

Abstract
Accumulating evidence strongly implicates the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) in the pathophysiology of multiple neurological disorders, but the downstream gene targets of PGC-1α in the brain have remained enigmatic. Previous data demonstrate that PGC-1α is primarily concentrated in inhibitory neurons and that PGC-1α is required for the expression of the interneuron-specific Ca2+-binding protein parvalbumin (PV) throughout the cortex. To identify other possible transcriptional targets of PGC-1α in neural tissue, we conducted a microarray on neuroblastoma cells overexpressing PGC-1α, mined results for genes with physiological relevance to interneurons, and measured cortical gene and protein expression of these genes in mice with underexpression and overexpression of PGC-1α. We observed bidirectional regulation of novel PGC-1α-dependent transcripts spanning synaptic [synaptotagmin 2 (Syt2) and complexin 1 (Cplx1)], structural [neurofilament heavy chain (Nefh)], and metabolic [neutral cholesterol ester hydrolase 1 (Nceh1), adenylate kinase 1 (Ak1), inositol polyphosphate 5-phosphatase J (Inpp5j), ATP synthase mitochondrial F1 complex O subunit (Atp5o), phytanol-CoA-2hydroxylase (Phyh), and ATP synthase mitrochondrial F1 complex α subunit 1 (Atp5a1)] functions. The neuron-specific genes Syt2, Cplx1, and Nefh were developmentally upregulated in an expression pattern consistent with that of PGC-1α and were expressed in cortical interneurons. Conditional deletion of PGC-1α in PV-positive neurons significantly decreased cortical transcript expression of these genes, promoted asynchronous GABA release, and impaired long-term memory. Collectively, these data demonstrate that PGC-1α is required for normal PV-positive interneuron function and that loss of PGC-1α in this interneuron subpopulation could contribute to cortical dysfunction in disease states.
Keywords: Barnes maze, cortical development, Huntington disease, ppargc1a, schizophrenia, strontium
Introduction
A growing body of literature strongly implicates the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) in the pathophysiology of neurological disorders. Initial reports demonstrated that PGC-1α expression is reduced in brain and muscle of patients with Huntington disease (HD; Cui et al., 2006; Weydt et al., 2006; Chaturvedi et al., 2009; Kim et al., 2010), and subsequent studies found polymorphisms and/or postmortem expression abnormalities in PGC-1α in patients with Huntington (Taherzadeh-Fard et al., 2009; Weydt et al., 2009, 2014), Alzheimer (Qin et al., 2009), and Parkinson (Zheng et al., 2010; Clark et al., 2011; Shin et al., 2011) diseases. Despite the proposed therapeutic potential of targeting PGC-1α in such disorders, little is known about the primary downstream transcriptional targets of PGC-1α in the CNS. Thus, the consequences of decreased expression and/or function of PGC-1α in the brain and its active role in neurological disorders have remained enigmatic.
As a transcriptional coactivator, PGC-1α regulates gene expression by binding to specific transcription factors and recruiting histone acetyltransferases to simultaneously influence chromatin structure and initiate transcription (Lin, 2009). In peripheral tissues, PGC-1α has been coined the “master regulator” of metabolism because of its ability to induce transcription of genes involved in mitochondrial biogenesis and antioxidant production (Lin et al., 2005). Whereas it has been mainly assumed that PGC-1α regulates similar genes in the brain (Rona-Voros and Weydt, 2010), only modest reductions have been found in well established peripheral PGC-1α target genes in brain from PGC-1α−/− animals (Lin et al., 2004; St-Pierre et al., 2006; Lucas et al., 2010).
Previous work has shown that PGC-1α becomes primarily concentrated in GABAergic neurons during early postnatal development of the rodent brain (Cowell et al., 2007), suggesting that PGC-1α serves to regulate gene expression specifically within this neuronal population. In support of this idea, we discovered that the presence of PGC-1α is required for expression of the Ca2+-binding protein parvalbumin (PV) in interneurons throughout the cerebrum (Lucas et al., 2010). Interestingly, PGC-1α−/− mice exhibit asynchronous and delayed neurotransmitter release from presynaptic terminals in the hippocampus (Lucas et al., 2010), which is uncharacteristic of normally functioning PV-positive interneurons and difficult to attribute to a loss of PV alone (Hefft and Jonas, 2005; Manseau et al., 2010). We thus hypothesized that PGC-1α must have additional transcriptional targets relevant to interneuron physiology.
We show here that PGC-1α is required for a developmental gene program regulating synaptotagmin 2 (Syt2) and complexin 1 (Cplx1), two high-affinity Ca2+ sensors essential for synchronous neurotransmitter release, and the structural protein neurofilament heavy chain (Nefh). In accordance with their expression in interneurons and the known physiological roles of these transcripts, conditional deletion of PGC-1α from PV-positive neurons decreased the expression of PGC-1α, PV, Syt2, Cplx1, and Nefh; increased asynchronous GABA release onto pyramidal cells; and impaired long-term memory. These data suggest that PGC-1α provides a transcriptional framework to mediate synchronous neurotransmitter release and that loss of PGC-1α in interneurons could contribute to cortical dysfunction in disease states.
Materials and Methods
Cell culture and microarray.
SH-SY5Y neuroblastoma cells were transfected using an adenovirus encoding green fluorescent protein (GFP) alone or GFP and PGC-1α in tandem (Cowell et al., 2009). Cells were collected 48 h later in Trizol reagent (n = 3 per group). RNA was isolated by the Trizol method following the manufacturer's (Invitrogen) instructions. Briefly, cells were triturated in Trizol reagent. RNA was isolated using the chloroform–isopropanol method and purified with an RNease MinElute Cleanup kit (Qiagen). RNA concentration was adjusted to 200 ng/μl before submitting to the W.M. Keck Biotechnology Resource Laboratory at Yale University for microarray analysis.
An Illumina human gene expression array was used. The expression data were normalized using quantile normalization. Independent-samples t tests were applied to compare the differences in gene expression between GFP- and PGC-1α-transfected samples. Transcripts with a twofold or greater expression change induced by PGC-1α overexpression and a t test p value of <0.01 were filtered out for further statistical analyses. False discovery rate correction for multiple testing was then set at <0.05 to identify transcripts for additional studies.
To mine microarray data for genes with physiological relevance in the mouse brain, we cross-referenced genes significantly upregulated by PGC-1α with a list of genes that overlapped neuroanatomically with ppargc1a and pvalb using the Neuroblast feature of the Allen Brain Atlas (www.brain-map.org). The Allen Brain Atlas is an online library of in situ hybridization images of ∼20,000 genes in the adult mouse brain, and the Neuroblast feature is a search tool to help identify genes with similar 3D spatial gene expression profiles. A mathematical algorithm is applied to each in situ image to determine “expression energy” for each gene with respect to individual cubes within a 200 μm2 grid (Lein et al., 2007; also see http://mouse.brain-map.org/pdf/NeuroBlast.pdf). Pearson's correlation coefficient is then used to measure similarity between expression energy of each gene within the same cube.
Animals.
All experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham. Two independent lines of PGC-1α−/− mice (Lin et al., 2004; Leone et al., 2005) were used for experiments, and PGC-1α+/+, PGC-1α+/−, and PGC-1α−/− mice were obtained from offspring of PGC-1α+/− breeding pairs. All experiments were conducted with male and female mice from the line described by Lin et al. (2004) on postnatal day 30 (P30) unless otherwise specified. Conditional deletion of PGC-1α was produced by crossing mice with LoxP sites flanking the exon 3–5 region of the PGC-1α gene (Lin et al., 2004; gift from Bruce Spiegleman, Dana-Farber Cancer Institute, Boston, MA) with mice expressing cre recombinase driven by the PV (Hippenmeyer et al., 2005; The Jackson Laboratory) or nestin (gift from Albert La Spada, University of San Diego, San Diego, CA) promoter. To determine specificity and efficiency of the cre-mediated recombination pattern, PV-Cre mice were crossbred to mutant tomato/mutant green reporter mice (Muzumdar et al., 2007; The Jackson Laboratory). For conditional knock-out experiments, littermates expressing cre recombinase without loxP sites were used as controls. All mice were maintained on a C57BL6/J genetic background and housed two to five in a cage at 26 ± 2°C room temperature with food and water ad libitum.
AAV preparation and injections.
Mouse PGC-1α (Addgene Plasmid #1026, donated by B. Speigelman; Monsalve et al., 2000) was cloned into the pTR-SB-smCBA-V2 vector and packaged into AAV2/9 (University of Florida Vector Core). C57BLl/6 mice received 2 μl injections of AAV- PGC-1α or AAV-GFP into the anterior cortex (AP, +1.34; ML, ±1.75; DV, −1.75 mm from bregma); three to four mice were given injections per group. Tissue was removed for qRT-PCR analysis 14 d later.
Gene expression analyses.
Mice were anesthetized with isoflurane before they were killed by decapitation. Brains were rapidly removed and dissected by gross anatomical markers. Tissue pieces were collected in centrifuge tubes, flash frozen on dry ice, and stored at −80°C. Before processing, samples were incubated in RNAlater-ICE (Ambion) according to the manufacturer's instructions. Tissue was homogenized with Tissue-Tearor (Biospec) in Trizol reagent, and RNA was isolated and reverse transcribed as described above. RNA concentrations and purity were quantified using NanoDrop 2000 (Thermo Fisher Scientific). Equivalent amounts of RNA (1 μg) were treated with DNase I (Promega) at 37°C for 30 min, and DNase was inactivated at 65°C for 15 min. RNA was reverse transcribed using the High-Capacity cDNA Archive kit (Applied Biosystems). Gene expression was measured with Taqman qRT-PCR using the mouse-specific Applied Biosystems primers listed in Table 1.
Table 1.
