

Abstract
Objective
A fundamental metric in the diagnosis of arthropathies is the pattern of joint involvement, including differences in proximal versus distal joints and patterns of symmetric or asymmetric disease. The basis for joint selectivity among arthritides and/or within a defined disease such as rheumatoid arthritis remains enigmatic. Coagulation and fibrinolytic activity are observed in both experimental animals with inflammatory joint disease and patients with inflammatory arthritis. However, the contribution of specific hemostatic factors to joint disease is not fully defined. We sought to determine the contribution of the fibrinolytic protease, plasminogen, to tumor necrosis factor α (TNFα)–driven arthritis in distinct joints in mice.
Methods
The impact of plasminogen and/or fibrinogen genetic deficiencies on arthritis progression was evaluated in Tg197 mice genetically predisposed to spontaneous, nonabating, and erosive polyarthritis due to exuberant human TNFα expression.
Results
Elimination of plasminogen in Tg197 mice significantly exacerbated the incidence and severity of arthritis within the paw joints, but simultaneously and dramatically diminished the entire spectrum of pathologies within the knee joints of the same animals. These opposing outcomes were both mechanistically linked to fibrin(ogen), in that superimposing fibrinogen deficiency reversed both the proarthritic phenotype in the paws and arthritis resistance in the knees of plasminogen-deficient mice. Intriguingly, the change in disease severity in the knees, but not the paws, was associated with a plasminogen-dependent reduction in matrix metalloproteinase 9 activity.
Conclusion
Plasminogen is a key molecular determinant of inflammatory joint disease capable of simultaneously driving or ameliorating arthritis pathogenesis in distinct anatomic locations in the same subject.
Rheumatoid arthritis (RA) is a chronic autoimmune disease of the joints that affects ~1.5 million people in the US (1). RA is a complex, heterogeneous disease with considerable variation among patients in disease progression and severity. It is well documented that RA affects multiple joints of the body, but certain joints, particularly the peripheral joints of the wrists, proximal interphalangeal joints, metacarpophalangeal joints, and ankle joints, are more susceptible to disease manifestation. Less commonly, RA may also manifest in large joints such as the knees, hips, and shoulders (2,3). Despite the well-characterized predilection for arthritis development in peripheral joints, the precise basis for differential joint involvement in RA, including underlying molecular determinants, is unknown.
Fibrin deposition along articular cartilage surfaces, within inflamed synovium, and as a component of rice bodies in the synovial fluid of affected joints is a consistent feature of RA patients and experimental animals with inflammatory arthritis (4–6). Furthermore, the levels of markers of coagulation system activity, including thrombin–antithrombin complexes and thrombin fragment F1+2, are significantly elevated in both the plasma and synovial fluid of RA patients (7). Plasminogen activation (PA) system components (e.g., plasminogen, tissue-type plasminogen activator [tPA], and urokinase-type plasminogen activator [uPA]) that are essential for fibrinolysis are also thought to modify arthritis pathogenesis (8–11). Elevated levels of fibrin degradation products, including D-dimer, in the synovial fluid of RA patients suggest ongoing PA system–mediated fibrinolysis (7,12). Studies using gene-targeted mice (6,13) and pharmacologic anticoagulants (8) have strongly implied that coagulation and fibrinolytic system activities are not merely secondary consequences of inflammatory events but actively participate in disease pathogenesis. However, studies of mice with genetically imposed deficiencies in PA system components have yielded seemingly mixed, and at times opposing, results regarding the putative roles of the fibrinolytic system in arthritis pathogenesis (9–11). Thus, the contribution of the PA system to arthritis is likely context dependent and influenced by other factors, including disease etiology, mechanisms of pathogenesis, and external events such as local injury.
Numerous studies have implicated a critical role for the proinflammatory cytokine tumor necrosis factor α (TNFα) as a master effector of RA pathology. TNFα levels are markedly elevated in the sera and synovial fluid of patients with RA (14). The central contribution of TNFα to arthritis pathogenesis has been documented through multiple gain-of-function and loss-of-function approaches in animal models of inflammatory arthritis (15–20). Notably, the early finding that human TNFα–transgenic mice develop a spontaneous, nonabating, erosive form of polyarthritis provided direct evidence that sustained TNFα expression was sufficient to drive inflammatory arthritis (21). The clinical significance of TNFα is highlighted by the fact that neutralizing agents against TNFα (e.g., infliximab, etanercept, and adalimumab) are generally, although not universally, effective in the treatment of inflammatory arthritides including RA.
In this study, we directly explored the role of the plasminogen–fibrinogen axis in the pathogenesis of inflammatory arthritis in human TNFα–transgenic Tg197 mice that develop spontaneous polyarthritis in the absence of any exogenous manipulation (e.g., intraarticular injection, immunization with heterologous antigens) (21). Our results demonstrate for the first time that plasminogen is a powerful modifier of TNFα-induced inflammatory joint disease; however, the impact of plasmin(ogen) on arthritis is shown to be highly context dependent and can either drive or ameliorate arthritis, even within the same animal, as a function of precise anatomic location, the availability of fibrin(ogen), and potentially, the activation status of matrix metalloproteinase 9 (MMP-9).
MATERIALS AND METHODS
Mice
Mice with genetic deficiencies in plasminogen and/or fibrinogen Aα chain as well as Tg197-transgenic mice have been described previously (21,23). All mice were back-crossed at least 7 generations to the C57BL/6J background. Interbreeding heterozygous Tg197 mice with mice carrying mutant Plg and Fib alleles produced littermates of Plg+/− Fib+/− (Plg+), Plg−/−Fib+/− (Plg−), Plg+/−Fib−/− (Fib−), and Plg−/−Fib−/− (Plg−Fib−) with (Tg197+) and without (Tg197−) the TNFα transgene for analysis. The Cincinnati Children’s Hospital Animal Care and Use Committee approved all of the study protocols.
Arthritis scoring
Mice were analyzed for macroscopic signs of disease at 8 and 10 weeks of age by multiple investigators who were blinded to animal genotype. Mice were assigned a score of 0–5 per paw based on the number of digits affected. The total number of digits affected per animal was determined, for a maximum score of 20, and expressed as the arthritis index score (6). The degree of arthritis severity was assessed by evaluating swelling in each paw using a scale of 0–2, for a total possible score of 8 per animal, as previously described (6,13).
