

Abstract
The gastrointestinal (GI) tract presents a major site of immune modulation by HIV, resulting in significant morbidity. Most GI processes affected during HIV infection are regulated by the enteric nervous system. HIV has been identified in GI histologic specimens in up to 40% of patients, and the presence of viral proteins, including the trans-activator of transcription (Tat), has been reported in the gut indicating that HIV itself may be an indirect gut pathogen. Little is known of how Tat affects the enteric nervous system. Here we investigated the effects of the Tat protein on enteric neuronal excitability, proinflammatory cytokine release, and its overall effect on GI motility. Direct application of Tat (100 nm) increased the number of action potentials and reduced the threshold for action potential initiation in isolated myenteric neurons. This effect persisted in neurons pretreated with Tat for 3 d (19 of 20) and in neurons isolated from Tat+ (Tat-expressing) transgenic mice. Tat increased sodium channel isoforms Nav1.7 and Nav1.8 levels. This increase was accompanied by an increase in sodium current density and a leftward shift in the sodium channel activation voltage. RANTES, IL-6, and IL-1β, but not TNF-α, were enhanced by Tat. Intestinal transit and cecal water content were also significantly higher in Tat+ transgenic mice than Tat− littermates (controls). Together, these findings show that Tat has a direct and persistent effect on enteric neuronal excitability, and together with its effect on proinflammatory cytokines, regulates gut motility, thereby contributing to GI dysmotilities reported in HIV patients.
Keywords: AIDS, cytokines, HIV, myenteric, sodium channels
Introduction
The gastrointestinal (GI) tract harbors 80%–90% of the lymphocytes of the body. It presents an important component of AIDS disease whereby, in addition to chronic diarrhea, alterations in structure and function occur as a result of the destruction of mucosal immunity, enhanced viral replication, and prolonged inflammation (Veazey et al., 1998; Orandle et al., 2007; Lackner et al., 2009; Handley et al., 2012; Mohan et al., 2012). Indeed, the mucosal CD4+ T-cell destruction within the lamina propria is detectable at earlier times than in peripheral blood (Lim et al., 1993; Sasseville et al., 1996; Smit-McBride et al., 1998; Brenchley et al., 2004; Mattapallil et al., 2005; Mavigner et al., 2012). AIDS progression is driven by significant damage to gut-associated lymphoid tissue. Much of our understanding of neuropathogenesis in HIV/AIDS arises from studies in the brain, although less is known about the effects of the disease on peripheral neurons (Simpson and Olney, 1992; Pardo et al., 2001; Cornblath and Hoke, 2006; Lehmann et al., 2011; Johnson et al., 2013).
HIV transcription is mainly controlled by Tat (Karn, 1999). Tat is an early regulatory protein, 86–104 amino acids in length (14–16 kDa). Tat plays an important role in viral transcription and replication and has also been implicated in inducing the expression of a variety of cellular genes as well as acting as a neurotoxic protein (Nath et al., 1996; Karn, 1999). Tat is released by intact infected cells (Chang et al., 1997). In the CNS, the principal cell types harboring active infection are perivascular macrophages and microglia and, to a lesser extent, astroglia (Kaul et al., 2001; González-Scarano and Martín-García, 2005; Ellis et al., 2007; Yukl et al., 2013). Neurological defects persist even in HIV-infected individuals on combined antiretroviral therapy (cARTs) who are aviremic (McArthur et al., 2010). In the gut as in the CNS, neuronal injury is an indirect consequence of cellular and viral toxins released by infected cells. Once infected, limiting HIV replication alone may be inadequate to prevent neuronal impairment. With improved survival with cART, the manifestations of chronic exposure to low levels of viral and cellular toxins associated with HIV-1 infection become increasingly evident, especially in the ileum (Yukl et al., 2013). Tat production from preintegration HIV-1 DNA or during the early phase of transcription from integrated proviral DNA is largely unaffected by cART (Wu, 2004; Kilareski et al., 2009).
The enteric nervous system (ENS) is composed of neurons and glia within two major plexii: the myenteric (Auerbach's) plexus and the submucosal (Meissner's) plexus. Not only is the ENS important in regulating GI motility, secretion, and digestion, but it is also intimately involved with the regulation of epithelial barrier functions, including the development and maintenance of the immune system (De Winter and De Man, 2010; van de Pavert and Mebius, 2010; Neunlist et al., 2013). Despite clinical evidence of altered gastric motor function and enteric ganglionitis in simian immunodeficiency virus-infected macaques (Konturek et al., 1997; Orandle et al., 2007), the effects of HIV-1 on the ENS have not been well studied. In the present study, we examined the effects of Tat on neuronal excitability, proinflammatory cytokine release, and the modulation of GI motility.
Materials and Methods
All experiments were conducted in accordance with the procedures reviewed and approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University.
Isolation and culture of neurons from the adult mouse myenteric plexus.
Cells were isolated as described recently (Smith et al., 2012, 2013). Briefly, after killing the mice, the ileum was removed and placed in ice-cold Krebs solution (in mm) as follows: 118 NaCl, 4.6 KCl, 1.3 NaH2PO4, 1.2 MgSO4 25 NaHCO3, 11 glucose, and 2.5 CaCl2, bubbled with carbogen (95% O2/5% CO2). The ileum was divided into short segments and threaded longitudinally on a plastic rod. Strips of the longitudinal muscle containing the myenteric plexus (LMMP) were gently separated using a cotton-tipped applicator. LMMP strips were rinsed three times in 1 ml Krebs and centrifuged (350 × g, 30 s). LMMP strips were minced with scissors, digested in 1.3 mg/ml collagenase Type II (Worthington) and 0.3 mg/ml BSA in bubbled Krebs (37°C) for 1 h, and then incubated in 0.05% trypsin for 7 min. Following each digestion, cells were triturated and centrifuged (350 × g for 8 min). Cells were then plated on laminin (BD Biosciences) and poly-D-lysine-coated coverslips in Neurobasal A media containing B-27 supplement, 1% FBS, 10 ng/ml glial cell line-derived neurotrophic factor (GDNF, Neuromics), and penicillin/streptomycin. Half of the cell culture medium was replaced every 2–3 d with fresh neuron medium.