List of Applied Biosystems (ABI) primers with catalog numbers
| Gene name | ABI catalog # |
|---|---|
| Acsl6 | Mm00522786_m1 |
| Ak1 | Mm00445475_m1 |
| Atp5o | Mm01611862_g1 |
| Atp5a1 | Mm00431960_m1 |
| Atp5h | Mm02392026_g1 |
| Actinb | Mm00607939_s1 |
| Cdc42ep1 | Mm00840486_m1 |
| Cox6c | Mm00835813_g1 |
| Cox7b | Mm00835076_g1 |
| Cplx1 | Mm00514378_m1 |
| Gas6 | Mm00490378_m1 |
| Grin2c | Mm00439180_m1 |
| Impa1 | Mm00497770_m1 |
| Itgb1bp | Mm00492707_m1 |
| KCKK1 | Mm00434624_m1 |
| Lifr | Mm00442942_m1 |
| Nefh | Mm01191456_m1 |
| Nceh1 | Mm00626772_m1 |
| Ndufs8 | Mm00523063_m1 |
| Pacsin2 | Mm00449303_m1 |
| PDHA1 | Mm00468675_m1 |
| Inpp5j | Mm00552486_m1 |
| Ppargc1a | Mm00447183_m1 |
| Phyh | Mm00477734_m1 |
| Pvalb | Mm00443100_m1 |
| Slc39a14 | Mm01317439_m1 |
| Sparcl1 | Mm00447780_m1 |
| St8sia1 | Mm00456915_m1 |
| St8sia5 | Mm00457285_m1 |
| Stac2 | Mm00524631_m1 |
| Syt2 | Mm00436864_m1 |
| Uqcrfs1 | Mm00481849_m1 |
| Uqcrh | Mm00835199_g1 |
| Vamp2 | Mm00494119_g1 |
| 18S | Hs99999901_s1 |
Reaction protocols consisted of an initial ramp (2 min, 50°C; 10 min, 95°C) and 40 subsequent cycles (15 s, 95°C; 1 min, 60°C). Relative concentrations of the genes of interest were calculated in comparison with a standard curve calculated from dilutions of cDNA (1:5, 1:10, 1:20) from a pool of GFP (cell-culture studies) or littermate (animal studies) controls. Values were normalized to 18S rRNA (cell-culture studies, developmental time course animal studies) or β-actin (all other animal studies) for values for the same sample and expressed as a ratio to control samples ± SEM.
Western blot analyses.
Primary antibodies included PGC-1α (gift from Daniel Kelly, Sanford-Burnham Medical Research Institute, Orlando, FL; Cowell et al., 2007), Syt2 (znp-1; Zebrafish International Resource Center; Fox and Sanes, 2007; Su et al., 2010), Cplx1,2 (Synaptic Systems; Pabst et al., 2000), Nefh (Abcam), and actin (Millipore; Cowell et al., 2007). All primary antibodies, with the exception of PGC-1α, produce bands at the previously published molecular weights. The PGC-1α antibody produces many nonspecific bands in the mouse brain and was, therefore, only used for confirmation of overexpression in cell-culture studies.
Cells (cell-culture studies) were sonicated, and brain regions (animal studies) were homogenized in RIPA buffer (150 mm NaCl, 50 mm Tris, 1% Triton X-100, 1% SDS, 0.5% deoxycholic acid, pH 8.0) containing a protease inhibitor tablet. Total protein concentration was determined with a bicinchonicic acid protein assay kit (Thermo Fisher Scientific), and absorbance was measured at 540 nm. Protein was denatured in sample buffer (62.5 mm Tris-HCl, 20% glycerol, 2% SDS, 5% β-mercaptoethanol, pH 6.8) at 95°C. Equivalent amounts of protein were loaded into precast polyacrylamide NuPage gels (Invitrogen). One interblot control sample was loaded onto every gel to permit comparison among gels. Protein was transferred onto nitrocellulose membranes. Blots were blocked with 5% milk in Tris-buffered saline (TBS; pH 7.6) with 1% Tween (TBS-T) and probed with primary antibodies in 5% IgG-free bovine serum albumin (BSA; Jackson ImmunoResearch)/TBS-T overnight at 4°C and peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) in 5% milk TBS-T for 1 h at room temperature. Blots were incubated in chemiluminescent substrate (Thermo Fisher Scientific) and exposed to film. The optical density of bands was calculated after background subtraction using UN-SCAN-IT gel analysis software (Silk Scientific). All bands were normalized to the interblot control band, then to actin, and expressed as mean optical density ± SEM.
Immunofluorescence.
Animals were anesthetized with isoflurane and perfused intracardially with PBS and 4% paraformaldehyde (PFA) in PBS. Brains were removed, postfixed in 4% PFA for 24–72 h, cryoprotected in graded sucrose (5–20%), embedded in a mixture of 20% sucrose and Tissue-Tek O.C.T. compound (Sakura Finetek), and frozen at −80°C. Tissue blocks were sectioned at 20 μm, mounted onto charged slides (Thermo Fisher Scientific), and allowed to dry overnight before freezing at −80°C.
The same primary antibodies were used for immunofluorescence as Western blot analysis (see above) with the addition of GAD67 (Millipore) and PV (Sigma or Swant). Slides were thawed, washed in PBS, and blocked with 10% serum from the host of the secondary antibody in PBS for 1 h. Optimal staining of PV and Syt2 required antigen retrieval by incubation in citrate buffer (10 mm citric acid, pH 4.0) at 37°C for 10 min and at room temperature for 20 min. Sections were then incubated in the primary antibodies overnight with 3% BSA and 5% serum from the host of the secondary antibody in PBS with Triton-X100 (PBS-T; Sigma) at 4°C. Slides were washed in PBS-T and PBS and incubated for 2 h at room temperature with the corresponding fluorescence-conjugated secondary antibody (Jackson ImmunoResearch) with 5% serum for the host of the secondary antibody in PBS-T. Slides were washed in PBS. For colabeling with GAD67, a mouse-on-mouse kit (Vector Laboratories) was used following the manufacturer's instructions to reduce background staining. Sections were coverslipped with Prolong Antifade Gold (Invitrogen) and stored at 4°C. All images were captured with a Leica confocal microscope. All confocal settings, including laser intensity, gain, offset, and zoom, were held constant across all groups for a given protein.
Whole-cell recordings.
Three-month-old PGC-1αWT:PV-Cre and PGC-1αfl/fl:PV-Cre mice were anesthetized with isoflurane, and brains were rapidly removed. Brains were sectioned at 300 μm in ice-cold artificial CSF (in mm: 125 NaCl, 3.5 KCl, 0.5 CaCl2, 3.5 MgCl2, 26 NaHCO3, and 10 d-glucose) bubbled with 95%O2/5%CO2. Slices were incubated for 30 min at 37 ± 1°C and stored at room temperature (22 ± 1°C) until recording. Whole-cell patch-clamp recordings were acquired from visually identified pyramidal neurons in layer 5 of the motor cortex. Cells were voltage clamped at −70 mV. Patch electrodes (2–5 MΩ) were filled with an internal solution containing (in mm) 129 CsCl, 2 MgATP, 10 EGTA, 10 HEPES, 0.2 GTP, and 2 QX-314, pH 7.2. Recordings were obtained using a MultiClamp 700A amplifier (Molecular Devices) controlled by Clampex 8.0 software via a Digidata 1322A interface (Molecular Devices), filtered at 5 kHz, and digitized at 10 kHz. Input resistance and series resistance were monitored by applying a 10 mV voltage step.
Evoked IPSCs (eIPSCs) were pharmacologically isolated with CNXQ (6-cyano-7-nitroquinoxaline-2,3-dione; 10 μm) and APV (dl-2-amino-5-phosphonoovaleric acid; 50 μm). Miniature IPSCs were further isolated with tetrodotoxin (TTX; 1 μm). To assess alterations in asynchronous neurotransmitter release, extracellular calcium was replaced with the cation strontium (Rumpel and Behrends, 1999). Control evoked responses were initially recorded in 2 mm calcium. The bathing solution was then changed to one in which 2 mm strontium was substituted for calcium. After 5 min, evoked responses were again measured with each cell serving as its own control.
Behavioral analyses.
Behavioral analyses were conducted on littermates at 3 or 6 months of age during the lights-on period (6:00 A.M. to 6:00 P.M.). All experiments were conducted blind to the genotype of the animals. Rotarod, open-field, and qualitative behavioral analyses were performed as done by Lucas et al. (2012) on the same group of animals. To detect subtle differences in motor coordination, a more difficult rotarod task was implemented on a separate set of conditional knock-out animals. The training protocol remained unchanged from that of Lucas et al. (2012). On test day, mice underwent four trials of accelerating 4–40 rotations per minute during, and latency to fall was recorded. Mice still remaining on the rotarod were removed after 5 min, and mice were allowed to rest at least 10 min between trials. To assess learning and memory, a separate group of animals underwent Barnes maze testing. The Barnes maze consisted of a 122-cm-diameter circular platform with forty 5 cm diameter holes spaced 9 cm apart. The platform was raised 140 cm above the floor. An escape box (22.9 × 5.3 × 8.6 cm) filled with clean bedding was placed beneath one of the holes of the platform. Animals were trained over a 5 d period, and the escape box position remained consistent for a given animal on all test days. Each day, mice were placed in the center of the Barnes maze, and distance traveled to the escape box was recorded with EthoVision 3.1 video tracking software (Noldus). If an animal did not find the escape box after 4 min, it was manually placed into the box. Differences between genotypes were assessed on the fifth day.
Data analyses.