Histologic and immunohistochemical analysis
Formalin-fixed, EDTA-decalcified, paraffin-embedded mouse tissue specimens were sectioned (5 μm) and stained with either hematoxylin and eosin (H&E) or Safranin O. H&E-stained mouse knee joint sections were semiquantitatively evaluated for the following parameters by investigators who were blinded to animal genotype: inflammation (scale of 0–3), synovial hyperplasia (scale of 0–3), pannus (scale of 0–1), cartilage erosion (scale of 0–2), and bone loss (scale of 0–3). Scores were summed to determine the total histopathology index per knee per mouse (scale of 0–12). Representative fore paw and knee joint sections were also immunostained using rabbit anti-mouse fibrin(ogen) sera in conjunction with alkaline phosphatase (Vector Laboratories)/fast red visualization (Sigma) systems as previously described (6). All images were captured using a Zeiss Axioplan 2 microscope.
RNA isolation and quantitative reverse transcription–polymerase chain reaction (qRT-PCR)
Frozen (−70°C) hind paws or whole knee joints from mice were homogenized in TRIzol reagent (Invitrogen) using a tissumizer (Tekmar) to extract total RNA and further purified using an RNeasy Micro kit (Qiagen). Complementary DNA (cDNA) synthesis was performed using a High Capacity RNA-to-cDNA kit (Applied Biosystems). Quantitative RT-PCR was performed with a StepOnePlus instrument (Applied Biosystems) using TaqMan probes for mouse interleukin-6 (IL-6; Mm00446190_m1), IL-1β (Mm01336189_m1), IL-10 (Mm00439614_m1), interferon-γ (IFNγ; Mm00801778_m1), transforming growth factor β (TGFβ; Mm00441724_m1), human TNFα (Hs99999043_m1), MMP-9 (Mm00442991_m1), MMP-2 (Mm00439498_m1), Plat (Mm00476930_m1), and Plau (Mm_01274460_g1). Data were analyzed using the Pfaffl method in which β2-microglobulin (Mm00437762_m1) was used as the normalizing gene.
MMP gelatin zymography and Western blot analysis
Frozen (−70°C) hind paws or whole knee joints from mice were homogenized in cold buffer as previously described (24). A total of 20 μg of protein extract was mixed with zymogram sample buffer (Bio-Rad) and subjected to electrophoresis in 10% zymogram gels with gelatin (Bio-Rad). Gels were incubated in renaturation buffer (Bio-Rad) followed by development buffer (Bio-Rad) for 18 hours at 37°C. Following staining by Coomassie blue, gels were destained for 24–48 hours at room temperature and visualized. For Western blot analysis, protein extracts subjected to electrophoresis were transferred onto a nitrocellulose membrane. MMP-9 was detected using an anti–MMP-9 antibody (Ab19016; Millipore) in conjunction with a biotinylated secondary antibody, ABC peroxidase system (PK4000; Vector), and enhanced chemiluminescent substrate (Thermo Scientific) according to the manufacturer’s instructions.
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 6.0. Median scores for macroscopic and histopathologic index analyses were compared between genotypes using the nonparametric Mann-Whitney U test. Comparison of messenger RNA (mRNA) levels was performed using Student’s 2-tailed t-test. P values less than 0.05 were considered significant.
RESULTS
Genetic elimination of plasminogen results in opposing, joint-specific differences in TNFα-induced arthritis severity
To determine the contribution of plasminogen to inflammatory arthritis induction and severity in an experimental setting in which disease develops spontaneously without investigator-imposed trauma (e.g., intraarticular injection–associated injury), comparative studies of joint disease were performed in Tg197+ mice with and without plasminogen. The consequence of plasminogen deficiency on TNFα-driven arthritis in the mouse paws was readily apparent by simple inspection of 10-week-old mice. Whereas Plg+/Tg197+ mice exhibited some swelling in the phalangeal joints, lesions appeared much larger and more frequent in the phalangeal joints of Plg−/Tg197+ mice (Figure 1A). A similar qualitative increase in swelling was noted in the wrist and ankle joints of Plg−/Tg197+ mice relative to Plg+/Tg197+ mice (results not shown).
Figure 1.

Plasminogen deficiency exacerbates tumor necrosis factor α–induced arthritis in mouse fore paws and hind paws. A, Representative images of hind paws from 10-week-old Tg197− and Tg197+ mice with and without plasminogen. Note the profound swelling in the phalangeal joints (arrows) of Plg−/Tg197+ mice compared to Plg+/Tg197+ mice. B and C, Scatterplots of the arthritis index (B) and arthritis severity scores (C) in the paws of Plg+/Tg197+ mice and Plg−/Tg197+ mice. Symbols represent individual mice; horizontal lines show the median. P values were determined by Mann-Whitney U test. D, Representative hematoxylin and eosin–stained metacarpophalangeal joint sections from 10-week-old Tg197− and Tg197+ mice with and without plasminogen. Note that Plg−/Tg197+ mice displayed substantially more joint destruction, including apparent obliteration of articular surfaces and bone, characterized by extensive synovial hyperplasia (arrowheads) and pannus formation (asterisks) relative to Plg+/Tg197+ mice. E, Representative Safranin O–stained metacarpophalangeal joint sections from 10-week-old Tg197− and Tg197+ mice with and without plasminogen. Note the substantial loss of proteoglycans (arrows) within the articular cartilage in both Plg+/Tg197+ and Plg−/Tg197+ mice. Bars in D and E = 100 μm.
Quantitative macroscopic evaluation of 10-week-old mice confirmed that plasminogen deficiency increases arthritis incidence in fore paw and hind paw joints, as evidenced by significantly increased arthritis index scores (Figure 1B). Similarly, Plg−/Tg197+ mice had a significant increase in arthritis severity relative to Plg+/Tg197+ mice (Figure 1C). A similar genotype-dependent pattern of worsened arthritic disease was observed in the paws of 8-week-old Plg−/Tg197+ mice relative to Plg+/Tg197+ mice, albeit with predictably lower overall disease activity scores in comparison to 10-week-old mice (data available from the author upon request).