All chemicals and reagents were obtained from Sigma-Aldrich, unless otherwise noted, except cell culture reagents, which were purchased from Invitrogen and the HIV Tat1–86 protein (Immunodiagnostics). Male, Swiss Webster mice (25–30 g, Harlan Sprague Dawley) or male and female doxycycline (DOX)-inducible, HIV-Tat1–86 transgenic mice (25–30 g) were used. The HIV-Tat1–86 transgenic mouse model was developed on a C57BL/6J hybrid background and was described in detail previously (Bruce-Keller et al., 2008). Tat expression, which is under the control of a tetracycline responsive, GFAP-selective promoter, was induced with a specially formulated chow containing 6 mg/g DOX (Harlan, product #TD.09282), fed to both the Tat− controls and the inducible Tat+ mice. Because of uncertainty regarding the timing and extent to which Tat would be expressed by enteric GFAP-expressing astroglia following induction with DOX, mice were fed with DOX-containing chow for variable durations lasting from 3 d to 2 weeks to induce the expression of the tat gene. Previously published studies show increased astrocyte activation and an increased percentage of neurons expressing active caspase-3 in the striatum, as early as 2 d after continuous exposure to DOX (Bruce-Keller et al., 2008), suggesting that Tat could be expressed and elicit bystander effects in neurons within several days of induction. Longer durations of Tat induction were also assessed based on findings that Tat expression was delayed in the spinal cord compared with the striatum (Fitting et al., 2012) and regional variations might exist.
Immunocytochemistry.
Isolated cells cultured for 4 d to 2 weeks on coverslips were fixed in 4% formaldehyde for 30 min. Cells were permeabilized with 0.01% Triton X-100 in PBS (30 min) and nonspecific immunoreactivity blocked with 10% goat serum (1 h). Preparations were incubated with the primary antibody overnight at 4°C. Primary antibodies used were as follows: neuronal specific anti-βIII-tubulin (rabbit, Abcam ab18207–100, 1:100), anti-Nav1.7 (mouse, Stressmarq, 1:300), and/or Nav1.8 (mouse, Stressmarq, 1:300). After 3 washes in PBS, cells were incubated with the appropriate secondary antibodies; goat anti-rabbit Alexa-488 Dye (Invitrogen, 1:1000, 1 h, room temperature) and goat anti-mouse Alexa-594 Dye (Invitrogen, 1:1000, 1 h, room temperature). Visualization was performed using an Olympus Fluoview Confocal Microscope and software (version 5.0).
Whole-cell patch clamp.
Myenteric neurons were studied after 1–4 d in culture. Coverslips containing cells were placed in an experimental chamber and perfused (1–2 ml/min) with an external physiological solution containing the following (in mm): 135 NaCl, 5.4 KCl, 0.3 NaH2PO4, 5 HEPES, 1 MgCl2, 2 CaCl2, and 5 glucose. Patch electrodes (2–4 mΩ) were pulled from borosilicate glass capillaries (Sutter Instruments) and filled with internal solution containing the following (in mm): 100 K-aspartic acid, 30 KCl, 4.5 ATP, 1 MgCl2, 10 HEPES, and 0.1 EGTA. In sodium channel experiments, K-aspartic acid was replaced by Cs-aspartate to block any outward potassium currents. Whole-cell patch-clamp recordings were made with an Axopatch 200B amplifier (Molecular Devices) at room temperature, and pulse generation and data acquisition were achieved with Clampex and Clampfit 10.2 software (Molecular Devices). Neuronal excitability was determined in current-clamp mode with current provided in 13 sweeps of 0.5 s duration ranging from −0.03 nA to 0.09 nA in 0.01 nA increments. Current–voltage relationships were determined in voltage-clamp mode, in 16 0.5-s sweeps beginning at −100 mV and increasing in 10 mV intervals to 50 mV. Current amplitudes were normalized to cell capacitance (pF) to determine current density. The threshold of action potentials was determined as the voltage at which the d(V)/d(T) function deviated from zero. Action potential height was determined by measuring the threshold to the peak of the action potential. Voltage dependence of steady-state inactivation and activation was determined and fit via Boltzmann's distribution as described previously (Akbarali and Giles, 1993).
ELISA.
Supernatant from cells treated with Tat overnight was used to detect the cytokines TNF-α and IL-6, and the chemokine RANTES using ELISA (R&D Systems).
RT-PCR.
RNA was isolated from ileum LMMP using TRIzol (Invitrogen) following the manufacturer's protocol. The RNA was quantified using a spectrophotometer; 5 μg of the extracted RNA was used for cDNA preparation, using random primer (Bioline) and Superscript II (Invitrogen) according to the manufacturer's protocol. Real-time PCR was performed with the cDNA prepared from each of the tissue with specific primers for NaV1.3, NaV1.7, NaV1.8, and NaV1.9 using SYBR Green chemistry (QIAGEN). The 18 s rRNA was used as the internal control. The primers used were as follows: reverse NaV1.8, 5′-CAA AAC CCT CTT GCC AGT ATCT-3′; forward NaV1.8, 5′ GTG TGC ATG ACC CGA ACT GAT-3′; forward NaV1.7, 5′-GCC TTG TTT CGG CTA ATG AC-3′; reverse NaV1.7, 5′-TCC CAG AAA TAT CAC CAC GAC-3′; reverse NaV1.3, 5′-TTG AGA GAA TCA CCA CCA CA-3′; and forward NaV1.3, 5′-TTG GCT CCA AAA AAC CTC AG-3′. Experiments were performed in triplicate from three separate samples. IL-1β mRNA levels were determined by RT-PCR in LMMP from Tat transgenic mice. The IL-1β primers used were as follows: 5′-GCA GAC AGC TCA ATC TCT AGG AG-3′ (forward) and 5′-TCT CTT TGA ACA GAA TGT GCC ATG-3′ (reverse). Tat primers used were as follows: 5′-GGA GCC AGT AGA TCC TAG CC-3′ (forward) and 5′-GTT CTT CGT CGC TGT CTC CG-3′ (reverse). The mean normalized fold expression ± SD is plotted as a histogram.
GI motility studies.
Upper GI transit was determined by feeding age- and weight-matched tat transgenic mice with DOX chow for 2 weeks to activate the tat transgene after which they were fasted overnight. On the test day, mice were administered a charcoal gavage; 30 min later, they were killed and the ileum removed carefully without artificially stretching the tissue. The total length and the leading edge of the charcoal meal were measured. Upper GI tract motility was determined by calculating percentage charcoal transit/total ileum length. In a separate series of experiments, age-matched mice were fed with DOX chow for 1 week after which the DOX chow was substituted for a regular chow for 3 weeks. This paradigm was used to avoid confounding effects of the loss of GI microbiota by DOX treatment. PCR was performed to confirm the expression of the tat gene in the ileum after 3 weeks of DOX removal. Gastric stasis was determined by dissecting out the stomachs of age-matched Tat+ and Tat− mice 2 mm proximal to the lower esophageal sphincter and 2 mm distal to the pyloric sphincter. The stomachs were then immediately weighed and bathed in oxygenated physiologic Krebs solution. The stomach contents were emptied by flushing, blotted onto paper towel, and weighed again. The rate of gastric stasis was determined using the following equation (Asakawa et al., 2003; Welsh et al., 2013):
 |
Overall GI transit was determined by counting the fecal pellet output. On test day (day 30), mice were placed in clean cages and the number of fecal pellets were collected and counted for 2 h (Anitha et al., 2012). The water content of the cecum was determined by dissecting out the cecum, collecting the cecal content, immediately weighing it, drying it by heating at 60°C for 1 h, and reweighing the dried content. The water content was determined by the following equation:
 |
Data analysis.