All statistical analyses were conducted with SPSS software (IBM). For initial tests of microarray results in PGC-1α+/+ versus PGC-1α−/− mice, two-tailed t tests with Bonferroni's correction for multiple testing were used. Subsequent gene and protein expression analyses were analyzed with one-tailed t tests based on a priori hypotheses based on initial results in PGC-1α+/+ versus PGC-1α−/− mice. Developmental time course data were analyzed with one-way ANOVA followed by Tukey's HSD. For behavioral analyses, repeated-measures ANOVA (rotarod), χ2 test for independence (qualitative data), or two-tailed t tests (open field, Barnes maze) were implemented. Electrophysiology data were analyzed with Kolmogorov–Smirnov tests (cumulative probability) or one-tailed t tests (miniature and evoked IPSCs). All data were confirmed to have normal distributions before parametric tests were applied. Data are reported as mean ± SEM.
Results
Identification of neuronal PGC-1α-dependent genes
With the initial goal of evaluating the expression patterns of interneuron-specific markers in PGC-1α−/− mice, we found that PGC-1α is required for the expression of the Ca2+-binding protein PV in the cerebrum (Lucas et al., 2010). In light of PGC-1α's well established role as a transcriptional coactivator, we hypothesized that PGC-1α is also required for the expression of other interneuron-specific transcripts. We surmised that a true PGC-1α-dependent gene would be both upregulated by PGC-1α overexpression and reduced in PGC-1α−/− animals. First, to identify novel PGC-1α-responsive genes in an unbiased manner, we conducted an Illumina human gene expression microarray analysis in SH-SY5Y neuroblastoma cells transfected with GFP alone or with PGC-1α and GFP in tandem. Of the ∼27,000 tested transcripts, 1067 were increased at least twofold by expression of PGC-1α (Fig. 1A). Then, to identify transcripts with a possible role in PV-positive interneurons, we mined the array data for transcripts with statistically similar gene expression profiles as PGC-1α or PV based on Neuroblast analyses using the Allen Brain Atlas (see Materials and Methods). The final list comprised 32 putative targets (Fig. 1B).
Figure 1.

Identification of novel PGC-1α-dependent genes in neural tissue. A, Microarray heat map of neuroblastoma cells overexpressing PGC-1α and GFP in tandem or control samples overexpressing GFP alone. Red/green indicates an increase/decrease in gene expression of the ∼27,000 tested transcripts. n = 3 per group. B, Microarray data were mined for significantly increased genes with physiological relevance to interneurons based on their neuroanatomical overlap with (1) PGC-1α or (2) PV (indicated by superscript number behind gene name; see Materials and Methods). Selected transcripts were then measured by qRT-PCR in cortical homogenates from PGC-1α+/+ and PGC-1α−/− mice with the hypothesis that a true PGC-1α-dependent gene would be both increased by overexpression and decreased by deletion. Two-tailed t tests with Bonferroni's correction for multiple testing. The gray highlight indicates statistically significant decreases. n = 6–7 per group. *Values expressed as PGC-1α:GFP ratio; **values expressed as PGC-1α−/−/PGC-1α+/+ ratio after normalization to β-actin. Data are presented as mean ± SEM.
The expression of these 32 transcripts was measured with qRT-PCR in anterior cortex from PGC-1α+/+ and PGC-1α−/− littermates (Lin et al., 2004) at P30, an age that precedes the appearance of vacuoles in this region but postdates the developmental peak of PGC-1α expression at P14 (Cowell et al., 2007). Ten transcripts spanning synaptic (PV, Syt2, and Cplx1), structural (Nefh), and metabolism-related [neutral cholesterol ester hydrolase 1 (Nceh1), adenylate kinase 1 (Ak1), inositol polyphosphate 5-phosphatase J (Inpp5j), ATP synthase mitochondrial F1 complex O subunit (Atp5o), phytanol-CoA-2hydroxylase (Phyh), and ATP synthase mitrochondrial F1 complex α subunit 1 (Atp5a1)] functions were significantly reduced in PGC-1α−/− compared with PGC-1α+/+ cortex after Bonferroni's correction for multiple comparisons (Fig. 1B, 2A). These transcripts were subsequently measured in three other lines of mice: (1) a second line of germline PGC-1α−/− mice in which the biologically active portion of the N terminus of PGC-1α is present (Leone et al., 2005); (2) nervous system conditional knock-out mice (Lucas et al., 2012); and (3) mice with inducible overexpression of PGC-1α. All 10 transcripts were significantly reduced in the nervous system conditional PGC-1α knock-out mice (Fig. 2C) and significantly increased by PGC-1α overexpression (Fig. 2D). Interestingly, N-terminus expression of PGC-1α was sufficient for expression of metabolism-related transcripts except Phyh but not the neuron-specific transcripts Syt2, Cplx1, and Nefh (Fig. 2B).
Figure 2.

Regulation of novel transcripts by ablation and overexpression of PGC-1α in vivo. Expression of putative transcriptional targets of PGC-1α was analyzed by qRT-PCR in cortical homogenates of four mouse lines. A, Complete germline knock-out of PGC-1α significantly decreased expression of the neuron-specific genes PV, Syt2, Cplx1, and Nefh and the metabolism-related genes Phyh, Inpp5j, Nceh1, Ak1, Atp5o, and Atp5a1. B, Incomplete germline deletion in which the biologically active N terminus of PGC-1α is still present significantly decreased neuron-specific but not metabolism-related transcripts. C, Conditional deletion of PGC-1α in the CNS recapitulated the transcriptional changes in the complete germline knock-out. D, Acute overexpression of PGC-1α in the cortex significantly increased the expression of all novel PGC-1α-dependent genes. Numbers per group are indicated on the bar histograms. *p < 0.05; ** p < 0.005; ***p < 0.0005, two-tailed t tests followed by Bonferroni's correction for multiple testing (A) and one-tailed t tests based on a priori hypotheses based on A (B–D). Data are presented as mean ± SEM.
Three of the 10 novel PGC-1α-dependent genes (Syt2, Cplx1, and Nefh) have well defined roles in the nervous system. Syt2 and Cplx1 are components of the soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE) complex and are essential for fast, synchronous neurotransmitter release triggered by Ca2+ (Reim et al., 2001; Pang et al., 2006; Tang et al., 2006; Sun et al., 2007; Xu et al., 2007; Xue et al., 2007; Maximov et al., 2009; Yang et al., 2010). Nefh is a member of the intermediate filament family that forms part of the cytoskeleton scaffolding complex and plays a role in axonal structure as well as the speed of axonal neurotransmission (Zhu et al., 1998; Lariviere and Julien, 2004). We limited subsequent analyses to these three genes, as neuron-specific transcripts regulated by PGC-1α have not been well defined.
Induction of Syt2, Cplx1, and Nefh expression by PGC-1α overexpression was first confirmed by Western blot analysis of neuroblastoma cells transfected with PGC-1α (Fig. 3A). Then, we also confirmed that Syt2 (t(13) = 15.65, p = 2.15 × 10−6), Cplx1 (t(13) = 3.63, p = 0.002), and Nefh (t(13) = 4.06, p = 0.001) protein expression was significantly reduced in the cortex of PGC-1α−/− compared with PGC-1α+/+ mice (Fig. 3B). Together, these studies indicate that PGC-1α is both necessary and sufficient for expression of Syt2, Cplx1, and Nefh gene and protein expression in vitro and in vivo.
Figure 3.

Control of protein expression by PGC-1α. A, Western blot analysis shows that overexpression of PGC-1α in neuroblastoma cells increases the protein expression of PGC-1α, Syt2, Cplx1, and Nefh, which are negligibly expressed in GFP control cells. B, Western blot analysis demonstrates that protein expression of Syt2, Cplx1, and Nefh, but not the non-PGC-1α-dependent gene Cplx2, were significantly decreased in PGC-1α−/− compared with PGC-1α+/+ cortex. Numbers per group are indicated on the bar histogram. **p < 0.005; ***p < 0.0005, one-tailed t tests. Data are presented as mean ± SEM.
Spatial and temporal distribution of novel PGC-1α-dependent genes
Expression of PGC-1α follows a distinct developmental time course in the rodent cortex with a peak in gene expression at P14 and concentration of protein in nuclei of neurons that express high levels of GAD67 (Cowell et al., 2007). If PGC-1α is a key driver of Syt2, Cplx1, and Nefh, we hypothesized that these putative downstream targets would have a similar spatial and temporal distribution as PGC-1α. To test our hypothesis, we conducted a developmental time course of transcript and protein expression of novel putative targets in the cortex of wild-type mice at P7, P14, P21, P30, and P90. Using qRT-PCR, we found that Syt2, Cplx1, and Nefh exhibited a pronounced upregulation of gene expression between P7 and P14, at which point expression stabilized throughout the late postnatal period and into adulthood (Fig. 4A). Western blot analysis revealed that protein expression followed a similar pattern as that of the transcript, albeit with cortical expression of all three proteins significantly increasing throughout the early postnatal period compared with adult expression at P90 (Fig. 4B,C).
Figure 4.