Histologic analyses performed in 10-week-old mice confirmed the pattern of increased macroscopic joint disease in Plg−/Tg197+ mice. Control Tg197− mice developed no evidence of gross arthritis, and microscopic evaluation of metacarpophalangeal joints established that joint architecture was histologically indistinguishable in Plg− and Plg+ mice (Figure 1D). Thus, plasminogen deficiency per se neither compromises normal joint development nor leads to arthritis. In contrast, metacarpophalangeal joints from Plg+/Tg197+ mice displayed significant joint pathology, including synovial hyperplasia, inflammatory cell infiltration, pannus formation, cartilage degradation, and bone erosion. Consistent with the macroscopic analyses, the entire spectrum of microscopic joint pathologies was significantly exacerbated in the metacarpophalangeal joints of Plg−/Tg197+ mice (Figure 1D). Indeed, in most of the affected joints of Plg−/Tg197+ mice there was a complete, or near-complete, loss of cartilage structure, which was replaced by hyperplastic granulation tissue (Figures 1D and E).
Tg197 mice have been reported to develop polyarthritis, with advanced disease evolving in joints throughout the animal (21,25). To determine whether plasminogen deficiency resulted in increased arthritis severity in joints other than those of the paws, we performed parallel histologic analyses of knee joints obtained from the same Tg197+ and Tg197− mice with and without plasminogen. Similar to joints of the paws, plasminogen deficiency alone did not alter normal knee joint development or architecture in mice through 10 weeks of age (Figure 2A). Remarkably, and in direct opposition to our findings in paw joints, histologic examination of knee joint sections from these same 10-week-old mice revealed that plasminogen deficiency strongly impeded the development of arthritis in these larger and more proximal joints. Whereas Plg+/Tg197+ mice developed severe joint pathology, including massive inflammatory infiltrates, synovial hyperplasia, profound cartilage degradation, and bone erosion, Plg−/Tg197+ mouse knee joints exhibited little evidence of arthritis and were often nearly indistinguishable from knee joints harvested from Tg197− mice (Figures 2A and B).
Figure 2.

Plasminogen deficiency ameliorates arthritis in the knees of tumor necrosis factor α–transgenic mice. A, Representative hematoxylin and eosin (H&E)–stained knee joint sections from 10-week-old Tg197− and Tg197+ mice with and without plasminogen. Note that widespread joint pathology, including synovial hyperplasia (arrowhead) and pannus (asterisk), was prominent in Plg+/Tg197+ mice, whereas joint architecture was near normal in the Plg−/Tg197+ mice. In the absence of the Tg197 transgene, mouse knee joints were uniformly unremarkable, regardless of the presence or absence of plasminogen. B, Representative Safranin O–stained knee joint sections from 10-week-old Tg197− and Tg197+ mice with and without plasminogen. Note the presence of contiguous red staining indicating intact articular cartilage (arrow) in the Plg−/Tg197+ mice, in contrast to Plg+/Tg197+ mice with extensive articular cartilage destruction. C, Quantitative microscopic analysis of individual disease metrics from H&E-stained knee joint sections from Plg+/Tg197+ mice and Plg−/Tg197+ mice. Bars show the mean ± SEM (n = 13 knees per group). # = P < 0.001; ** = P < 0.01; * = P < 0.05, by Student’s t-test. D, Scatterplot of composite histopathology index analysis of H&E-stained knee joint sections. Symbols represent individual mice; horizontal bars show the median. P values were determined by Mann-Whitney U test. Bars in A and B = 200 μm.
Semiquantitative analysis of pathologic disease features within the knee joints confirmed a significant diminution in arthritis severity in the knees of Plg−/Tg197+ mice (Figures 2C and D). Importantly, amelioration of disease in the knees of Plg−/Tg197+ mice was not restricted to a single pathologic feature. Each disease parameter was significantly decreased compared to Plg+/Tg197+ mice (Figure 2C), resulting in a significantly reduced median composite histopathology index score for Plg−/Tg197+ mouse knee joints compared to Plg+/Tg197+ mouse knee joints (4 and 9, respectively; P < 0.0001) (Figure 2D). Taken together, these findings indicate that plasmin(ogen) effectively drives local arthritis pathology within the mouse knee joints while at the same time limiting disease development in the paw joints. A similar pattern of disease diminution in the knee joints of Plg−/Tg197+ mice compared to Plg+/Tg197+ mice was observed at 8 weeks of age (data available from the author upon request).
As an initial investigation of the molecular basis of plasminogen-dependent differences in TNFα-driven arthritis, we examined the local expression of the plasminogen activators, tPA (Plat) and uPA (Plau), in the 2 joint locations. No differences were observed in the levels of mRNA for tPA or uPA in the paws of Plg−/Tg197+ mice relative to Plg+/Tg197+ mice (data available from the author upon request). Of note, in the knees, the expression of both tPA and uPA was significantly elevated in Plg+/Tg197+ mice relative to Plg+/Tg197− nonarthritic controls (data available from the author upon request). This finding is consistent with previous data indicating that uPA expression is significantly elevated in the knee and hip joints of patients with RA relative to patients with osteoarthritis (26). A modest but statistically significant reduction in local tPA expression in the knees of Plg−/Tg197+ mice relative to Plg+/Tg197+ mice was observed. However, uPA expression was virtually identical in the knees of Plg+ and Plg−/Tg197+ mice. Although the precise contribution of uPA and tPA to TNFα-driven arthritis remains to be formally established, our data suggest that the differences in disease outcome are likely not a simple function of expression changes in PA system components.
The proarthritic and antiarthritic properties of plasminogen are mechanistically tied to the presence of fibrinogen
Provisional fibrin matrices are a key, albeit not sole, biologically meaningful plasmin substrate in vivo (27–29). Consistent with previous reports highlighting fibrin as a conspicuous feature of inflamed joint tissue in humans and experimental animals (5,6,13,30), robust fibrin deposition was readily detected in hyperplastic synovium and along the articular surfaces in metacarpophalangeal and proximal interphalangeal joints obtained from 10-week-old Plg−/Tg197+ mice, particularly joints with microscopic evidence of severe disease (Figure 3A). Intriguingly, despite the general absence of other significant disease manifestations in the knees of Plg−/Tg197+ mice, substantial fibrin deposition was readily observed along the synovial lining, within synovial tissues, and covering the articular cartilage surfaces (Figure 3B).
Figure 3.