Results are presented as mean ± SEM for the number of cells (n). Statistical tests were performed using GraphPad Prism 5.0 software using one- or two-way ANOVA or two-tailed paired or Student's t test. Values of p < 0.05 were regarded as significant.
Results
Tat increased enteric neuronal excitability
Enteric neuronal excitability was assessed using current-clamp experiments. The rheobase for action potential generation, the number of action potentials evoked, and the threshold potential were studied as determinants of neuronal excitability. The average resting membrane potential of neurons was −50.3 ± 0.6 mV (n = 65). Current injection evoked single action potentials in all control cells and multiple action potentials at rheobase in some cells (6 of 19). Alternatively, when neurons were continuously perfused with 100 nm Tat for ≤30 min, they became increasingly excitable with a reduced rheobase and multiple action potentials elicited at rheobase (n = 5) (Fig. 1A). In neurons pretreated with Tat (100 nm) for 17–48 h in vitro, the current required to evoke an action potential (rheobase) and the threshold potential for the upstroke of the action potential were significantly reduced. In addition, 19 of the 20 cells evoked multiple action potentials (Table 1). Increases in neuronal excitability were also revealed by the presence of spontaneous action potentials in 4 cells at 0 nA current injection (Fig. 1B). There was no significant difference in resting membrane potential between Tat-treated cells and controls (Table 1). To determine that the increase in neuronal excitability was a Tat-specific effect, we treated neurons with heat-inactivated Tat. Importantly, the excitability of neurons treated with heat-inactivated Tat did not significantly differ from controls (Table 1). Next, we assessed the in vivo effects of Tat induction on enteric neuronal excitability. Neurons were isolated from Tat transgenic mice (Tat+) and control (Tat−) littermates expressing only the reverse tetracycline trans-activator gene (Bruce-Keller et al., 2008). To confirm that Tat is expressed in the ileum of these mice, RT-PCR was performed on LMMP isolated from both Tat+ and Tat− mice that had been treated with DOX for the preceding 4 d. The tat gene was highly expressed in Tat+ mice (Fig. 1C). Neurons isolated from Tat+ mice after 4 d of DOX treatment fired multiple action potentials at significantly lower rheobase and threshold potentials than those isolated from Tat− mice (Fig. 1D; Table 1). These data suggest that HIV-1 Tat, either following direct exposure to recombinant Tat protein in vitro or following exposure via induction in Tat transgenic mice in vivo, resulted in enhanced neuronal excitability.
Figure 1.

Tat increased enteric neuronal excitability. A, Representative traces showing current-clamp recordings of a neuron in the absence and in the presence of 100 nm Tat. Increased neuronal excitability in response to 100 nm Tat is evidenced by an increase in the number of action potentials evoked. Action potentials were initiated after 30 pA current injection in the absence of Tat and 10 pA after continuous perfusion with 100 nm Tat; n = 5. B, Spontaneous action potentials were recorded in 4 neurons after Tat exposure. C, RT-PCR experiments showing that tat gene is expressed in Tat+ LMMP and absent in Tat− LMMP. *p < 0.05 (t test). D, Neurons isolated from Tat+ transgenic mice were also more excitable than Tat− mice. Gray line indicates response recorded at rheobase.
Table 1.
Tat-mediated increase in neuronal excitability is Tat specific and long lastinga
| Cells with multiple APs/N | Em (mV) | Rheobase (pA) | AP threshold (mV) | AP height | |
|---|---|---|---|---|---|
| Control | 6/19 | −50.6 ± 1.5 | 20 ± 5 | −11.5 ± 2.7 | 77.7 ± 5.8 |
| 100 nm Tat | 19/20* | −49.2 ± 1.2 | 10 ± 2* | −16.1 ± 1.2* | 87.9 ± 3.8* |
| Inactivated 100 nm Tat | 1/5 | −50.2 ± 1.4 | 20 ± 6 | −13.2 ± 1.4 | 71.7 ± 8.7 |
| Tat− | 1/10 | −48.9 ± 4.6 | 24 ± 3 | −16.3 ± 1.3 | 51.9 ± 3.7 |
| Tat+ | 11/11* | −52.5 ± 1.0 | 10 ± 0* | −21.0 ± 0.8* | 71.8 ± 2.4* |
aWhole-cell patch-clamp experiments in current-clamp mode on neurons pretreated with 100 nm Tat for 17 to 48 h. These neurons fired a statistically significant higher number of multiple action potentials and at lower rheobase and action potential thresholds compared with controls. Neurons treated with heat-inactivated Tat were less excitable compared with Tat pretreated and as excitable as controls (untreated neurons). Neurons isolated from Tat+ mice were also more excitable than Tat− mice neurons.
*p < 0.05 (paired t test).
Tat produced a leftward shift of the Boltzmann's activation curve of sodium channels
Sodium currents were measured in voltage-clamp studies with cesium (Cs) in the pipette solution as reported previously (Smith et al., 2012). The current–voltage relationship of sodium currents was shifted to the left upon overnight Tat (100 nm) exposure (Fig. 2A). There was no difference in the channel density. To determine the effects of short-term exposure (17 h) of neurons to Tat on the voltage dependence of steady-state activation/inactivation of sodium channels, we used a double-pulse protocol in which a variable conditioning pulse was applied from −100 mV to 50 mV in 10 mV increments, for 50 ms followed by a test pulse to 0 mV. For a conventional time- and voltage-dependent Hodgkin–Huxley conductance, the steady-state inactivation curve describes the relative number of available sodium channels as a function of voltage. The resultant sigmoidal curve was fit to a Boltzmann distribution (Fig. 2B). The V0.5 of activation, the voltage at which half of the channels are activated, was −29.08 ± 0.6 mV with a slope factor of 4.5 ± 0.5 mV for control neurons and −38.5 ± 0.8 mV with a slope factor of 1.9 ± 1.0 mV for Tat-treated neurons (Fig. 2B). There was a significant leftward shift in the activation curve of sodium channels, indicating that the channels were activated at more negative membrane potentials. Consistent with this, sodium currents activated 10 mV more negative (−50 mV) in Tat (100 nm) pretreated cells (t test, p < 0.05) (Fig. 2A,B). There was no significant change in the inactivation curve upon treatment with Tat. The V0.5 for inactivation was −36.9 ± 0.8 mV for controls and −39.5 ± 0.1 mV for Tat-treated neurons (Fig. 2B,D).
Figure 2.