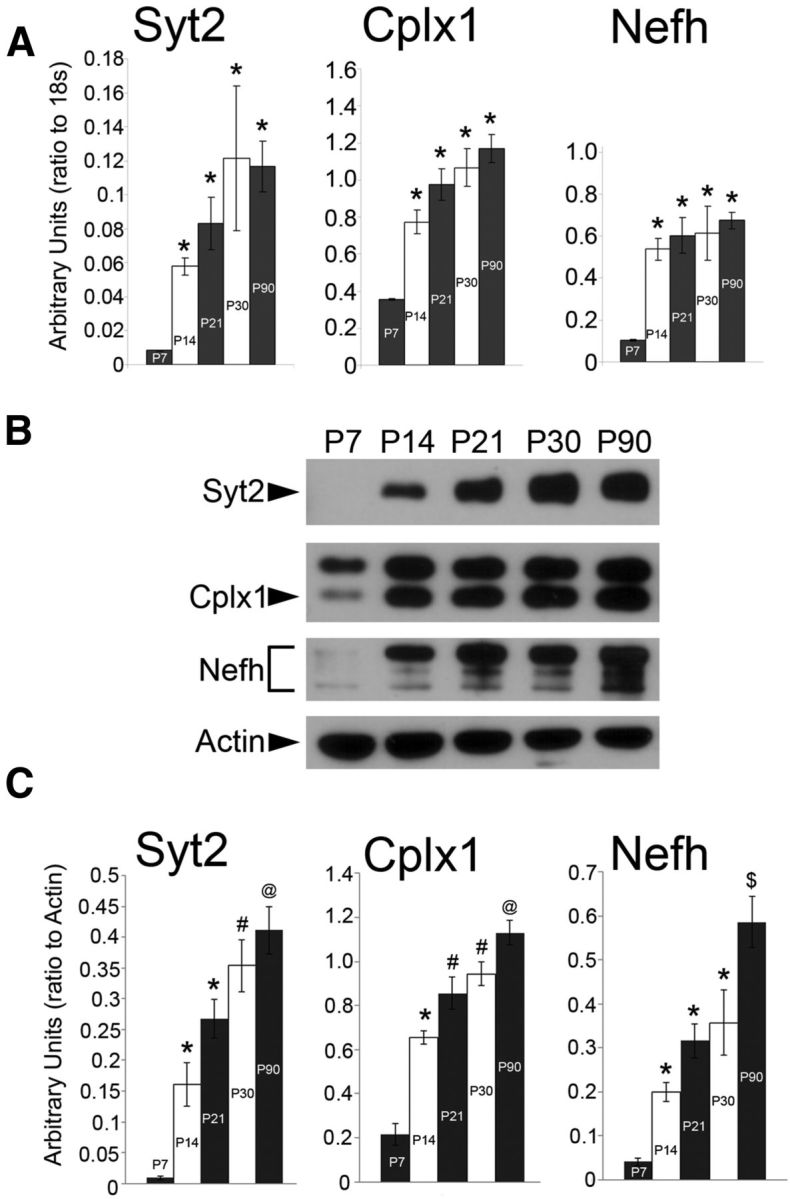
Neuron-specific PGC-1α-dependent genes have a similar temporal expression pattern as PGC-1α. A, qRT-PCR shows that cortical gene expression of Syt2, Cplx1, and Nefh is significantly upregulated between P7 and later ages. B, Representative Western blots of cortical Syt2, Cplx1, and Nefh protein expression in development. C, Protein expression tends to increase throughout the late postnatal period into adulthood (P90). n = 4 per group. *p < 0.05 versus P7; #p < 0.05 versus P7–P14; @p < 0.05 versus P7–P21; $p < 0.05 versus P7–P30, one-way ANOVA followed by Tukey's HSD. Data are presented as mean ± SEM.
As PGC-1α is primarily concentrated in inhibitory interneurons in the cortex, a series of immunofluorescence double-labeling experiments was conducted to determine whether Syt2, Cplx1, and Nefh are also localized to cortical GAD67- and/or PV-positive neurons in PGC-1α+/+ and PGC-1α−/− mice. Expression of Syt2 was predominantly localized to cortical layers V/VI; punctate in nature; and abundantly, but not exclusively, expressed in neuronal processes. Some Syt2 labeling consisted of clear somatic staining, and double-labeling with PV revealed that the majority of these somatically labeled cells (Fig. 5A, arrowheads) were PV-positive in PGC-1α+/+ mice. Immunoreactivity for Syt2 and PV was almost undetectable in the cortex of PGC-1α−/− mice; the remaining faintly labeled PV cells in layer V/VI did not express detectable levels of Syt2 (Fig. 5A). PV, rather than GAD67, was used for Syt2 double-labeling experiments because of primary antibody species constraints.
Figure 5.
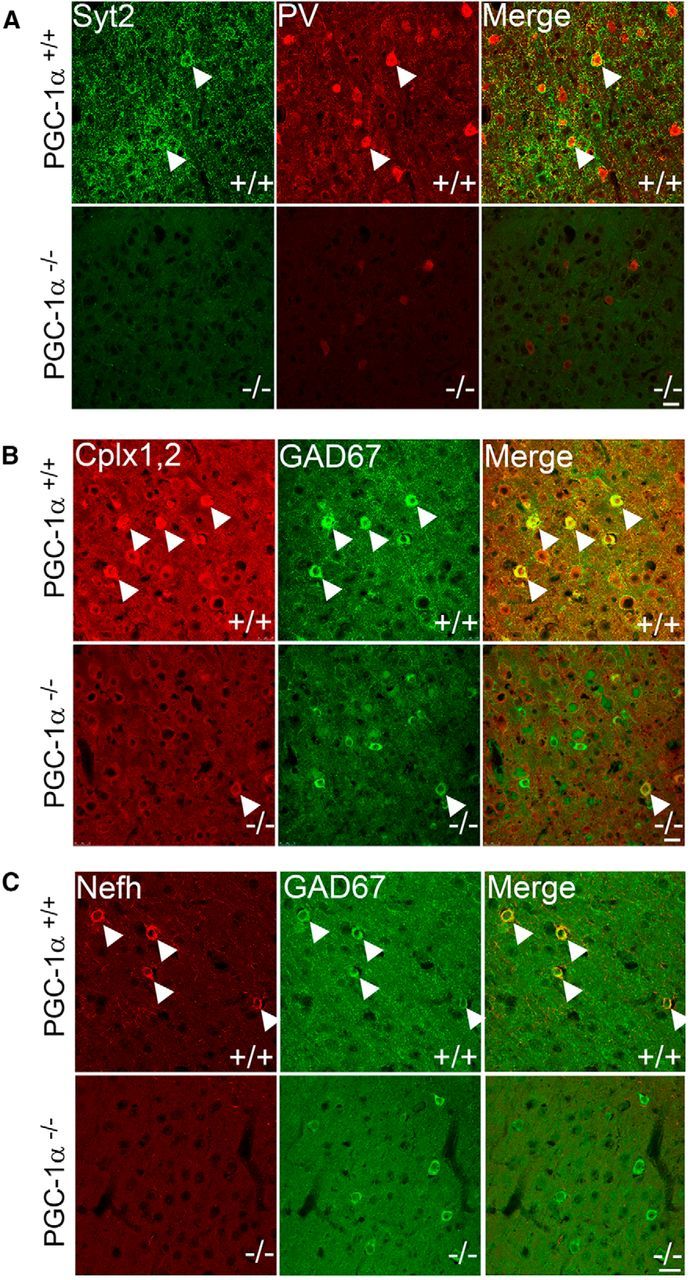
Reduction of cortical interneuron-specific expression of PGC-1α-dependent proteins in PGC-1α−/− mice at P30. Immunofluorescence colabeling was conducted to determine cellular localization of Syt2, Cplx1, and Nefh. A, Syt2 (green) exhibited strong cortical labeling in the soma (arrowheads) of neurons that also express PV (red) in PGC-1α+/+ mice. Expression of both Syt2 and PV were drastically reduced in PGC-1α−/− mice. B, Cplx1,2 (red) exhibited strong cortical labeling in the soma (arrowheads) of neurons that also express GAD67 (green) in PGC-1α+/+ mice. Expression of Cplx1,2 was drastically reduced but still colocalized to GAD67 in PGC-1α−/− mice. C, Nefh (red) expression was predominantly localized to neuronal processes. However, in superficial cortical layers, clear somatic staining was observed specifically in cells that also expressed GAD67 (green) in PGC-1α+/+ mice. No somatic staining of Nefh was observed in PGC-1α−/− mice. Confocal microscope settings were kept consistent between genotypes for a given protein. Scale bars, 25 μm (representative of all images). n = 5 per group.
Cplx expression was predominantly localized to cortical layers IV/V and exhibited high staining in neuronal cell bodies and processes. Double labeling with GAD67 in PGC-1α+/+ mice revealed that the majority of somatically labeled Cplx-positive cells also expressed GAD67 (Fig. 5B, arrowheads). Almost no somatic staining of Cplx was apparent in PGC-1α−/− mice in this region, but the few faintly labeled cells were still GAD67 positive (Fig. 5B, arrowheads). As the antibody for Cplx recognizes both Cplx1 and Cplx2, we were unable to conclude with certainty whether Cplx1, Cplx2, or both are localized to cortical interneurons. However, Western blot analysis with the same antibody did not reveal significant reductions in Cplx2 in cortical homogenates in PGC-1α−/− mice (Fig. 3B). Given that Cplx staining was greatly reduced in PGC-1α−/− animals and that Cplx1 has previously been reported to be the main isoform in layer IV/V by in situ hybridization (Freeman and Morton, 2004), it is likely that the observed colocalization of Cplx immunoreactivity with GAD67 is specific for Cplx1 in PGC-1α+/+ mice.
Nefh expression was almost exclusively expressed in neuronal processes. However, in cortical layer II/III, clear somatic staining was present. In PGC-1α+/+ mice, all somatically labeled cells were GAD67 positive (Fig. 5C). No somatically labeled Nefh-positive cells were present in PGC-1α−/− mice (Fig. 5C), but labeled neuronal processes were still present in deeper cortical layers (data not shown).
Deletion of PGC-1α in PV-positive interneurons results in decreased transcription of novel PGC-1α-dependent genes
Based on our previous observation for the requirement of PGC-1α for PV expression and PGC-1α's concentration in GABAergic neurons, we hypothesized that conditional deletion of PGC-1α specifically within PV-positive neurons would recapitulate the gene expression changes and behavioral phenotype of PGC-1α−/− mice. Conditional deletion was accomplished by crossing mice with loxP sites flanking critical exons of the PGC-1α gene (Lin et al., 2004) with mice expressing cre recombinase driven by the PV promoter (Hippenmeyer et al., 2005). Activity of cre recombinase was verified by crossing the PV-Cre mouse with a mutant tomato/mutant green reporter mouse strain (Muzumdar et al., 2007). Consistent with a prior study (Carlén et al., 2012), we determined that recombination occurs in most cortical PV-positive interneurons by 3 months of age (Fig. 6A). Recombination was confirmed in DNA isolated from whole brains of PGC-1αWT:PV-Cre, PGC-1αfl/+:PV-Cre, and PGC-1αfl/fl:PV-Cre mice (Fig. 6B). PGC-1αfl/fl:PV-Cre mice were born at the expected Mendelian ratio and developed normally. Unlike PGC-1α−/− or PGC-1αfl/fl:Nestin-Cre mice (Lucas et al., 2012), PGC-1αfl/fl:PV-Cre mice did not develop spongiform lesions in the striatum or cortex (Fig. 6C).