Fibrin deposition is a consistent histologic feature of joint tissue in plasminogen-deficient mice despite the opposing differences in arthritis disease severity in the paw and knee joints. A, Immunohistochemical staining for fibrin or fibrinogen in representative proximal interphalangeal joint sections isolated from 10-week-old Tg197− and Tg197+ mice with and without plasminogen. Note the robust fibrin deposition (red staining) in the hyperplastic synovium and eroding cartilage surfaces in Plg−/Tg197+ mice. B, Immunohistochemical staining for fibrin deposition in representative knee joint sections isolated from 10-week-old Tg197− and Tg197+ mice with and without plasminogen. Intense fibrin staining was consistently observed in the knee joints of Plg−/Tg197+ mice, particularly along articular surfaces, despite the general lack of microscopic features of arthritis. Not surprisingly, fibrin deposition was also observed within the appreciably more disrupted knee joints of Plg+/Tg197+ mice, often dispersed within the hyperplastic synovial tissue. Bars = 200 μm
Given both the prevalence of fibrin in the joints of Plg−/Tg197+ mice and the dual capacity of fibrin to support both thromboinflammatory pathologies and reparative processes, we sought to determine whether the liabilities (observed in mouse paws) or benefits (observed in mouse knees) of plasminogen deficiency on TNFα-induced arthritis were mechanistically linked to fibrin(ogen). Consistent with our prior independent findings, fibrinogen deficiency alone did not alter the degree of TNFα-driven arthritis in the paws or knees of 10-week-old mice (Figures 4A and B) (6). Nevertheless, consistent with the concept that persistent fibrin deposition promotes the progression of degradative inflammatory arthritis in the paws, Plg−Fib−/Tg197+ mice developed significantly less arthritis in distal joints than did mice in the Plg−/Tg197+ cohorts (Figures 4A and B). Thus, the loss of fibrin(ogen) in this context effectively rescued mice from the disadvantages associated with plasminogen deficiency. Remarkably, microscopic analyses of H&E-stained mouse knee joints revealed a significant increase in knee joint pathology (i.e., inflammation, synovial hyperplasia, pannus formation, cartilage degradation, and bone erosion) (Figure 4C) in Plg−Fib−/Tg197+ mice compared to Plg−/Tg197+ mice (histopathology index 5.25 versus 1; P < 0.001) to a level not statistically different from that in Plg+/Tg197+ mice (histopathology index 5.25 versus 8, respectively) (Figure 4D). Thus, elimination of fibrin(ogen) abolished the advantages associated with plasminogen deficiency in the mouse knees.
Figure 4.

Genetic elimination of fibrinogen reverses both the proarthritic and antiarthritic phenotypes observed in the paws and knees of plasminogen-deficient Tg197 mice. A and B, Scatterplots of the arthritis index (A) and arthritis severity scores (B) in the paws of Tg197+ mice with single and combined deficits in plasminogen and fibrinogen. Note that plasminogen deficiency resulted in worsened arthritis in the paws, and superimposing fibrinogen deficiency significantly lessened arthritis in this location. The loss of fibrinogen alone had no significant impact on arthritis in the paws relative to control Tg197+ mice. C, Histology scores of inflammatory joint disease parameters in the knees of Tg197+ mice with single and combined deficits in plasminogen and fibrinogen. Bars show the mean ± SEM. * = P < 0.001; # = P < 0.01, by Student’s t-test. D, Scatterplot of histopathology index composite scores in the knees of Tg197+ mice with single and combined deficits in plasminogen and fibrinogen. In contrast to the paws, plasminogen deficiency strongly lessened arthritis in the knees, and superimposing fibrinogen deficiency significantly worsened arthritis in this location. The loss of fibrinogen alone had no significant impact on arthritis in the knees relative to control Tg197+ mice. In A and B, symbols represent individual mice; in D, symbols represent individual knees. Horizontal bars show the median. P values were determined by Mann-Whitney U test. NS = not significant.
Correlation of local proinflammatory and anti-inflammatory cytokine expression with arthritis severity
Consistent with the pattern of increased arthritis severity in the paws of Plg−/Tg197+ mice relative to Plg+/Tg197+ mice, qRT-PCR analyses of mRNA isolated from the paws revealed a marked parallel increase in the expression of the proinflammatory cytokines IL-1β, IL-6, and IFNγ (Figures 5A–C) and the anti-inflammatory cytokine IL-10 (Figure 5D). No change in expression based on plasminogen or fibrinogen genotype difference was observed for TGFβ (Figure 5E) in the paws of Tg197+ mice. Furthermore, consistent with the amelioration of arthritis pathologies in the paws of Plg−Fib−/Tg197+ mice, the expression of proinflammatory and antiinflammatory cytokines in the paws of these animals was diminished to levels comparable to those in control Plg+/Tg197+ mice (Figures 5A–D).
Figure 5.

Proinflammatory and antiinflammatory cytokine expression within joints is dependent on both plasminogen and fibrinogen and correlates with arthritis severity in Tg197 mice. Results of quantitative reverse transcriptase–polymerase chain reaction analysis of interleukin-1γ (IL-1γ), IL-6, interferon-β (IFNβ), IL-10, and transforming growth factor β (TGFβ) messenger RNA isolated from the paws (A–E) and knees (F–J) of 10-week-old Tg197+ and Tg197− mice with selected deficits in plasminogen and/or fibrinogen are shown. Cytokine expression was low in the arthritis-free Tg197− mice, regardless of hemostatic factor status. In Tg197+ mice, expression of each of these endogenous inflammatory mediators was readily detected in both the paws and knees. Importantly, plasminogen acted as both a positive and a negative determinant of endogenous cytokine expression in Tg197+ mice, depending on joint location. Note that the pattern of endogenous cytokine expression closely followed the location-dependent and hemostatic factor–dependent worsened or diminished overall arthritis pathology. Bars show the mean ± SEM (n = 4 mice in the Tg197− control group; n = 6–8 mice in all other groups). P values were determined by Student’s t-test. NS = not significant.
Complementary qRT-PCR analyses focusing on the knee joints from the same set of experimental cohorts revealed a genotype-dependent pattern of cytokine expression quite distinct from that seen in the paws. In the knee joints, Plg−/Tg197+ mice exhibited significantly lower IL-1β, IL-6, IFNγ, and IL-10 expression compared to Plg+/Tg197+ mice (Figures 5F–I). The one exception was the expression of TGFβ, which was significantly higher in the knee joints of Plg−/Tg197+ mice relative to Plg+/Tg197+ mice. Additionally, superimposed fibrinogen deficiency in Plg−/Tg197+ mice re-established high proinflammatory cytokine expression within knee tissues, effectively removing the potential local benefit of plasminogen deficiency in limiting inflammatory changes in this anatomic location. Notably, plasminogen deficiency did not alter the local expression of transgene-derived human TNFα in the paws or knees of Tg197+ mice or systemic levels of human TNFα protein (data available from the author upon request). Together, these results suggest that the differences in disease severity in the paws and knees of Plg−/Tg197+ mice cannot be explained by alterations in the local production of TNFα, but other distinct mechanisms involving key downstream effectors (i.e., IL-1β and IL-6) are important in mediating disease pathogenesis.