Tat changes the steady-state voltage dependence of activation of sodium channels. Na+ channel activation/inactivation was examined using whole-cell patch-clamp experiments in voltage-clamp mode and a double-pulse protocol in the presence and absence of 100 nm Tat and without Tat (control). Cs+ is present in the internal solution to block outward K+ current. A, Current density/voltage curve of controls (untreated cells) and Tat-pretreated neurons. B, Boltzmann curve analysis of inactivation and activation of Na+ indicates a leftward shift of the activation curve in response to Tat. C, Significant difference in V0.5 of activation. D, There was no significant difference in V0.5 of inactivation. *p < 0.05 (t test).
Tat selectively modulates sodium channel isoforms
To date, nine mammalian pore-forming α-subunit isoforms (Nav1.1-Nav1.9) of sodium channels have been cloned and functionally characterized with a 10th (Nax) isoform appearing to be gated by sodium ion concentration. Nav1.1, Nav1.2, Nav1.3 Nav1.4, and Nav1.7 are TTX- sensitive, whereas Nav1.5, Nav1.8, and Nav1.9 are TTX-resistant. In the presence of internal Cs+, fast inward sodium currents were elicited positive to 40 mV from Vh −60 mV. Currents were markedly reduced in the presence of 1 μm TTX but not completely abolished. TTX-sensitive components were obtained by subtracting TTX resistant sodium currents from the total sodium currents (Fig. 3A). The mRNA for Nav1.3, Nav1.7, Nav1.8, and Nav1.9 were examined by quantitative PCR of LMMP that were treated with Tat (100 nm) for 30 min or 2 d. There was no difference in Nav1.7 and Nav1.8 mRNA expression after 30 min Tat treatment (Fig. 3B,C). After 2 d treatment with Tat, the mRNA for Nav1.7 and Nav1.8 were significantly enhanced. Nav1.3 and Nav1.9 were not significantly different between Tat treated and controls (Fig. 3D). These data suggest that Tat significantly increases the transcription of sodium channel isoforms upon longer treatment and that Tat's effect is isoform specific. Similar findings were seen in LMMP preparations from Tat transgenic mice (Fig. 3E), whereas mice containing the tat gene (i.e., Tat+ mice) expressed significantly higher Nav1.7 and Nav1.8 mRNA levels compared with control transgenic mice (Tat−).
Figure 3.

Tat transcriptionally modulates Nav1.7 and Nav1.8. A, Representative raw traces of voltage-clamp experiments showing the presence of TTX-sensitive and -resistant sodium channels in neurons isolated from the adult mouse ileum. B, C, Quantitative PCR of LMMP preparations after 30 min of Tat treatment. Nav1.7 and Nav1.8 mRNA levels are unchanged. D, After long-term exposure (2–3 d) to Tat, Nav1.7 and Nav1.8 mRNA levels are significantly increased, but Nav1.3 and Nav1.9 levels are not affected by Tat pretreatment. E, Significantly higher Nav1.7 and Nav1.8 mRNA are observed in Tat+ compared with Tat− LMMPs. *p < 0.05 (ANOVA followed by Bonferroni's post hoc test).
Nav1.7 and Nav1.8 are expressed on enteric neurons
To examine the distribution of Nav1.7 and Nav1.8 channels in enteric neurons, cells were grown on coverslips for 7 d to allow for significant neurite outgrowth. The subcellular distribution of Nav1.7 and Nav1.8 was assessed by immunohistochemistry. Nav1.7 was predominantly expressed at the cell membrane of neuronal perikarya, whereas Nav1.8 was mainly expressed in association with the neurites (Fig. 4A,B). Because neurite outgrowth may affect adequate voltage clamp, we measured sodium currents in cells that were within 1–3 d after isolation (Fig. 4C). These cells had phase-bright round cell bodies.
Figure 4.
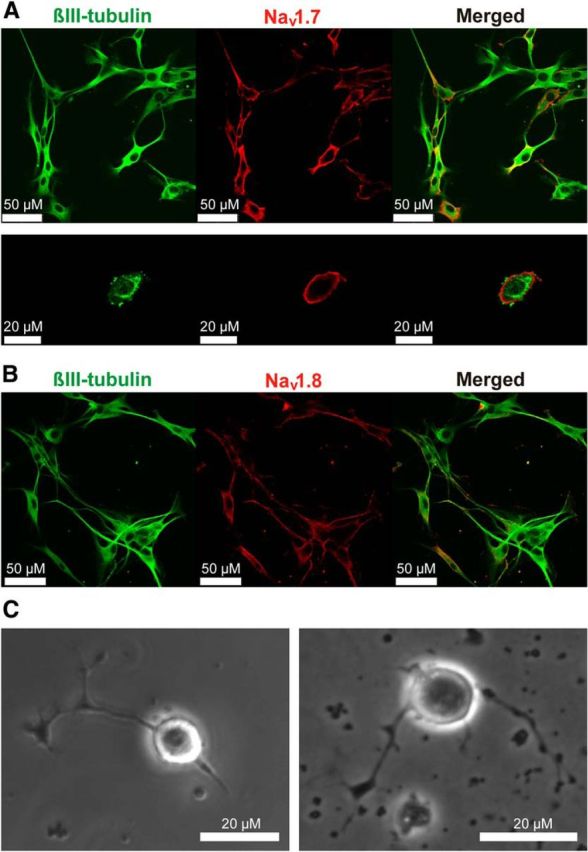
Nav1.7 and Nav1.8 are expressed on enteric neurons and are upregulated by Tat. Representative immunocytochemistry showing β-III tubulin (green), a neuronal marker, and the sodium channel isoforms (red) Nav1.7 (A) and Nav1.8 (B). Sodium channel isoforms colocalize with neurons and are associated with the cell membrane. C, Phase-contrast microscopic images showing neuronal cells with rounded cell bodies used for patch-clamp analysis.
Tat upregulates sodium current density
The current–voltage relationship of sodium currents showed significantly higher sodium current densities in neurons isolated from Tat+ compared with those isolated from Tat− mice. This higher current density was associated with a loss of voltage-control evident in the current–voltage relationship (Fig. 5A). Reducing extracellular Na+ was not sufficient to avoid the loss of voltage control (Fig. 5B). Adequate voltage clamp was also not achievable in Tat+ neurons that were in culture for only 1 d and mostly round in shape (to avoid axonal sprouting artifacts), and using wider pipette tips (1.5–2 mΩ). This indicated that long-term exposure to Tat in vivo enhanced sodium channel expression.
Figure 5.