Figure 6.
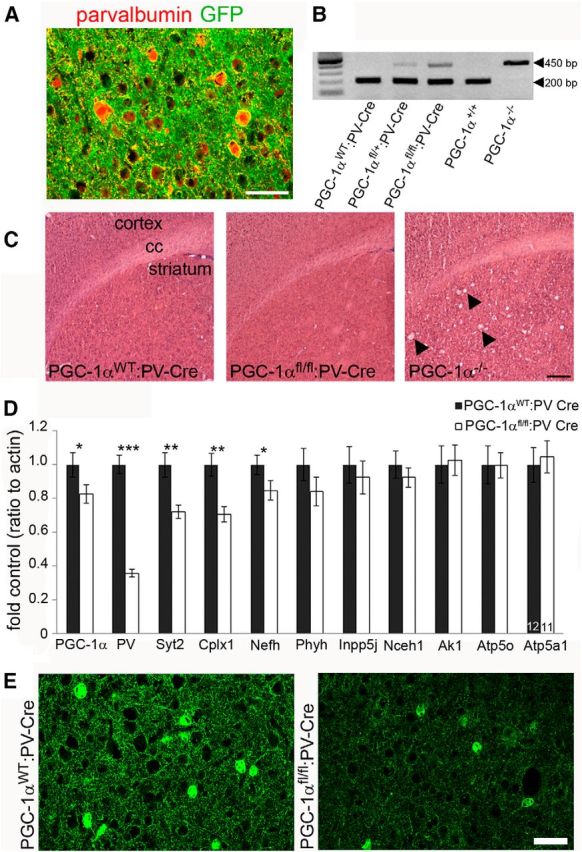
Conditional deletion of PGC-1α in PV-positive interneurons reduces cortical expression of neuron-specific PGC-1α-dependent genes. A, To determine specificity of cre recombinase, PV-Cre mice were crossbred to reporter mice that express GFP after recombination. Somatic GFP expression (green) was restricted to PV (red)-positive cell bodies. Scale bar, 25 μm. B, Efficacy of Cre-mediated recombination was confirmed with qualitative PCR on genomic DNA isolated from whole brains. PGC-1αfl/+:PV-Cre and PGC-1αfl/fl:PV-Cre but not PGC-1αWT:PV-Cre exhibit recombination of the PGC-1α gene (450 bp PGC-1α−/− band). C, Representative photos of hematoxylin and eosin staining demonstrating that PGC-1αfl/fl:PV-Cre mice (6 months) do not develop spongiform lesions in the striatum like PGC-1α−/− mice (P30; arrowheads). Scale bar, 2 mm. cc, Corpus collosum. n = 3 per group. D, qRT-PCR revealed that transcript expression of PGC-1α, PV, Syt2, Cplx1, and Nefh was significantly decreased in PGC-1αfl/fl:PV-Cre compared with PGC-1αWT:PV-Cre cortex. *p < 0.05; **p < 0.005; ***p < 0.0005, one-tailed t tests. E, Protein expression of PV was confirmed to be decreased by immunofluorescence staining (n = 3–4 per group). Scale bar, 50 μm. Numbers per group are indicated on the bar histogram. Data are presented as mean ± SEM.
To determine whether deletion of PGC-1α specifically in PV-positive neurons was sufficient to produce transcriptional changes, we used qRT-PCR to analyze gene expression of PGC-1α-dependent genes in cortical homogenates. Expression of PGC-1α (t(23) = 1.94, p = 0.03) and its neuron-specific dependent transcripts PV (t(21) = 10.98, p = 2.0 × 10−9), Syt2 (t(23) = 3.41, p = 0.001), Cplx1 (t(23) = 3.74, p = 0.0005), and Nefh (t(23) = 1.94 p = 0.03) was significantly decreased in PGC-1αfl/fl:PV-Cre compared with PGC-1αWT:PV-Cre littermates (Fig. 6D). Expression of the metabolism-related transcripts Nceh1, Ak1, Inpp5j, Atp5o, Phyh, and Atp5a1, although significantly reduced in PGC-1αfl/fl:Nestin-Cre cortex (Fig. 2C), was unaltered in PGC-1αfl/fl:PV-Cre mice. Reductions in PV expression were confirmed at the protein level using immunofluorescence. PV-positive cell bodies were still present in PGC-1αfl/fl:PV-Cre, yet the intensity of the staining in processes was markedly reduced (Fig. 6E), similar to the germline PGC-1α−/− mouse (Fig. 5A). Of note, expression of Syt2, Cplx1, and Nefh was not reduced to the same extent as in germline and nervous system conditional PGC-1α−/− mice, indicating that PGC-1α-mediated regulation of these transcripts is not exclusive to PV-positive interneurons.
Conditional PGC-1α deletion in PV-positive interneurons produces cortical GABAergic asynchrony
As our prior electrophysiological results in germline PGC-1α−/− mice (Lucas et al., 2010; Dougherty et al., 2014b) may have been affected by alterations in pyramidal cell dendritic spine formation and/or maintenance (Cheng et al., 2012), we sought to determine the physiological impact of PGC-1α deletion specifically in aspiny cortical PV-positive interneurons. We evaluated basal inhibitory synaptic transmission and evoked IPSCs in layer V pyramidal neurons from PGC-1αfl/fl:PV-Cre mice. Given the well established requirement of Syt2 and Cplx1 for synchronous release of neurotransmitter from the presynaptic terminal (Reim et al., 2001; Pang et al., 2006; Tang et al., 2006; Sun et al., 2007; Xu et al., 2007; Maximov et al., 2009; Yang et al., 2010), we hypothesized that the synaptic phenotype of PGC-1αfl/fl:PV-Cre mice would include asynchronous GABA release onto cortical pyramidal cells. Whole-cell voltage-clamp recordings were obtained in anterior cortex from mice 3–5 months of age. We found no change in the frequency or amplitude of spontaneous IPSCs (data not shown). However, consistent with PGC-1α-induced presynaptic loss of Syt2 and/or Cplx1, we observed an increase in frequency (t(15) = 2.02, p = 0.04) of miniature IPSCs without a change in amplitude (Fig. 7A,B), demonstrating an increase in vesicular GABA release in PGC-1αfl/fl:PV-Cre cortex. Typical examples of miniature IPSCs recorded in the presence of TTX are shown in Figure 7C.
Figure 7.

Ablation of PGC-1α in PV-positive interneurons increases asynchronous GABA release in the cortex. Recordings of IPSCs in cortical layer V pyramidal cells were conducted to determine the physiological consequences of PGC-1α deletion. A, The frequency of miniature IPSCs was significantly increased in PGC-1αfl/fl:PV-Cre compared with PGC-1αWT:PV-Cre cortex. B, Miniature IPSC amplitude was not affected. C, Representative traces of spontaneous miniature IPSCs. D, Representative traces showing evoked IPSCs from PGC-1αWT:PV-Cre and PGC-1αfl/fl:PV-Cre mice recorded in normal concentrations of extracellular calcium (left) and after replacing calcium with equimolar concentrations of strontium (right). Traces represent 10 stimulus sweeps from the same cell. Late asynchronous events after strontium application (arrowheads; left) were present in PGC-1αfl/fl:PV-Cre neurons under normal physiological conditions (arrowheads; right). E, Evoked IPSC amplitudes were significantly decreased in PGC-1αfl/fl:PV-Cre compared with PGC-1αWT:PV-Cre mice. F, No changes were observed in charge transfer (evoked IPSC area under the curve), a measure of total GABA release. Numbers per group are indicated on the bar histograms. *p < 0.05; **p < 0.005, one-tailed t tests. Data are presented as mean ± SEM.
To assess alterations in synaptic synchrony that could occur with the combined loss of PV, Syt2, and Cplx1 expression in PV-positive interneurons, we replaced calcium in the extracellular recording medium with strontium during the measurement of eIPSCs. Bath application of strontium results in asynchronous release as a result of changes in molecular buffering and produces a reduction in eIPSC amplitude and an increase in delayed synaptic events (Behrends and ten Bruggencate, 1998; Rumpel and Behrends, 1999). Representative traces of eIPSCs in the presence of calcium and strontium are shown in Figure 7D; eIPSC amplitudes were reduced after replacement of calcium with strontium. The decrease in eIPSC amplitude was markedly greater in PGC-1αfl/fl:PV-Cre compared with PGC-1αWT:PV-Cre pyramidal cells (Fig. 7D). In both PGC-1αWT:PV-Cre and PGC-1αfl/fl:PV-Cre mice, there was an increase in late IPSC frequency after stimulation in the presence of strontium (Fig. 7D, right, arrowheads). Similar late IPSC events were observed in PGC-1αfl/fl:PV-Cre slices before strontium replacement of calcium (Fig. 7D, left, arrowheads), indicating that asynchronous neurotransmitter release occurs in these animals under normal physiological conditions. Replacement of calcium with strontium severely attenuated the amplitude of evoked responses in PGC-1αfl/fl:PV-Cre cells (t(18) = 4.89, p = 0.0008; Fig. 7E). However, asynchronous events were still observed. Importantly, these alterations in peak amplitude were not caused by a change in total GABA release (no change in charge transfer; Fig. 7F). Together, these data suggest that the reduction in amplitude of eIPSCs represents increased sensitivity to strontium-mediated asynchrony, supporting the hypothesis that a functional consequence of PGC-1α ablation from PV-positive interneurons includes abnormalities in synchronous neurotransmitter release.
