspase activation through release of cytochrome c from mitochondria in apoptotic cell death of macrophages infected with Actinobacillus actinomycetemcomitans. FEMS Microbiol Lett. 2004;233:29–35. doi: 10.1016/j.femsle.2004.01.034. [DOI] [PubMed] [Google Scholar]
- 25.Tsujimoto Y. Cell death regulation by the Bcl-2 protein family in the mitochondria. J Cell Physiol. 2003;195:158–167. doi: 10.1002/jcp.10254. [DOI] [PubMed] [Google Scholar]
- 26.Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun. 2003;304:437–444. doi: 10.1016/s0006-291x(03)00615-6. [DOI] [PubMed] [Google Scholar]
- 27.Ohtsuka T, Ryu H, Minamishima YA, Macip S, Sagara J, Nakayama KI, Aaronson SA, Lee SW. ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nat Cell Biol. 2004;6:121–128. doi: 10.1038/ncb1087. [DOI] [PubMed] [Google Scholar]
- 28.Pearson AS, Spitz FR, Swisher SG, Kataoka M, Sarkiss MG, Meyn RE, McDonnell TJ, Cristiano RJ, Roth JA. Up-regulation of the proapoptotic mediators Bax and Bak after adenovirus-mediated p53 gene transfer in lung cancer cells. Clin Cancer Res. 2000;6:887–890. [PubMed] [Google Scholar]
- 29.Frederick MJ, Holton PR, Hudson M, Wang M, Clayman GL. Expression of apoptosis-related genes in human head and neck squamous cell carcinomas undergoing p53-mediated programmed cell death. Clin Cancer Res. 1999;5:361–369. [PubMed] [Google Scholar]
- 30.Mohiuddin I, Cao X, Fang B, Nishizaki M, Smythe WR. Significant augmentation of pro-apoptotic gene therapy by pharmacologic bcl-xl down-regulation in mesothelioma. Cancer Gene Ther. 2001;8:547–554. doi: 10.1038/sj.cgt.7700332. [DOI] [PubMed] [Google Scholar]
- 31.Thornborrow EC, Manfredi JJ. The tumor suppressor protein p53 requires a cofactor to activate transcriptionally the human BAX promoter. J Biol Chem. 2001;276:15598–15608. doi: 10.1074/jbc.M011643200. [DOI] [PubMed] [Google Scholar]
- 32.Srivastava M, Ahmad N, Gupta S, Mukhtar H. Involvement of Bcl-2 and Bax in photodynamic therapy-mediated apoptosis. Antisense Bcl-2 oligonucleotide sensitizes RIF 1 cells to photodynamic therapy apoptosis. J Biol Chem. 2001;276:15481–15488. doi: 10.1074/jbc.M006920200. [DOI] [PubMed] [Google Scholar]
- 33.Ahn SG, Kim HS, Jeong SW, Kim BE, Rhim H, Shim JY, Kim JW, Lee JH, Kim IK. Sox-4 is a positive regulator of Hep3B and HepG2 cells' apoptosis induced by prostaglandin (PG)A(2) and delta(12)-PGJ(2) Exp Mol Med. 2002;34:243–249. doi: 10.1038/emm.2002.34. [DOI] [PubMed] [Google Scholar]
- 34.Takehara T, Liu X, Fujimoto J, Friedman SL, Takahashi H. Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology. 2001;34:55–61. doi: 10.1053/jhep.2001.25387. [DOI] [PubMed] [Google Scholar]
- 35.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 36.Chun E, Lee KY. Bcl-2 and Bcl-xL are important for the induction of paclitaxel resistance in human hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2004;315:771–779. doi: 10.1016/j.bbrc.2004.01.118. [DOI] [PubMed] [Google Scholar]