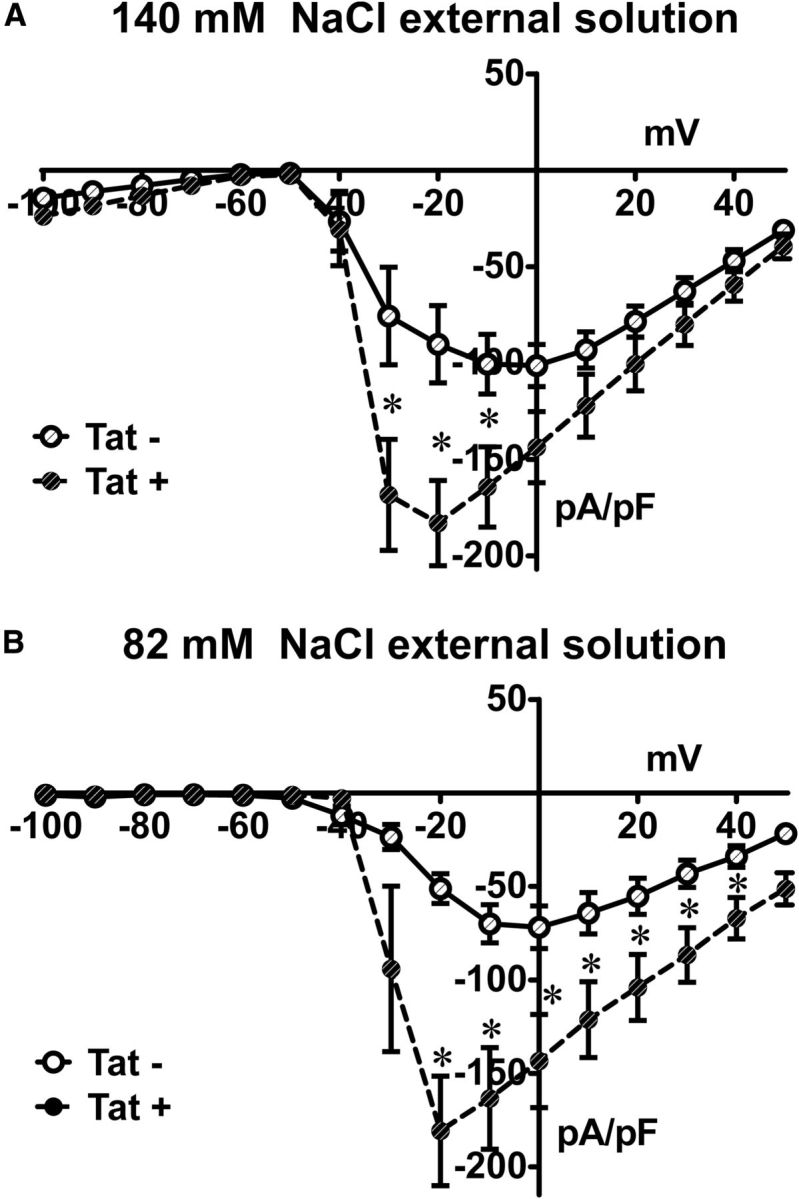
Tat+ neurons had higher sodium current densities compared with Tat−. Current density/voltage curves of Tat+ and Tat− neurons at 140 mm (A) and 82 mm (B) bath solution NaCl concentration. Tat+ neurons had higher sodium current densities. *p < 0.05 (t test).
Tat upregulates proinflammatory cytokines
To determine Tat's effect on inflammation in the GI tract, proinflammatory cytokines were measured in Tat-treated and control (untreated) enteric neuronal cells. Cultured neurons/glia cocultures were exposed to Tat overnight, after which the supernatant was removed and analyzed by ELISA to quantitatively assess the release of the cytokines TNF-α, IL-6, and the chemokine RANTES. Tat significantly increased RANTES and IL-6 release (p < 0.05) but did not affect TNF-α levels (Fig. 6A–C). Similarly, RANTES and IL-6 expression was enhanced in the ileum of Tat+ (p < 0.05), whereas TNF-α levels were unaffected. In addition, Tat+ transgenic LMMP preparations had threefold higher IL-1β mRNA levels than Tat− LMMPs (Fig. 6D).
Figure 6.

Tat selectively upregulates proinflammatory cytokines. ELISA assay, assessing proinflammatory cytokine release. A–C, Tat increases RANTES and IL-6 levels but has no effect on TNF-α levels. D, PCR showing increased levels of IL-1β mRNA. *p < 0.05 (Student's t test).
Tat increases GI motility
To determine the effects of Tat on GI motility, upper GI tract motility was determined by calculating percentage charcoal transit/total ileum length as described above. The distance traveled in Tat+ mice was modestly but significantly greater than control Tat− mice (p < 0.05) (Fig. 7A). To avoid the interference of the DOX diet used to induce Tat expression on GI effects and to allow for recolonization of the gut by GI bacteria, age-matched mice were fed DOX chow for 1 week after which the DOX chow was substituted for a regular chow for 3 weeks. PCR experiments showed that the tat gene expression was maintained for 3 weeks without DOX in the ileum of Tat+ mice and absent in the Tat− ileum (Fig. 7B). There was no significant difference in the rate of stomach stasis in Tat+ mice compared with Tat− mice (Fig. 7C). On the other hand, Tat+ mice contained more fluids in their cecum than Tat− mice, which had significantly lower cecal water content than Tat+ mice (Fig. 7D). Analysis of the number of stool pellets showed that Tat+ mice had faster stool output rates compared with Tat− mice (Fig. 7E).
Figure 7.

A, Tat increased GI motility in Tat transgenic mice. Tat+ mice showed higher upper GI transit rates compared with Tat−. GI transit was measured as distance traveled by charcoal meal. Quantitative PCR showing expression of Tat following 1 week of DOX treatment followed by 3 weeks of regular chow diet. B, Tat was expressed in Tat+ and absent in Tat− mouse ilea after 3 weeks without DOX. C, Gastric emptying shows no significant difference in stomach stasis rates between Tat+ and Tat− mice. D, Cecal water content is significantly higher in Tat+ mice than in Tat−. E, The number of stool pellets was significantly higher in Tat+ than Tat− mice. *p < 0.05 (Student's t test).
Discussion
The GI effects of HIV infection include alterations in structural integrity, diarrhea, and motility disorders. These effects are not mutually exclusive and may involve both direct effects of the HIV and viral proteins, and indirect effects resulting from opportunistic infections following breakdown of the epithelial barrier and immunosuppression within the lamina propria. Neuronal injury largely results from the effects of neurotoxic factors, including viral proteins Tat, gp120, and Nef (van Marle et al., 2004). The present study shows that enteric neurons can be directly affected by Tat. Tat exposure leads to increased neuronal excitability that occurred within 30 min. Two effects of Tat on sodium currents were observed that might account for enhanced excitability: (1) a shift in the activation kinetics of the sodium channel and (2) enhanced expression of Nav1.7 and Nav1.8. Tat also enhanced the release of proinflammatory cytokine release and GI motility. To our knowledge, this is the first study that evaluates the effects of Tat on enteric neuronal excitability and the first study to highlight the role of sodium channel expression and kinetics in Tat-mediated effects. The direct effects of Tat on neuronal excitability have been previously examined in human fetal brain cells, cultured rat cerebral cortical neurons, and in DRG neurons (Nath et al., 1996; Brailoiu et al., 2008; Chi et al., 2011). In fetal brain cells, Tat induced a depolarization in membrane potentials that were likely due to activation of a nonselective cation channel. In DRG neurons, the increased excitability may partly result from an inhibition of Cdk5, although the concentration of Tat used (20 μm) was much higher than used in our present study (100 nm). The 100 nm concentration was chosen for our studies from a range that elicited functional deficits in glia and neurons similar to those occurring in HIV-1 and that are considered to reflect levels seen under pathological conditions (Kruman et al., 1998; Nath et al., 1999; El-Hage et al., 2005, 2008; Perry et al., 2010). In enteric neurons, we found that increased excitability is seen within 15 min and remains when examined in Tat transgenic mice following induction of the gene for over 2 weeks. However, the mechanisms may differ among neuron types. After short-term exposure (17 h) to Tat, the steady-state, voltage dependence of activation of sodium channel currents is shifted to the left, whereas, with prolonged exposure (2 weeks DOX), there is an additional enhancement of sodium current density due to the increased expression of Nav1.7 and Nav1.8. The increase in sodium current density resulted in significant loss of voltage control. This might be due to enhanced expression of Nav1.8, which appears to be localized on neuronal projections and thus contributes to the inability to adequately control the voltage (Cummins et al., 2009).