Impaired long-term memory in mice lacking PGC-1α in PV-positive neurons
Given the proposed role of PV-positive interneurons in memory formation (Donato et al., 2013), we hypothesized that deficits in long-term memory formation would be impaired in mice lacking PGC-1α in PV-positive neurons. As germline PGC-1α−/− mice exhibit severe motor impairments that preclude their testing in paradigms of cognitive functioning, we first wanted to determine whether motor functioning was compromised in PGC-1αfl/fl:PV-Cre mice. Using the same behavioral protocols as Lucas et al. (2012), we found that deletion of PGC-1α in PV-positive cells was not sufficient to elicit the behavioral deficits present in PGC-1α−/− mice, such as hindlimb clasping, tremor, or rotarod impairment, at 3 months of age (Fig. 8A,B). As our previously published rotarod paradigm may not be sufficient to detect subtle deficits in motor coordination, we implemented a more difficult rotarod task in which mice run on the rotarod at an accelerating speed of 4–40 rotations per minute for 5 min. PGC-1αfl/fl:PV-Cre mice were able to perform this paradigm as well as their PGC-1αWT:PV-Cre littermates (Fig. 8C), demonstrating that ablation of PGC-1α in PV-positive cells does not result in motor deficits.
Figure 8.

Impaired long-term memory but not motor function in mice lacking PGC-1α in PV-positive neurons. A, Unlike PGC-1α−/− mice, PGC-1αfl/fl:PV-Cre mice do not have decreased latencies to fall on a rotarod task in which mice were removed after 60 s. B, Unlike PGC-1α−/− mice, PGC-1αfl/fl:PV-Cre do not exhibit increased instance of tremor or hindlimb clasping. C, PGC-1αfl/fl:PV-Cre mice do not perform differently from PGC-1αWT:PV-Cre littermates on a rotarod task in which mice were removed after 5 min. D, E, PGC-1αfl/fl:PV-Cre do not display differences in locomotor activity (D) or thigmotaxis (E) compared with PGC-1αWT:PV-Cre mice in a novel open field. F, PGC-1αfl/fl:PV-Cre mice traveled significantly farther to reach the escape box on the fifth day of the Barnes maze compared with their PGC-1αWT:PV-Cre littermates. Data from PGC-1α−/− mice are reproduced with permission from PLoS One (Lucas et al., 2012). Numbers per group are indicated on the bar histograms. *p < 0.05 for PGC-1α−/− versus PGC-1αWT:PV-Cre; #p < 0.05 for PGC-1α−/− versus PGC-1αfl/fl:PV-Cre; $p < 0.05 for PGC-1αWT:PV-Cre versus PGC-1αfl/fl:PV-Cre, repeated-measures ANOVA followed by planned comparisons with Holm-Bonferroni test (A), χ2 test for independence (B), and two-tailed t tests (D–F). Data are presented as mean ± SEM.
We next trained male PGC-1αWT:PV-Cre and PGC-1αfl/fl:PV-Cre mice on the Barnes maze task in which mice are trained to use spatial cues to find an escape box for 4 consecutive days. On test day (day 5), PGC-1αfl/fl:PV-Cre mice traveled significantly further to reach the escape box than their PGC-1αWT:PV-Cre littermates (t(15) = 2.587, p = 0.03; Fig. 8F). This deficit in Barnes maze performance was not attributable to differences in overall locomotor activity, as distance traveled and anxiety-like behavior in an open field did not differ between PGC-1αfl/fl:PV-Cre and PGC-1αWT:PV-Cre mice (Fig. 8D,E). These data suggest that impaired memory formation is a primary behavioral consequence of decreased PGC-1α expression in PV-positive cells.
Discussion
While it has been primarily assumed that the transcriptional coactivator PGC-1α is a “master regulator” of metabolism in central and peripheral tissues, we show here that, in addition to metabolism-related transcripts, PGC-1α also controls a distinct transcriptional program designed to mediate synchronous release of neurotransmitter in the brain. Previous work has demonstrated that PGC-1α localizes to the nuclei of GABAergic neurons (Cowell et al., 2007), and our laboratory has recently discovered that PGC-1α is required for the expression of the Ca2+-binding protein PV within GABAergic interneurons in the cerebrum (Lucas et al., 2010). To determine other putative transcriptional targets of PGC-1α in this neuronal population, we conducted a microarray in neuroblastoma cells overexpressing PGC-1α and mined microarray results for genes to test in PGC-1α−/− mice based on their potential relevance to interneuron physiology using unbiased criteria (see Materials and Methods). We found significant reductions in cortical transcript expression of neuron-specific transcripts Syt2, Cplx1, and Nefh in PGC-1α−/− mice. We then confirmed that the expression of Syt2, Cplx1, and Nefh, like that of PGC-1α, is postnatally upregulated and localized to cortical interneurons within the first month of life. In support of the cellular specificity of our immunofluorescence studies, conditional deletion of PGC-1α within PV-positive neurons decreased transcript expression of PGC-1α, PV, Syt2, Cplx1, and Nefh and increased asynchronous GABA release onto pyramidal cells in the cortex. Based on the defined roles for these genes in the regulation of neurotransmitter release and neuronal function, we propose that PGC-1α regulates a developmental program for the synchronous release of neurotransmitter from neurons, including PV-positive interneurons, and that disruptions of PGC-1α expression in disease states could contribute to abnormalities in neuronal function in cortex.
PGC-1α mediates synchronous neurotransmitter release by coordinated regulation of PV, Syt2, and Cplx1
In our prior investigation, we found that most physiological abnormalities of inhibition in the hippocampus of PGC-1α−/− mice resembled abnormalities reported for PV knock-out mice. However, inhibitory PGC-1α−/− synapses were also characterized by delayed and asynchronous neurotransmitter release (Lucas et al., 2010). Accumulation of residual Ca2+ in the presynaptic terminal during high-frequency action potential trains is thought to mediate asynchronous release (Atluri and Regehr, 1998; Lu and Trussell, 2000; Hagler and Goda, 2001). In this way, the Ca2+-buffering properties of PV itself are predicted to provide a means to limit Ca2+ transient buildup, and thus asynchronous release, after an action potential train (Collin et al., 2005). However, the effects of endogenous levels of PV on asynchronous release pale in comparison with the application of exogenous buffers such as EGTA (Manseau et al., 2010), so the finding that PGC-1α−/− synapses also have reduced expression of Syt2 and Cplx1, Ca2+-sensitive SNARE proteins required for fast, synchronous release of neurotransmitter from the presynaptic terminal, further explains their electrophysiological phenotype.
Our findings that PGC-1α provides a transcriptional mechanism to mediate synchronous release in PV-positive interneurons by coordinately regulating PV, Syt2, and Cplx1 is of extreme importance considering the substantial evidence that properly functioning PV-positive interneurons play a critical role in the generation and maintenance of gamma oscillations in the cortex and hippocampus (Tamás et al., 2000; Fuchs et al., 2007; Cardin et al., 2009; Sohal et al., 2009; Korotkova et al., 2010; Carlén et al., 2012). Although beyond the scope of this study, it is appealing to speculate that disruption of synchronous release of neurotransmitter by PV-positive interneurons caused by a loss of PGC-1α would lead to abnormalities in gamma oscillations.
In addition to PV, Syt2, Cplx1, and Nefh, it is possible that PGC-1α controls the expression of metabolic transcripts in PV-positive interneurons. However, the novel metabolism-related PGC-1α-dependent genes Nceh1, Ak1, Inpp5j, Atp5o, Phyh, and Atp5a1, while reduced in the cortex from PGC-1α−/− mice and increased by acute cortical overexpression of PGC-1α, were unchanged in PGC-1αfl/fl:PV-Cre cortex, suggesting that these transcripts may not be concentrated in PV-positive interneurons. Our electrophysiology results indicate that, if present, any metabolic deficiencies caused by deletion of PGC-1α from PV-positive interneurons is not sufficient to impair GABA release meaningfully. Mitochondrial inhibition has been shown to greatly reduce the firing rate of fast-spiking basket cells and decrease the frequency of inhibitory postsynaptic potentials in the hippocampus (Whittaker et al., 2011), effects we have observed in PGC-1α−/− cortex (Dougherty et al., 2014b). However, in PGC-1αfl/fl:PV-Cre cortex, we observed an increase rather than a decrease in inhibitory events, which is not consistent with mitochondrial defects and alludes to a specific alteration in vesicle fusion events.
Developmental regulation of novel PGC-1α-dependent genes
Expression of PGC-1α in cerebral regions increases significantly between P7 and P14, a time period marked by an overall increase in the number of synapses and synaptic vesicles in the cortex (Jones and Cullen, 1979; Blue and Parnavelas, 1983). Accordingly, we and others have found that transcript and protein expression of Syt2, Cplx1, and Nefh substantially increase during this period (Shaw and Weber, 1982; Alcántara et al., 1993; de Lecea et al., 1995; Berton et al., 1997), indicating that PGC-1α may be required for the developmental induction of these genes. In light of the knock-out and overexpression data suggesting that PGC-1α may be both necessary and sufficient for the expression of these genes, we hypothesize that PV, Syt2, Cplx1, and Nefh are direct targets of PGC-1α. Chromatin immunoprecipitation experiments are required to identify the specific promoter regions occupied by PGC-1α-containing complexes to test this hypothesis.