Reduced MMP-9 activity specifically in the knee joints, but not in the paws, of plasminogen-deficient Tg197 mice
To determine whether plasmin(ogen) influences TNFα-driven arthritis through mechanisms linked to MMP activation, we examined the local expression and activity of the gelatinases MMP-9 and MMP-2 in the paws and knees of Tg197+ and Tg197− mice. MMP-2 and MMP-9 were the focus of investigation since studies of knockout mice linked both of these MMPs to joint destruction in the context of inflammatory arthritis, with MMP-9 driving proarthritic activity and MMP-2 supporting antiarthritic activities (31). Intriguingly, plasminogen deficiency generally correlated with significantly elevated levels of mRNA for MMP-9 and MMP-2 in both the paws (Figures 6A and B) and the knees (Figures 6C and D) of mice.
Figure 6.
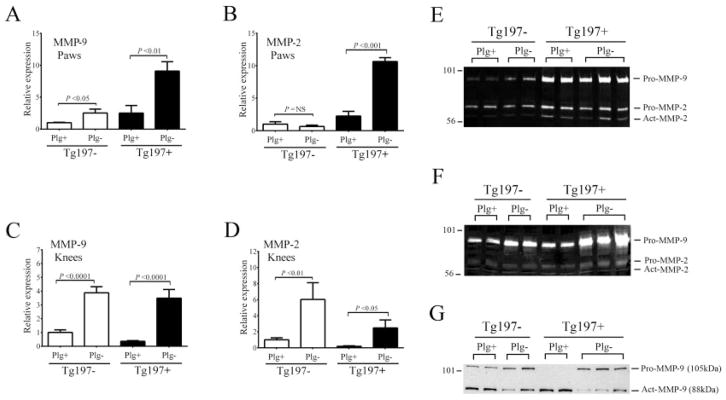
Plasminogen deficiency results in diminished matrix metalloproteinase 9 (MMP-9), but not MMP-2, activity in the knees but not the paws of Tg197 mice. A–D, Results of quantitative reverse transcriptase–polymerase chain reaction analysis of MMP-9 and MMP-2 mRNA isolated from the paws (A and B) and knees (C and D) of 10-week-old Plg+ and Plg− Tg197 mice. Bars show the mean ± SEM. P values were determined by Student’s t-test. E and F, Gelatin zymography of protein extracts prepared from the paws (E) and knees (F) of Tg197 mice to detect MMP-9 and MMP-2 activity. G, Western blot analysis using an anti–MMP-9 antibody, which detects both proMMP-9 (105 kd) and active MMP-9 (act MMP-9; 88 kd). Note that active MMP-9 is readily detected in the knees of Plg+/Tg197+ mice, but that levels of active MMP-9 are significantly reduced in the knees of Plg−/Tg197+ mice, despite higher overall levels of proMMP-9 protein. NS = not significant.
To investigate the local, joint-specific protein and activity levels of MMP-9 and MMP-2, gelatin zymography and Western blot analyses were performed on total protein extracts prepared from the paws and knees of Plg+ and Plg− mice. As expected, zymography revealed that MMP-9 and MMP-2 activities within the paws were visibly more prominent in Tg197+ mice compared to Tg197− mice regardless of the plasminogen genotype (Figure 6E). Furthermore, no plasminogen-dependent differences were observed in the ratios of pro- and active MMP-9 or MMP-2 in the paws of Tg197+ mice (Figure 6E). Western blot analyses revealed little to no signal for either pro- or active MMP-9 in any of the extracts derived from paw tissue (data not shown). Importantly, within the mouse knee joints a dramatically different pattern was observed. First, we observed an obvious overall increase in proMMP-9 protein in the knees of Plg−/Tg197+ mice compared to Plg+/Tg197+ mice, as shown by both zymography (Figure 6F) and Western blot analysis (Figure 6G). No differences were observed in proMMP-2 and active MMP-2 activities between the two genotypes based on zymography (Figure 6E). More impressive was that the Western blot analysis indicated that the majority of MMP-9 in the knees of Plg+/Tg197+ mice was in the 88-kd active form, whereas in Plg−Tg197+ mouse knees the majority of MMP-9 was in the 105-kd pro(inactive) form. Indeed, there was much less active MMP-9 (88 kd) in the knees of Plg−/Tg197+ mice compared to Plg+/Tg197+ mice. These results are consistent with impaired conversion of zymogen proMMP-9 into enzymatically active MMP-9 in the knees (but not the paws) of Plg−/Tg197+ mice.
DISCUSSION
The distinct anatomic locations (e.g., distal versus proximal joints) and the symmetry/asymmetry of joint disease are uniformly accepted as important metrics in the differential diagnosis of various forms of arthritis, such as RA, psoriatic arthritis, ankylosing spondylitis (AS), and inflammatory bowel disease–associated arthritis (32). The concept of differential joint involvement in RA has long been appreciated; clinically significant RA is considerably more common in small peripheral joints (e.g., metacarpophalangeal and proximal interphalangeal joints) than in larger and/or more proximal joints, such as the knees and the hips, and often occurs in a symmetric manner (2). However, the basis for such selective localization of the inflammatory arthropathy, including the precise molecular, anatomic, or mechanical determinants involved, has remained largely speculative, and is effectively an unresolved knowledge gap.
Clinical data suggest that fibrinolytic system components could contribute to joint-specific arthritis. For example, circulating levels of plasminogen activator inhibitor 1 and D-dimer were found to be significantly higher in patients with RA than in those with AS (33). Similarly, the expression of soluble uPA receptor in plasma was found to be significantly higher in patients with RA than in patients with reactive arthritis or Sjögren’s syndrome (34). Indeed, in this study we show that plasminogen constitutes a location-dependent modifier of arthritis, where even the direction of disease (tolerance versus sensitivity) is dictated by the combination of plasminogen and precise microenvironment. To our knowledge, this is the first identification of a molecular determinant dictating joint selectivity, capable of advancing or hindering arthritic disease within a single subject in distinct locations.