Voltage-gated sodium channels play an important role in regulating neuronal excitability by producing the fast depolarization responsible for the upstroke of the action potential (Ekberg and Adams, 2006; Mantegazza et al., 2010). When neurons were exposed to Tat for a longer time (2 weeks DOX), Tat increased sodium current densities in neurons isolated from Tat+ compared with Tat− mice. These changes in sodium channel expression may have significant consequences in enhanced excitability following bacterial exposure as would occur with HIV infection. More recently, Chiu et al. (2013) demonstrated that bacterial proteins activate Nav1.8-expressing nociceptive neurons in the DRG, thus modulating pain sensation. Following epithelial barrier breakdown, intrinsic sensory neurons within the ENS may similarly be affected by bacterial proteins, thus defining a potential site of interaction between HIV and opportunistic bacteria.
Cytokines can also modulate enteric neuronal excitability. IL-6 and IL-1β have previously been shown to increase enteric neuronal excitability (Xia et al., 1999). Experimental and clinical data show that GI inflammation is a serious and common problem in HIV-infected individuals (Kotler et al., 1993; Kotler, 2005; Estes et al., 2010; Brenchley, 2013). Higher levels of the proinflammatory cytokines IL-6, IL-10, and IFN-γ have been reported in HIV patients (Kedzierska and Crowe, 2001). Studies in the CNS showed that Tat increased proinflammatory cytokine release by a glia-mediated mechanism (El-Hage et al., 2008). In our studies, Tat selectively increased IL-6, Il-10, IL-1β, and RANTES release but had no significant effect on TNF-α levels. The mechanisms underlying Tat-mediated increases in proinflammatory cytokine release in the gut are not well understood. The increase in cytokine release may also contribute to and/or exacerbate Tat-induced increases in neuronal excitability.
Increases in both neuronal excitability and in the release of proinflammatory cytokines have been reported to correlate with increased GI motility and to contribute to the diarrhea observed in many GI disorders. GI dysmotilities and diarrhea are persistent problems in the cART era (Mitra et al., 2001; Mathur et al., 2013). Upper GI transit was slightly but significantly higher in Tat+ mice maintained on DOX for 2 weeks. Because DOX treatment alters gut microbiota, which can affect GI function, we examined gastric stasis, cecum water content, and fecal output rate after recolonization of colonic bacteria. Under these conditions, GI transit was markedly enhanced. Further studies are required to determine whether the increase in cecal water content is due to increased secretion or reduced reabsorption.
Together, these studies have analyzed the effects of Tat in single neurons and shown that Tat increases enteric neuronal excitability by modulating sodium channels. The increase in neuronal excitability together with an increase in the release of proinflammatory cytokines could account for the augmented GI motility observed in Tat+ mice. These findings correlate with increases in GI motility and diarrhea observed in HIV-infected individuals, suggesting a possible mechanism by which this may be occurring. Interest in the brain–gut axis has also been prompted by findings that many CNS diseases may have an ENS component. For example, GI disturbances and the presence of Lewy bodies in enteric neurons of Parkinson disease patients occur decades before advent of motor symptoms (Pan-Montojo et al., 2010, 2012). The ileum, in particular, is one of the earliest sites of infection (Levesque et al., 2009) and displays a higher proportion of HIV-infected cells than other regions of the GI tract (Yukl et al., 2013). These studies point to more effective therapeutic targets in the design of future HIV therapies.
Footnotes
This work was supported by National Institutes of Health Grants F31 NS087952 to J.N., K99 DA033878 to S.F., R01 DK046367 to H.I.A., R01 DA024009 to W.L.D. and H.I.A., K02 DA027374, and R01 DA018633 to K.F.H.
The authors declare no competing financial interests.