Disease relevance of novel PGC-1α-dependent genes
PGC-1α−/− mice were originally reported to develop severe neurodegeneration with behavioral abnormalities indicative of neurological disorders such as hyperactivity and hindlimb clasping (Lin et al., 2004; Lucas et al., 2012). Remarkably, 4 of the 10 novel PGC-1α-dependent genes have been implicated in motor function. Mice lacking Syt2, Cplx1, or Phyh recapitulate one or more of the behavioral manifestations of germline PGC-1α−/− mice (Glynn et al., 2005; Pang et al., 2006; Wierzbicki, 2007), and Nefh has long been used as a biomarker for neurodegenerative disorders, particularly those with a loss of motor function component (Constantinescu et al., 2009). Given the proposed involvement of PGC-1α in neurological disorders, the finding that PGC-1α may regulate four genes independently involved in motor functioning could increase its potential therapeutic value.
Our conditional knock-out studies give further insight to the brain regions where PGC-1α is required for proper motor functioning. Global CNS deletion of PGC-1α mediated by Nestin-Cre was able to completely recapitulate the neurodegeneration and motor phenotype of PGC-1α−/− mice, including impaired rotarod performance and increased instance of himdlimb clasping and tremor (Lucas et al., 2012). We hypothesized that if PGC-1α expression is truly restricted to PV-expressing neurons, then loss of PGC-1α solely within this neuronal population would recapitulate the phenotype of the whole-body and CNS-specific knock-out. However, PV-Cre-mediated deletion of PGC-1α did not cause motor coordination deficits. This suggests that PGC-1α expression in cells that normally express PV, such as the globus pallidus, reticular nucleus of the thalamus, substantia nigra pars reticulata, Purkinje cells of the cerebellum, and PV-positive interneurons throughout the CNS, is not required for normal motor function.
A large amount of evidence suggests the involvement of PGC-1α in the pathophysiology of HD, so it is interesting that several recent studies have found abnormalities in PV expression and PV-positive interneuron function in mouse models of HD. Decreased expression of PV has been reported in the cortex and striatum of mouse models of the disease (Fusco et al., 1999; Luthi-Carter et al., 2002; Giampàet al., 2009), and recent evidence from our laboratory indicates that selective expression of mutant huntingtin within this neuronal population is sufficient to elicit the motor hyperactivity characterized by HD (Dougherty et al., 2014a). Whereas deletion of PGC-1α in PV-positive cells did not cause hyperactivity, it is still possible that dysregulation of PGC-1α-dependent transcripts contributes to interneuron dysfunction in this disorder.
A large amount of evidence also exists suggesting the involvement of PV interneurons in the pathology of schizophrenia (Lewis et al., 2005). In fact, downregulation of PGC-1α in cortical PV-positive interneurons was recently observed in a mouse model of schizophrenia (Jiang et al., 2013). As several genetic association studies have linked the chromosomal location of the PGC-1α gene (4p15.1) with the occurrence of schizophrenia and bipolar disorder (Blackwood et al., 1996; Christoforou et al., 2007, 2011; Le Hellard et al., 2007; Dempster et al., 2011), it is attractive to speculate that either deficiencies in PGC-1α and/or alterations in its activity contributes to interneuron dysfunction in this disorder, considering the well established role of properly functioning PV-positive interneurons in cognitive processes such as long-term memory, which is impaired in PGC-1αfl/fl:PV-Cre mice. Future studies investigating the mechanisms by which PGC-1α directs cell-specific programs of gene regulation will enable targeted manipulation of gene programs for maintenance of neurotransmitter release in multiple pathological settings.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants K01MH077955-05 (R.M.C.) and R01NS070009-05 (R.M.C.) and by the Civitan Emerging Scholar (E.K.L.) and Civitan McNulty Investigator (R.M.C.) awards. This work was made possible by the NIH Microarray Consortium and the W.M. Keck Biotechnology Resource Laboratory at Yale University for microarray processing and data analysis, the University of Florida Vector Core, the Evelyn F. McKnight UAB Neurobiology Behavior Core, the UAB Neuroscience Behavioral Assessment Core (NIH Grant P30NS047466), and the Zebrafish International Resource Center for generation of the synaptotagmin 2 antibody (NIH–NCRR Grant RR12546). Microarray data are in the process of being deposited into GEO NCBI database. We thank Scott Phillips at the UAB Intellectual and Disabilities Research Center (NIH Grant P30HD038985) for AAV design, Grace Nix for mouse immunofluorescence staining, and David Standaert for access to his confocal microscope.
The authors declare no competing financial interests.
References
- Alcántara S, Ferrer I, Soriano E. Postnatal development of parvalbumin and calbindin D28K immunoreactivities in the cerebral cortex of the rat. Anat Embryol. 1993;188:63–73. doi: 10.1007/BF00191452. [DOI] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG. Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci. 1998;18:8214–8227. doi: 10.1523/JNEUROSCI.18-20-08214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends JC, ten Bruggencate G. Changes in quantal size distributions upon experimental variations in the probability of release at striatal inhibitory synapses. J Neurophysiol. 1998;79:2999–3011. doi: 10.1152/jn.1998.79.6.2999. [DOI] [PubMed] [Google Scholar]
- Berton F, Iborra C, Boudier JA, Seagar MJ, Marquèze B. Developmental regulation of synaptotagmin I, II, III, and IV mRNAs in the rat CNS. J Neurosci. 1997;17:1206–1216. doi: 10.1523/JNEUROSCI.17-04-01206.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood DH, He L, Morris SW, McLean A, Whitton C, Thomson M, Walker MT, Woodburn K, Sharp CM, Wright AF, Shibasaki Y, St Clair DM, Porteous DJ, Muir WJ. A locus for bipolar affective disorder on chromosome 4p. Nat Genet. 1996;12:427–430. doi: 10.1038/ng0496-427. [DOI] [PubMed] [Google Scholar]
- Blue ME, Parnavelas JG. The formation and maturation of synapses in the visual cortex of the rat. II. Quantitative analysis. J Neurocytol. 1983;12:697–712. doi: 10.1007/BF01181531. [DOI] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlén M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, Rühlmann C, Jones SR, Deisseroth K, Sheng M, Moore CI, Tsai LH. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2012;17:537–548. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RK, Adhihetty P, Shukla S, Hennessy T, Calingasan N, Yang L, Starkov A, Kiaei M, Cannella M, Sassone J, Ciammola A, Squitieri F, Beal MF. Impaired PGC-1alpha function in muscle in Huntington's disease. Hum Mol Genet. 2009;18:3048–3065. doi: 10.1093/hmg/ddp243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Wan R, Yang JL, Kamimura N, Son TG, Ouyang X, Luo Y, Okun E, Mattson MP. Involvement of PGC-1alpha in the formation and maintenance of neuronal dendritic spines. Nat Commun. 2012;3:1250. doi: 10.1038/ncomms2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforou A, Le Hellard S, Thomson PA, Morris SW, Tenesa A, Pickard BS, Wray NR, Muir WJ, Blackwood DH, Porteous DJ, Evans KL. Association analysis of the chromosome 4p15–p16 candidate region for bipolar disorder and schizophrenia. Mol Psychiatry. 2007;12:1011–1025. doi: 10.1038/sj.mp.4002003. [DOI] [PubMed] [Google Scholar]
- Christoforou A, McGhee KA, Morris SW, Thomson PA, Anderson S, McLean A, Torrance HS, Le Hellard S, Pickard BS, StClair D, Muir WJ, Blackwood DH, Porteous DJ, Evans KL. Convergence of linkage, association and GWAS findings for a candidate region for bipolar disorder and schizophrenia on chromosome 4p. Mol Psychiatry. 2011;16:240–242. doi: 10.1038/mp.2010.25. [DOI] [PubMed] [Google Scholar]
- Clark J, Reddy S, Zheng K, Betensky RA, Simon DK. Association of PGC-1alpha polymorphisms with age of onset and risk of Parkinson's disease. BMC Med Gen. 2011;12:69. doi: 10.1186/1471-2350-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin T, Chat M, Lucas MG, Moreno H, Racay P, Schwaller B, Marty A, Llano I. Developmental changes in parvalbumin regulate presynaptic Ca2+ signaling. J Neurosci. 2005;25:96–107. doi: 10.1523/JNEUROSCI.3748-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu R, Zetterberg H, Holmberg B, Rosengren L. Levels of brain related proteins in cerebrospinal fluid: an aid in the differential diagnosis of parkinsonian disorders. Parkinsonism Relat Disord. 2009;15:205–212. doi: 10.1016/j.parkreldis.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Cowell RM, Blake KR, Russell JW. Localization of the transcriptional coactivator PGC-1alpha to GABAergic neurons during maturation of the rat brain. J Comp Neurol. 2007;502:1–18. doi: 10.1002/cne.21211. [DOI] [PubMed] [Google Scholar]
- Cowell RM, Talati P, Blake KR, Meador-Woodruff JH, Russell JW. Identification of novel targets for PGC-1alpha and histone deacetylase inhibitors in neuroblastoma cells. Biochem Biophys Res Commun. 2009;379:578–582. doi: 10.1016/j.bbrc.2008.12.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- de Lecea L, del RíoJ A, Soriano E. Developmental expression of parvalbumin mRNA in the cerebral cortex and hippocampus of the rat. Brain Res Mol Brain Res. 1995;32:1–13. doi: 10.1016/0169-328X(95)00056-X. [DOI] [PubMed] [Google Scholar]
- Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, Kalidindi S, Picchioni M, Kravariti E, Toulopoulou T, Murray RM, Mill J. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–4796. doi: 10.1093/hmg/ddr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato F, Rompani SB, Caroni P. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature. 2013;504:272–276. doi: 10.1038/nature12866. [DOI] [PubMed] [Google Scholar]