Plasminogen- and uPA-deficient mice were previously shown to develop a less severe form of arthritis in the setting of both major histocompatibility complex haplotype–dependent collagen-induced arthritis (CIA) and immune complex–driven K/BxN serum transfer–induced arthritis (10,35). However, in the experimental setting of antigen-induced arthritis (AIA), a monarticular arthritis model involving intraarticular injection of antigen, both uPA-deficient and plasminogen-deficient mice developed markedly exacerbated joint inflammation (8). One element of commonality in these studies pointing in the direction of exacerbated arthritis is investigator-imposed local mechanical trauma (e.g., needle track injury associated with intraarticular injections).
The potential significance of local trauma as a determinant of inflammatory arthritis was recently observed in a setting of uPA deficiency wherein uPA-deficient mice were found to be generally protected against arthritis induced by intraperitoneal K/BxN serum transfer, but developed severe arthritis if the same challenge was accompanied by an intraarticular needle track injury (11). It was proposed that in the absence of local injury, uPA/plasminogen may aggravate disease by way of nonfibrin targets, but the same factors may limit arthritis by supporting a fibrinolysis-dependent “wound healing–like” process when the challenge also involves local trauma (11). Here, persistent fibrin deposition due to both the local injury and compromised fibrinolysis could potentiate joint inflammation. Consistent with this notion, we previously demonstrated that the capacity to generate fibrin and, more specifically, Mac-1–dependent leukocyte engagement of fibrin within joints, strongly promoted arthritis development in CIA (6). The present study directly and definitively identifies plasminogen as one factor that can differentially influence the manifestation of arthritis in distinct anatomic locations in a setting where the primary underlying etiologic challenge (i.e., exuberant TNFα expression) is precisely the same and involves no investigator-imposed local mechanical trauma.
Our results provide unexpected evidence that the capacity to generate fibrin can be either proarthritic or antiarthritic, depending on the microenvironment. The unanticipated, context-dependent antiarthritic capacity of fibrin(ogen) deposition was firmly illustrated by the significantly increased arthritis severity observed in the knees of Plg−Fib−/Tg197+ mice. One concept consistent with a fibrin-associated suppression of TNFα-driven arthritis in the knees is that provisional fibrin matrices in this location could be particularly beneficial in supporting vascular integrity, limiting counterproductive hemorrhagic events, and/or aiding reparative processes (22,23). Such a model would explain the worsened knee joint pathology when fibrinogen was eliminated in Plg−/Tg197+ mice. The negative impact of fibrin(ogen) deficiency on vascular integrity or tissue repair would be mechanistically distinct from, and superseded by, any direct advantage achieved by the elimination of plasminogen. Evidence in support of this notion, particularly in large, load-bearing joints subject to recurrent mechanical trauma, can be found in the propensity of patients with hemophilia to develop arthropathies in proximal joints, such as the knees, but not in the digits (36,37).
An additional mechanistic dimension of PA-dependent protection against arthritis is the absence of plasmin-mediated proteolysis of extracellular matrix proteins (e.g., laminin, fibronectin, and collagen) critical to normal joint architecture either directly or indirectly via MMP activation (e.g., MMP-1, MMP-2, and MMP-9) (38–40). Numerous studies have implied a crucial role for plasmin-mediated activation of MMP-9 in physiologic and pathologic processes such as neointima formation (41), wound healing (42), and an experimental model of the autoimmune skin disease, bullous pemphigoid (43). Our data provide strong evidence that proMMP-9 activation was impaired in the absence of plasminogen within the mouse knee joints but not in the paws, suggesting that MMP-9 is a putative plasmin target within the knee joints that drives inflammation and tissue damage.
There are several possible explanations for the differences in plasmin-mediated MMP-9 activation in the paws as compared to the knees of mice. One is simply that MMP-9 is present at high levels in the mouse knee joints and at very low levels in the paws, which is consistent with the results of our Western blot analyses. A second possibility is that factors other than plasmin activate proMMP-9 in the paw tissue, but plasmin is critical for activation of MMP-9 in the knee joints. However, zymography and Western blot analyses did not detect active MMP-9 in the paw tissue of mice, but active MMP-9 was readily detected in the knee joints. A third concept potentially accounting for joint-specific changes in MMP-9 activity is that differences in the pathologic function of MMP-9 may be dependent on its cellular source. Consistent with this concept, previous studies have suggested that whereas epithelial-derived MMP-9 exacerbates inflammation during colitis, MMP-9 from granulocytes ameliorates colitis symptoms (44). Thus, one intriguing hypothesis is that depending on the cellular source of MMP-9 in the two joint locations, plasmin-mediated MMP-9 activation could result in dramatically different disease outcomes.
Multiple additional plasmin targets are compatible with the finding that plasmin(ogen) promotes TNFα-driven inflammatory arthritis in some specific anatomic locations (e.g., the knees). For example, plasmin has been shown to support inflammatory cell trafficking as well as cell signaling events and cytokine secretion, in part through engagement of cell surface plasminogen receptors (e.g., Plg-RKT, annexin II, α-enolase) found on numerous cell types relevant to arthritis, including endothelial cells and inflammatory monocyte/macrophages (45–48). In this regard, inhibition of Plg-RKT with a monoclonal antibody was recently shown to markedly reduce macrophage migration in vivo (45,46). The knee joints of Plg−/Tg197+ mice were found to be remarkably free of inflammatory infiltrates, but whether this is a primary effect of plasminogen deficiency or a secondary consequence of overall limited arthritis remains to be resolved. Thus, the present data do not exclude the possibility that plasmin influences inflammatory arthritis through multiple distinct mechanisms.
In conclusion, these studies highlight a unique relationship between the PA system and TNFα-driven arthritis. Furthermore, these findings highlight the concept that the action of local hemostatic factors may be one basis for the differential development of inflammatory arthritis in distinct joints (e.g., large versus small). Accordingly, clinical interventions at the level of hemostatic factors may be a useful therapeutic strategy in the treatment of some, but not necessarily all, inflammatory arthropathies.
Acknowledgments
Supported by the NIH (grants HL-096126 to Dr. Degen and AR-056990 to Dr. Flick) and by the Cincinnati Rheumatic Diseases Center–Animal Models of Inflammatory Diseases Core at Cincinnati Children’s Hospital Medical Center (grant P30-AR-47363 to Dr. Flick).
We wish to thank Kathryn McElhinney, Keith Kombrinck, Elizabeth Blevins, and Whitney Miller for excellent technical assistance. We wish to thank Drs. Joseph Palumbo and Eric Mullins for helpful suggestions in the preparation of this manuscript. We wish to thank Dr. George Kollias for kindly providing the human TNFα–transgenic Tg197 mouse line.