References
- Akbarali HI, Giles WR. Ca2+ and Ca(2+)-activated Cl− currents in rabbit oesophageal smooth muscle. J Physiol. 1993;460:117–133. doi: 10.1113/jphysiol.1993.sp019462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anitha M, Vijay-Kumar M, Sitaraman SV, Gewirtz AT, Srinivasan S. Gut microbial products regulate murine gastrointestinal motility via toll-like receptor 4 signaling. Gastroenterology. 2012;143:1006–1016. doi: 10.1053/j.gastro.2012.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakawa A, Inui A, Kaga T, Katsuura G, Fujimiya M, Fujino MA, Kasuga M. Antagonism of ghrelin receptor reduces food intake and body weight gain in mice. Gut. 2003;52:947–952. doi: 10.1136/gut.52.7.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brailoiu GC, Brailoiu E, Chang JK, Dun NJ. Excitatory effects of human immunodeficiency virus 1 tat on cultured rat cerebral cortical neurons. Neuroscience. 2008;151:701–710. doi: 10.1016/j.neuroscience.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley JM. Mucosal immunity in human and simian immunodeficiency lentivirus infections. Mucosal Immunol. 2013;6:657–665. doi: 10.1038/mi.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen PL, Khoruts A, Larson M, Haase AT, Douek DC. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Turchan-Cholewo J, Smart EJ, Geurin T, Chauhan A, Reid R, Xu R, Nath A, Knapp PE, Hauser KF. Morphine causes rapid increases in glial activation and neuronal injury in the striatum of inducible HIV-1 tat transgenic mice. Glia. 2008;56:1414–1427. doi: 10.1002/glia.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HC, Samaniego F, Nair BC, Buonaguro L, Ensoli B. HIV-1 tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. AIDS. 1997;11:1421–1431. doi: 10.1097/00002030-199712000-00006. [DOI] [PubMed] [Google Scholar]
- Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, Wainger B, Strominger A, Muralidharan S, Horswill AR, Bubeck Wardenburg J, Hwang SW, Carroll MC, Woolf CJ. Bacteria activate sensory neurons that modulate pain and inflammation. Nature. 2013;501:52–57. doi: 10.1038/nature12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X, Amet T, Byrd D, Chang KH, Shah K, Hu N, Grantham A, Hu S, Duan J, Tao F, Nicol G, Yu Q. Direct effects of HIV-1 tat on excitability and survival of primary dorsal root ganglion neurons: possible contribution to HIV-1-associated pain. PLoS One. 2011;6:e24412. doi: 10.1371/journal.pone.0024412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornblath DR, Hoke A. Recent advances in HIV neuropathy. Curr Opin Neurol. 2006;19:446–450. doi: 10.1097/01.wco.0000245366.59446.57. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Rush AM, Estacion M, Dib-Hajj SD, Waxman SG. Voltage-clamp and current-clamp recordings from mammalian DRG neurons. Nat Protoc. 2009;4:1103–1112. doi: 10.1038/nprot.2009.91. [DOI] [PubMed] [Google Scholar]
- De Winter BY, De Man JG. Interplay between inflammation, immune system and neuronal pathways: effect on gastrointestinal motility. World J Gastroenterol. 2010;16:5523–5535. doi: 10.3748/wjg.v16.i44.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekberg J, Adams DJ. Neuronal voltage-gated sodium channel subtypes: key roles in inflammatory and neuropathic pain. Int J Biochem Cell Biol. 2006;38:2005–2010. doi: 10.1016/j.biocel.2006.06.008. [DOI] [PubMed] [Google Scholar]
- El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Synergistic increases in intracellular Ca 2+,and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia. 2005;50:91–106. doi: 10.1002/glia.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Bruce-Keller AJ, Yakovleva T, Bazov I, Bakalkin G, Knapp PE, Hauser KF. Morphine exacerbates HIV-1 tat-induced cytokine production in astrocytes through convergent effects on [Ca(2+)](i), NF-kappaB trafficking and transcription. PLoS One. 2008;3:e4093. doi: 10.1371/journal.pone.0004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8:33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]
- Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, Barclay GR, Smedley J, Pung R, Oliveira KM, Hirsch VM, Silvestri G, Douek DC, Miller CJ, Haase AT, Lifson J, Brenchley JM. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 2010;6:e1001052. doi: 10.1371/journal.ppat.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Scoggins KL, Xu R, Dever SM, Knapp PE, Dewey WL, Hauser KF. Morphine efficacy is altered in conditional HIV-1 tat transgenic mice. Eur J Pharmacol. 2012;689:96–103. doi: 10.1016/j.ejphar.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Scarano F, Martín-García J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Handley SA, Thackray LB, Zhao G, Presti R, Miller AD, Droit L, Abbink P, Maxfield LF, Kambal A, Duan E, Stanley K, Kramer J, Macri SC, Permar SR, Schmitz JE, Mansfield K, Brenchley JM, Veazey RS, Stappenbeck TS, Wang D, et al. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell. 2012;151:253–266. doi: 10.1016/j.cell.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TP, Patel K, Johnson KR, Maric D, Calabresi PA, Hasbun R, Nath A. Induction of IL-17 and nonclassical T-cell activation by HIV-tat protein. Proc Natl Acad Sci U S A. 2013;110:13588–13593. doi: 10.1073/pnas.1308673110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn J. Tackling tat. J Mol Biol. 1999;293:235–254. doi: 10.1006/jmbi.1999.3060. [DOI] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kedzierska K, Crowe SM. Cytokines and HIV-1: interactions and clinical implications. Antiviral Chem Chemother. 2001;12:133–150. doi: 10.1177/095632020101200301. [DOI] [PubMed] [Google Scholar]
- Kilareski EM, Shah S, Nonnemacher MR, Wigdahl B. Regulation of HIV-1 transcription in cells of the monocyte-macrophage lineage. Retrovirology. 2009;6:118. doi: 10.1186/1742-4690-6-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konturek JW, Fischer H, van der Voort IR, Domschke W. Disturbed gastric motor activity in patients with human immunodeficiency virus infection. Scand J Gastroenterol. 1997;32:221–225. doi: 10.3109/00365529709000198. [DOI] [PubMed] [Google Scholar]
- Kotler DP. HIV infection and the gastrointestinal tract. AIDS. 2005;19:107–117. doi: 10.1097/00002030-200501280-00002. [DOI] [PubMed] [Google Scholar]
- Kotler DP, Reka S, Clayton F. Intestinal mucosal inflammation associated with human immunodeficiency virus infection. Dig Dis Sci. 1993;38:1119–1127. doi: 10.1007/BF01295730. [DOI] [PubMed] [Google Scholar]
- Kruman II, Nath A, Mattson MP. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol. 1998;154:276–288. doi: 10.1006/exnr.1998.6958. [DOI] [PubMed] [Google Scholar]
- Lackner AA, Mohan M, Veazey RS. The gastrointestinal tract and AIDS pathogenesis. Gastroenterology. 2009;136:1965–1978. doi: 10.1053/j.gastro.2008.12.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann HC, Chen W, Borzan J, Mankowski JL, Höke A. Mitochondrial dysfunction in distal axons contributes to human immunodeficiency virus sensory neuropathy. Ann Neurol. 2011;69:100–110. doi: 10.1002/ana.22150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque MC, Moody MA, Hwang KK, Marshall DJ, Whitesides JF, Amos JD, Gurley TC, Allgood S, Haynes BB, Vandergrift NA, Plonk S, Parker DC, Cohen MS, Tomaras GD, Goepfert PA, Shaw GM, Schmitz JE, Eron JJ, Shaheen NJ, Hicks CB, et al. Polyclonal B cell differentiation and loss of gastrointestinal tract germinal centers in the earliest stages of HIV-1 infection. PLoS Med. 2009;6:e1000107. doi: 10.1371/journal.pmed.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SG, Condez A, Lee CA, Johnson MA, Elia C, Poulter LW. Loss of mucosal CD4 lymphocytes is an early feature of HIV infection. Clin Exp Immunol. 1993;92:448–454. doi: 10.1111/j.1365-2249.1993.tb03419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza M, Curia G, Biagini G, Ragsdale DS, Avoli M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010;9:413–424. doi: 10.1016/S1474-4422(10)70059-4. [DOI] [PubMed] [Google Scholar]