- Dougherty SE, Hollimon JJ, McMeekin LJ, Bohannon AS, West AB, Lesort M, Hablitz JJ, Cowell RM. Hyperactivity and cortical disinhibition in mice with restricted expression of mutant huntingtin to parvalbumin-positive cells. Neurobiol Dis. 2014a;62:160–171. doi: 10.1016/j.nbd.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty SE, Bartley AF, Lucas EK, Hablitz JJ, Dobrunz LE, Cowell RM. Mice lacking the transcriptional coactivator PGC-1alpha exhibit alterations in inhibitory synaptic transmission in the motor cortex. Neuroscience. 2014b;271:137–148. doi: 10.1016/j.neuroscience.2014.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MA, Sanes JR. Synaptotagmin I and II are present in distinct subsets of central synapses. J Comp Neurol. 2007;503:280–296. doi: 10.1002/cne.21381. [DOI] [PubMed] [Google Scholar]
- Freeman W, Morton AJ. Differential messenger RNA expression of complexins in mouse brain. Brain Res Bull. 2004;63:33–44. doi: 10.1016/j.brainresbull.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, Rozov A, Whittington MA, Traub RD, Rawlins JN, Monyer H. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 2007;53:591–604. doi: 10.1016/j.neuron.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Fusco FR, Chen Q, Lamoreaux WJ, Figueredo-Cardenas G, Jiao Y, Coffman JA, Surmeier DJ, Honig MG, Carlock LR, Reiner A. Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington's disease. J Neurosci. 1999;19:1189–1202. doi: 10.1523/JNEUROSCI.19-04-01189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GiampàGiampa C, Middei S, Patassini S, Borreca A, Marullo F, Laurenti D, Bernardi G, Ammassari-Teule M, Fusco FR. Phosphodiesterase type IV inhibition prevents sequestration of CREB binding protein, protects striatal parvalbumin interneurons and rescues motor deficits in the R6/2 mouse model of Huntington's disease. Eur J Neurosci. 2009;29:902–910. doi: 10.1111/j.1460-9568.2009.06649.x. [DOI] [PubMed] [Google Scholar]
- Glynn D, Drew CJ, Reim K, Brose N, Morton AJ. Profound ataxia in complexin I knockout mice masks a complex phenotype that includes exploratory and habituation deficits. Hum Mol Genet. 2005;14:2369–2385. doi: 10.1093/hmg/ddi239. [DOI] [PubMed] [Google Scholar]
- Hagler DJ, Jr, Goda Y. Properties of synchronous and asynchronous release during pulse train depression in cultured hippocampal neurons. J Neurophysiol. 2001;85:2324–2334. doi: 10.1152/jn.2001.85.6.2324. [DOI] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Rompala GR, Zhang S, Cowell RM, Nakazawa K. Social isolation exacerbates schizophrenia-like phenotypes via oxidative stress in cortical interneurons. Biol Psychiatry. 2013;73:1024–1034. doi: 10.1016/j.biopsych.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DG, Cullen AM. A quantitative investigation of some presynaptic terminal parameters during synaptogenesis. Exp Neurol. 1979;64:245–259. doi: 10.1016/0014-4886(79)90266-8. [DOI] [PubMed] [Google Scholar]
- Kim J, Moody JP, Edgerly CK, Bordiuk OL, Cormier K, Smith K, Beal MF, Ferrante RJ. Mitochondrial loss, dysfunction and altered dynamics in Huntington's disease. Hum Mol Genet. 2010;19:3919–3935. doi: 10.1093/hmg/ddq306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68:557–569. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- Lariviere RC, Julien JP. Functions of intermediate filaments in neuronal development and disease. J Neurobiol. 2004;58:131–148. doi: 10.1002/neu.10270. [DOI] [PubMed] [Google Scholar]
- Le Hellard S, Lee AJ, Underwood S, Thomson PA, Morris SW, Torrance HS, Anderson SM, Adams RR, Navarro P, Christoforou A, Houlihan LM, Detera-Wadleigh S, Owen MJ, Asherson P, Muir WJ, Blackwood DH, Wray NR, Porteous DJ, Evans KL. Haplotype analysis and a novel allele-sharing method refines a chromosome 4p locus linked to bipolar affective disorder. Biol Psychiatry. 2007;61:797–805. doi: 10.1016/j.biopsych.2006.06.029. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jäger S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Lin JD. Minireview: the PGC-1 coactivator networks: chromatin-remodeling and mitochondrial energy metabolism. Mol Endocrinol. 2009;23:2–10. doi: 10.1210/me.2008-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Trussell LO. Inhibitory transmission mediated by asynchronous transmitter release. Neuron. 2000;26:683–694. doi: 10.1016/S0896-6273(00)81204-0. [DOI] [PubMed] [Google Scholar]
- Lucas EK, Markwardt SJ, Gupta S, Meador-Woodruff JH, Lin JD, Overstreet-Wadiche L, Cowell RM. Parvalbumin deficiency and GABAergic dysfunction in mice lacking PGC-1alpha. J Neurosci. 2010;30:7227–7235. doi: 10.1523/JNEUROSCI.0698-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Dougherty SE, McMeekin LJ, Trinh AT, Reid CS, Cowell RM. Developmental alterations in motor coordination and medium spiny neuron markers in mice lacking pgc-1alpha. PloS One. 2012;7:e42878. doi: 10.1371/journal.pone.0042878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002;11:1911–1926. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- Manseau F, Marinelli S, Mendez P, Schwaller B, Prince DA, Huguenard JR, Bacci A. Desynchronization of neocortical networks by asynchronous release of GABA at autaptic and synaptic contacts from fast-spiking interneurons. PLoS Biol. 2010;8:e1000492. doi: 10.1371/journal.pbio.1000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Südhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsalve M, Wu Z, Adelmant G, Puigserver P, Fan M, Spiegelman BM. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol Cell. 2000;6:307–316. doi: 10.1016/S1097-2765(00)00031-9. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Pabst S, Hazzard JW, Antonin W, Südhof TC, Jahn R, Rizo J, Fasshauer D. Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J Biol Chem. 2000;275:19808–19818. doi: 10.1074/jbc.M002571200. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Melicoff E, Padgett D, Liu Y, Teich AF, Dickey BF, Lin W, Adachi R, Südhof TC. Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci. 2006;26:13493–13504. doi: 10.1523/JNEUROSCI.3519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, Pasinetti GM. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 2009;66:352–361. doi: 10.1001/archneurol.2008.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Südhof TC, Brose N, Rosenmund C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/S0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- Róna-Vörös K, Weydt P. The role of PGC-1alpha in the pathogenesis of neurodegenerative disorders. Curr Drug Targets. 2010;11:1262–1269. doi: 10.2174/1389450111007011262. [DOI] [PubMed] [Google Scholar]
- Rumpel E, Behrends JC. Sr2+-dependent asynchronous evoked transmission at rat striatal inhibitory synapses in vitro. J Physiol. 1999;514:447–458. doi: 10.1111/j.1469-7793.1999.447ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw G, Weber K. Differential expression of neurofilament triplet proteins in brain development. Nature. 1982;298:277–279. doi: 10.1038/298277a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Su J, Gorse K, Ramirez F, Fox MA. Collagen XIX is expressed by interneurons and contributes to the formation of hippocampal synapses. J Comp Neurol. 2010;518:229–253. doi: 10.1002/cne.22228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature. 2007;450:676–682. doi: 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taherzadeh-Fard E, Saft C, Andrich J, Wieczorek S, Arning L. PGC-1alpha as modifier of onset age in Huntington disease. Mol Neurodegener. 2009;4:10. doi: 10.1186/1750-1326-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamás G, Buhl EH, Lörincz A, Somogyi P. Proximally targeted GABAergic synapses and gap junctions synchronize cortical interneurons. Nat Neurosci. 2000;3:366–371. doi: 10.1038/73936. [DOI] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Südhof TC. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, Cui L, Beyer RP, Easley CN, Smith AC, Krainc D, Luquet S, Sweet IR, Schwartz MW, La Spada AR. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Weydt P, Soyal SM, Gellera C, Didonato S, Weidinger C, Oberkofler H, Landwehrmeyer GB, Patsch W. The gene coding for PGC-1alpha modifies age at onset in Huntington's disease. Mol Neurodegener. 2009;4:3. doi: 10.1186/1750-1326-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Soyal SM, Landwehrmeyer GB, Patsch W. A single nucleotide polymorphism in the coding region of PGC-1alpha is a male-specific modifier of Huntington disease age-at-onset in a large European cohort. BMC Neurol. 2014;14:1. doi: 10.1186/1471-2377-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker RG, Turnbull DM, Whittington MA, Cunningham MO. Impaired mitochondrial function abolishes gamma oscillations in the hippocampus through an effect on fast-spiking interneurons. Brain. 2011;134:e180. doi: 10.1093/brain/awr018. author reply e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki AS. Peroxisomal disorders affecting phytanic acid alpha-oxidation: a review. Biochem Soc Trans. 2007;35:881–886. doi: 10.1042/BST0350881. [DOI] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Südhof TC. Synaptotagmin-1, -2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–581. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Xue M, Reim K, Chen X, Chao HT, Deng H, Rizo J, Brose N, Rosenmund C. Distinct domains of complexin I differentially regulate neurotransmitter release. Nat Struct Mol Biol. 2007;14:949–958. doi: 10.1038/nsmb1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Kaeser-Woo YJ, Pang ZP, Xu W, Südhof TC. Complexin clamps asynchronous release by blocking a secondary Ca(2+) sensor via its accessory alpha helix. Neuron. 2010;68:907–920. doi: 10.1016/j.neuron.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA, Grünblatt E, Moran LB, Mandel SA, Riederer P, Miller RM, Federoff HJ, Wüllner U, Papapetropoulos S, Youdim MB, Cantuti-Castelvetri I, et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med. 2010;2:52ra73. doi: 10.1126/scitranslmed.3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Lindenbaum M, Levavasseur F, Jacomy H, Julien JP. Disruption of the NF-H gene increases axonal microtubule content and velocity of neurofilament transport: relief of axonopathy resulting from the toxin beta,beta'-iminodipropionitrile. J Cell Biol. 1998;143:183–193. doi: 10.1083/jcb.143.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]