Footnotes
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Flick had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Raghu, Thornton, Degen, Flick.
Acquisition of data. Raghu, Jone, Cruz, Rewerts, Frederick, Thornton, Flick.
Analysis and interpretation of data. Raghu, Thornton, Degen, Flick.
References
- 1.Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, et al. for the National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: part I. Arthritis Rheum. 2008;58:15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- 2.Abdel-Nasser AM, Rasker JJ, Valkenburg HA. Epidemiological and clinical aspects relating to the variability of rheumatoid arthritis. Semin Arthritis Rheum. 1997;27:123–40. doi: 10.1016/s0049-0172(97)80012-1. [DOI] [PubMed] [Google Scholar]
- 3.Alamanos Y, Voulgari PV, Drosos AA. Incidence and prevalence of rheumatoid arthritis, based on the 1987 American College of Rheumatology criteria: a systematic review. Semin Arthritis Rheum. 2006;36:182–8. doi: 10.1016/j.semarthrit.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Yang YH, Carmeliet P, Hamilton JA. Tissue-type plasminogen activator deficiency exacerbates arthritis. J Immunol. 2001;167:1047–52. doi: 10.4049/jimmunol.167.2.1047. [DOI] [PubMed] [Google Scholar]
- 5.Marty I, Peclat V, Kirdaite G, Salvi R, So A, Busso N. Amelioration of collagen-induced arthritis by thrombin inhibition. J Clin Invest. 2001;107:631–40. doi: 10.1172/JCI11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flick MJ, LaJeunesse CM, Talmage KE, Witte DP, Palumbo JS, Pinkerton MD, et al. Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin αMβ2 binding motif. J Clin Invest. 2007;117:3224–35. doi: 10.1172/JCI30134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.So AK, Varisco PA, Kemkes-Matthes B, Herkenne-Morard C, Chobaz-Peclat V, Gerster JC, et al. Arthritis is linked to local and systemic activation of coagulation and fibrinolysis pathways. J Thromb Haemost. 2003;1:2510–5. doi: 10.1111/j.1538-7836.2003.00462.x. [DOI] [PubMed] [Google Scholar]
- 8.Busso N, Peclat V, Van Ness K, Kolodziesczyk E, Degen J, Bugge T, et al. Exacerbation of antigen-induced arthritis in urokinase-deficient mice. J Clin Invest. 1998;102:41–50. doi: 10.1172/JCI2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cook AD, Braine EL, Campbell IK, Hamilton JA. Differing roles for urokinase and tissue-type plasminogen activator in collagen-induced arthritis. Am J Pathol. 2002;160:917–26. doi: 10.1016/S0002-9440(10)64914-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook AD, De Nardo CM, Braine EL, Turner AL, Vlahos R, Way KJ, et al. Urokinase-type plasminogen activator and arthritis progression: role in systemic disease with immune complex involvement. Arthritis Res Ther. 2010;12:R37. doi: 10.1186/ar2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Nardo CM, Lenzo JC, Pobjoy J, Hamilton JA, Cook AD. Urokinase-type plasminogen activator and arthritis progression: contrasting roles in systemic and monoarticular arthritis models. Arthritis Res Ther. 2010;12:R199. doi: 10.1186/ar3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busso N, Hamilton JA. Extravascular coagulation and the plasminogen activator/plasmin system in rheumatoid arthritis [review] Arthritis Rheum. 2002;46:2268–79. doi: 10.1002/art.10498. [DOI] [PubMed] [Google Scholar]
- 13.Flick MJ, Chauhan AK, Frederick M, Talmage KE, Kombrinck KW, Miller W, et al. The development of inflammatory joint disease is attenuated in mice expressing the anticoagulant pro-thrombin mutant W215A/E217A. Blood. 2011;117:6326–37. doi: 10.1182/blood-2010-08-304915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tetta C, Camussi G, Modena V, Di Vittorio C, Baglioni C. Tumour necrosis factor in serum and synovial fluid of patients with active and severe rheumatoid arthritis. Ann Rheum Dis. 1990;49:665–7. doi: 10.1136/ard.49.9.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, et al. Critical roles for interleukin 1 and tumor necrosis factor α in antibody-induced arthritis. J Exp Med. 2002;196:77–85. doi: 10.1084/jem.20020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci U S A. 1992;89:9784–8. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piguet PF, Grau GE, Vesin C, Loetscher H, Gentz R, Lesslauer W. Evolution of collagen arthritis in mice is arrested by treatment with anti-tumour necrosis factor (TNF) antibody or a recombinant soluble TNF receptor. Immunology. 1992;77:510–4. [PMC free article] [PubMed] [Google Scholar]
- 18.Lewthwaite J, Blake S, Hardingham T, Foulkes R, Stephens S, Chaplin L, et al. Role of TNFα in the induction of antigen induced arthritis in the rabbit and the anti-arthritic effect of species specific TNFα neutralising monoclonal antibodies. Ann Rheum Dis. 1995;54:366–74. doi: 10.1136/ard.54.5.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Probert L, Akassoglou K, Alexopoulou L, Douni E, Haralambous S, Hill S, et al. Dissection of the pathologies induced by transmembrane and wild-type tumor necrosis factor in transgenic mice. J Leukoc Biol. 1996;59:518–25. doi: 10.1002/jlb.59.4.518. [DOI] [PubMed] [Google Scholar]
- 20.Alexopoulou L, Pasparakis M, Kollias G. A murine transmembrane tumor necrosis factor (TNF) transgene induces arthritis by cooperative p55/p75 TNF receptor signaling. Eur J Immunol. 1997;27:2588–92. doi: 10.1002/eji.1830271018. [DOI] [PubMed] [Google Scholar]
- 21.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bugge TH, Flick MJ, Daugherty CC, Degen JL. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. 1995;9:794–807. doi: 10.1101/gad.9.7.794. [DOI] [PubMed] [Google Scholar]
- 23.Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ, Degen JL. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell. 1996;87:709–19. doi: 10.1016/s0092-8674(00)81390-2. [DOI] [PubMed] [Google Scholar]