- Mathur MK, Verma AK, Makwana GE, Sinha M. Study of opportunistic intestinal parasitic infections in human immunodeficiency virus/acquired immunodeficiency syndrome patients. J Global Infect Dis. 2013;5:164–167. doi: 10.4103/0974-777X.122012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434:1093–1097. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- Mavigner M, Cazabat M, Dubois M, L'Faqihi FE, Requena M, Pasquier C, Klopp P, Amar J, Alric L, Barange K, Vinel JP, Marchou B, Massip P, Izopet J, Delobel P. Altered CD4+ T cell homing to the gut impairs mucosal immune reconstitution in treated HIV-infected individuals. J Clin Invest. 2012;122:62–69. doi: 10.1172/JCI59011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur JC, Steiner J, Sacktor N, Nath A. Human immunodeficiency virus-associated neurocognitive disorders: mind the gap. Ann Neurol. 2010;67:699–714. doi: 10.1002/ana.22053. [DOI] [PubMed] [Google Scholar]
- Mitra AK, Hernandez CD, Hernandez CA, Siddiq Z. Management of diarrhoea in HIV-infected patients. Int J STD AIDS. 2001;12:630–639. doi: 10.1258/0956462011923840. [DOI] [PubMed] [Google Scholar]
- Mohan M, Kaushal D, Aye PP, Alvarez X, Veazey RS, Lackner AA. Focused examination of the intestinal lamina propria yields greater molecular insight into mechanisms underlying SIV induced immune dysfunction. PLoS One. 2012;7:e34561. doi: 10.1371/journal.pone.0034561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, Geiger JD. Identification of a human immunodeficiency virus type 1 tat epitope that is neuroexcitatory and neurotoxic. J Virol. 1996;70:1475–1480. doi: 10.1128/jvi.70.3.1475-1480.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 tat protein results in cytokine production in macrophages and astrocytes: a hit and run phenomenon. J Biol Chem. 1999;274:17098–17102. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- Nath A, Booze RM, Hauser KF, Mactutus CF, Bell JE, Maragos WF, Berger JR. Critical questions for neuroscientists in interactions of drugs of abuse and HIV infection. NeuroAIDS. 1999;2:1–12. [Google Scholar]
- Neunlist M, Van Landeghem L, Mahé MM, Derkinderen P, des Varannes SB, Rolli-Derkinderen M. The digestive neuronal-glial-epithelial unit: a new actor in gut health and disease. Nat Rev Gastroenterol Hepatol. 2013;10:90–100. doi: 10.1038/nrgastro.2012.221. [DOI] [PubMed] [Google Scholar]
- Orandle MS, Veazey RS, Lackner AA. Enteric ganglionitis in rhesus macaques infected with simian immunodeficiency virus. J Virol. 2007;81:6265–6275. doi: 10.1128/JVI.02671-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan-Montojo F, Anichtchik O, Dening Y, Knels L, Pursche S, Jung R, Jackson S, Gille G, Spillantini MG, Reichmann H, Funk RH. Progression of Parkinson's disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One. 2010;5:e8762. doi: 10.1371/journal.pone.0008762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan-Montojo F, Schwarz M, Winkler C, Arnhold M, O'Sullivan GA, Pal A, Said J, Marsico G, Verbavatz JM, Rodrigo-Angulo M, Gille G, Funk RH, Reichmann H. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep. 2012;2:898. doi: 10.1038/srep00898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo CA, McArthur JC, Griffin JW. HIV neuropathy: insights in the pathology of HIV peripheral nerve disease. J Peripheral Nerv Syst. 2001;6:21–27. doi: 10.1046/j.1529-8027.2001.006001021.x. [DOI] [PubMed] [Google Scholar]
- Perry SW, Barbieri J, Tong N, Polesskaya O, Pudasaini S, Stout A, Lu R, Kiebala M, Maggirwar SB, Gelbard HA. Human immunodeficiency virus-1 Tat activates calpain proteases via the ryanodine receptor to enhance surface dopamine transporter levels and increasetransporter-specific uptake and Vmax. J Neurosci. 2010;30:14153–14164. doi: 10.1523/JNEUROSCI.1042-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasseville VG, Du Z, Chalifoux LV, Pauley DR, Young HL, Sehgal PK, Desrosiers RC, Lackner AA. Induction of lymphocyte proliferation and severe gastrointestinal disease in macaques by a nef gene variant SIVmac239. Am J Pathol. 1996;149:163–176. [PMC free article] [PubMed] [Google Scholar]
- Simpson DM, Olney RK. Peripheral neuropathies associated with human immunodeficiency virus infection. Neurol Clin. 1992;10:685–711. [PubMed] [Google Scholar]
- Smith TH, Grider JR, Dewey WL, Akbarali HI. Morphine decreases enteric neuron excitability via inhibition of sodium channels. PLoS One. 2012;7:e45251. doi: 10.1371/journal.pone.0045251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TH, Ngwainmbi J, Grider JR, Dewey WL, Akbarali HI. An in-vitro preparation of isolated enteric neurons and glia from the myenteric plexus of the adult mouse. J Vis Exp. 2013;7:78. doi: 10.3791/50688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit-McBride Z, Mattapallil JJ, McChesney M, Ferrick D, Dandekar S. Gastrointestinal T lymphocytes retain high potential for cytokine responses but have severe CD4(+) T-cell depletion at all stages of simian immunodeficiency virus infection compared to peripheral lymphocytes. J Virol. 1998;72:6646–6656. doi: 10.1128/jvi.72.8.6646-6656.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–674. doi: 10.1038/nri2832. [DOI] [PubMed] [Google Scholar]
- van Marle G, Henry S, Todoruk T, Sullivan A, Silva C, Rourke SB, Holden J, McArthur JC, Gill MJ, Power C. Human immunodeficiency virus type 1 nef protein mediates neural cell death: a neurotoxic role for IP-10. Virology. 2004;329:302–318. doi: 10.1016/j.virol.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, Rosenzweig M, Johnson RP, Desrosiers RC, Lackner AA. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280:427–431. doi: 10.1126/science.280.5362.427. [DOI] [PubMed] [Google Scholar]
- Welsh C, Enomoto M, Pan J, Shifrin Y, Belik J. Tetrahydrobiopterin deficiency induces gastroparesis in newborn mice. Am J Physiol Gastrointest Liver Physiol. 2013;305:G47–G57. doi: 10.1152/ajpgi.00424.2012. [DOI] [PubMed] [Google Scholar]
- Wu Y. HIV-1 gene expression: lessons from provirus and non-integrated DNA. Retrovirology. 2004;1:13. doi: 10.1186/1742-4690-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Hu HZ, Liu S, Ren J, Zafirov DH, Wood JD. IL-1beta and IL-6 excite neurons and suppress nicotinic and noradrenergic neurotransmission in guinea pig enteric nervous system. J Clin Invest. 1999;103:1309–1316. doi: 10.1172/JCI5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukl SA, Shergill AK, Ho T, Killian M, Girling V, Epling L, Li P, Wong LK, Crouch P, Deeks SG, Havlir DV, McQuaid K, Sinclair E, Wong JK. The distribution of HIV DNA and RNA in cell subsets differs in gut and blood of HIV-positive patients on ART: implications for viral persistence. J Infect Dis. 2013;208:1212–1220. doi: 10.1093/infdis/jit308. [DOI] [PMC free article] [PubMed] [Google Scholar]