- 24.Sullivan BP, Kassel KM, Jone A, Flick MJ, Luyendyk JP. Fibrin-(ogen)-independent role of plasminogen activators in acetamino-phen-induced liver injury. Am J Pathol. 2012;180:2321–9. doi: 10.1016/j.ajpath.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li P, Schwarz EM. The TNF-α transgenic mouse model of inflammatory arthritis. Springer Semin Immunopathol. 2003;25:19–33. doi: 10.1007/s00281-003-0125-3. [DOI] [PubMed] [Google Scholar]
- 26.Busso N, Peclat V, So A, Sappino AP. Plasminogen activation in synovial tissues: differences between normal, osteoarthritis, and rheumatoid arthritis joints. Ann Rheum Dis. 1997;56:550–7. doi: 10.1136/ard.56.9.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bezerra JA, Bugge TH, Melin-Aldana H, Sabla G, Kombrinck KW, Witte DP, et al. Plasminogen deficiency leads to impaired remodeling after a toxic injury to the liver. Proc Natl Acad Sci U S A. 1999;96:15143–8. doi: 10.1073/pnas.96.26.15143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng VL, Sabla GE, Melin-Aldana H, Kelley-Loughnane N, Degen JL, Bezerra JA. Plasminogen deficiency results in poor clearance of non-fibrin matrix and persistent activation of hepatic stellate cells after an acute injury. J Hepatol. 2001;35:781–9. doi: 10.1016/s0168-8278(01)00212-4. [DOI] [PubMed] [Google Scholar]
- 29.Tsirka SE, Bugge TH, Degen JL, Strickland S. Neuronal death in the central nervous system demonstrates a non-fibrin substrate for plasmin. Proc Natl Acad Sci U S A. 1997;94:9779–81. doi: 10.1073/pnas.94.18.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plows D, Kontogeorgos G, Kollias G. Mice lacking mature T and B lymphocytes develop arthritic lesions after immunization with type II collagen. J Immunol. 1999;162:1018–23. [PubMed] [Google Scholar]
- 31.Itoh T, Matsuda H, Tanioka M, Kuwabara K, Itohara S, Suzuki R. The role of matrix metalloproteinase-2 and matrix metalloproteinase-9 in antibody-induced arthritis. J Immunol. 2002;169:2643–7. doi: 10.4049/jimmunol.169.5.2643. [DOI] [PubMed] [Google Scholar]
- 32.Firestein GS, Budd RC, Gabriel SE, McInnes IB, O’Dell JR, editors. Kelley’s textbook of rheumatology. 9. Philadelphia: Elsevier/Saunders; 2013. [Google Scholar]
- 33.Wallberg-Jonsson S, Rantapaa-Dahlqvist S, Nordmark L, Ranby M. Mobilization of fibrinolytic enzymes in synovial fluid and plasma of rheumatoid arthritis and spondyloarthropathy and their relation to radiological destruction. J Rheumatol. 1996;23:1704–9. [PubMed] [Google Scholar]
- 34.Slot O, Brunner N, Locht H, Oxholm P, Stephens RW. Soluble urokinase plasminogen activator receptor in plasma of patients with inflammatory rheumatic disorders: increased concentrations in rheumatoid arthritis. Ann Rheum Dis. 1999;58:488–92. doi: 10.1136/ard.58.8.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Ny A, Leonardsson G, Nandakumar KS, Holmdahl R, Ny T. The plasminogen activator/plasmin system is essential for development of the joint inflammatory phase of collagen type II-induced arthritis. Am J Pathol. 2005;166:783–92. doi: 10.1016/S0002-9440(10)62299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valentino LA. Blood-induced joint disease: the pathophysiology of hemophilic arthropathy. J Thromb Haemost. 2010;8:1895–902. doi: 10.1111/j.1538-7836.2010.03962.x. [DOI] [PubMed] [Google Scholar]
- 37.Norian JM, Ries MD, Karp S, Hambleton J. Total knee arthroplasty in hemophilic arthropathy. J Bone Joint Surg Am. 2002;84-A:1138–41. doi: 10.2106/00004623-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 38.Lijnen HR, Van Hoef B, Lupu F, Moons L, Carmeliet P, Collen D. Function of the plasminogen/plasmin and matrix metalloproteinase systems after vascular injury in mice with targeted inactivation of fibrinolytic system genes. Arterioscler Thromb Vasc Biol. 1998;18:1035–45. doi: 10.1161/01.atv.18.7.1035. [DOI] [PubMed] [Google Scholar]
- 39.Carmeliet P, Moons L, Lijnen R, Baes M, Lemaitre V, Tipping P, et al. Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat Genet. 1997;17:439–44. doi: 10.1038/ng1297-439. [DOI] [PubMed] [Google Scholar]
- 40.Gong Y, Hart E, Shchurin A, Hoover-Plow J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J Clin Invest. 2008;118:3012–24. doi: 10.1172/JCI32750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lijnen HR. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost. 2001;86:324–33. [PubMed] [Google Scholar]
- 42.Lund LR, Romer J, Bugge TH, Nielsen BS, Frandsen TL, Degen JL, et al. Functional overlap between two classes of matrix-degrading proteases in wound healing. EMBO J. 1999;18:4645–56. doi: 10.1093/emboj/18.17.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Z, Li N, Diaz LA, Shipley M, Senior RM, Werb Z. Synergy between a plasminogen cascade and MMP-9 in autoimmune disease. J Clin Invest. 2005;115:879–87. doi: 10.1172/JCI23977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castaneda FE, Walia B, Vijay-Kumar M, Patel NR, Roser S, Kolachala VL, et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology. 2005;129:1991–2008. doi: 10.1053/j.gastro.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 45.Lighvani S, Baik N, Diggs JE, Khaldoyanidi S, Parmer RJ, Miles LA. Regulation of macrophage migration by a novel plasminogen receptor Plg-R KT. Blood. 2011;118:5622–30. doi: 10.1182/blood-2011-03-344242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miles LA, Lighvani S, Baik N, Andronicos NM, Chen EI, Parmer CM, et al. The plasminogen receptor, Plg-R(KT), and macrophage function. J Biomed Biotechnol. 2012;2012:250464. doi: 10.1155/2012/250464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miles LA, Dahlberg CM, Plescia J, Felez J, Kato K, Plow EF. Role of cell-surface lysines in plasminogen binding to cells: identification of α-enolase as a candidate plasminogen receptor. Biochemistry. 1991;30:1682–91. doi: 10.1021/bi00220a034. [DOI] [PubMed] [Google Scholar]
- 48.Kim J, Hajjar KA. Annexin II: a plasminogen-plasminogen activator co-receptor. Front Biosci. 2002;7:d341–8. doi: 10.2741/kim. [DOI] [PubMed] [Google Scholar]