

Abstract
Background
We examined the activation pattern of extracellular signal-regulated kinase 1 and 2 (ERK1/2) and its dependence on D1 versus D2 dopamine receptors in hemiparkinsonian rats treated with 3,4-dihydroxyphenyl-L-alanine (L-DOPA).
Methods
6-Hydroxydopamine-lesioned rats were treated acutely or chronically with L-DOPA in combination with antagonists for D1 or D2 receptors. Development of dyskinesia was monitored in animals receiving chronic drug treatment. Phosphorylation of ERK1/2, mitogen- and stress-activated protein kinase-1 (MSK-1), and the levels of FosB/ΔFosB expression were examined immunohistochemically.
Results
L-DOPA treatment caused phosphorylation of ERK1/2 in the dopamine-denervated striatum after acute and chronic administration. Similar levels were observed in matrix and striosomes, and in enkephalin-positive and dynorphin-positive neurons. The severity of dyskinesia was positively correlated with phospho-ERK1/2 levels. Phosphorylation of ERK1/2 and MSK-1 was dose-dependently blocked by SCH23390, but not by raclopride. SCH23390 also inhibited the development of dyskinesia and the induction of FosB/ΔFosB.
Conclusions
L-DOPA produces pronounced activation of ERK1/2 signaling in the dopamine-denervated striatum through a D1-receptor-dependent mechanism. This effect is associated with the development of dyskinesia. Phosphorylated ERK1/2 is localized to both dynorphinergicand enkephalinergic striatal neurons, suggesting a general role of ERK1/2 as a plasticity molecule during L-DOPA treatment.
Keywords: Gene transcription, motor complications, Parkinson’s disease, rodent, signaling pathways, therapy
The dopamine (DA) precursor 3,4-dihydroxyphenyl-L-alanine (L-DOPA) is still the most effective and commonly used treatment for Parkinson’s disease (PD), although its long-term use is hampered by serious complications, such as abnormal involuntary movements (dyskinesia), in a majority of patients (Marsden 1994; Nutt 1990). The primary cause of L-DOPA–induced dyskinesia is not known, but chronic dyskinesiogenic treatment with L-DOPA is closely paralleled by large changes in striatal gene expression in both rodent and primate models of Parkinson’s disease (Aubert et al. 2006; Brotchie et al. 1998; Cenci et al. 1998; Doucet et al. 1996; Konradi et al. 2004). The intracellular signaling pathways mediating this aberrant gene response have yet to be determined.
Extracellular signal-regulated kinases 1 and 2 (ERK1/2) are critical mediators of activity-dependent plasticity as demonstrated by their role in long-term potentiation (English and Sweatt 1996; Impey et al. 1998; Mazzucchelli et al. 2002), classical conditioning (Crow et al. 1998), and memory formation (Atkins et al. 1998; Brambilla et al. 1997). Phosphorylation of ERK1/2 reflects a balance between the activities of upstream kinases and inactivating phosphatases (Keyse 2000). Active ERK1/2 mediate changes in gene expression via phosphorylation of histone kinase proteins, such as mitogen- and stress-activated protein kinase-1 (MSK-1; Deak et al. 1998; Dunn et al. 2005) and activation of nuclear transcription factors, including Elk-1 and CREB (Marshall 1995).
Previous studies reported that selective agonists for D2-type DA receptors enhanced ERK1/2 phosphorylation in the DA-denervated striatum (Cai et al. 2000; Zhen et al. 2002). By contrast, Gerfen and collaborators (2002) showed that agonists of D1- but not D2 receptors induced ERK1/2 phosphorylation in striatal medium-sized neurons of hemiparkinsonian rats. Both reports suggested that increased ERK1/2 activity in striatal neurons is a possible mechanism of L-DOPA–induced dyskinesia (LID).
Here we examine the DA receptor-dependence of ERK1/2 phosphorylation in hemiparkinsonian rats treated with L-DOPA, and we compare it with ΔFosB, an established molecular marker of dyskinesia in this model (Andersson et al. 1999; Cenci et al. 1999; Westin et al. 2001). The fosB gene contains a SRE regulatory element in its promotor sequence (Lazo et al. 1992), which is a potential target for ERK1/2-dependent regulation. We provide the first evidence of an association between the striatal levels of ERK1/2 phosphorylation and the severity of the dyskinetic motor response evoked by L-DOPA. Furthermore, we show that the striatal activation of ERK1/2-dependent signaling by L-DOPA relies on D1- and not D2-type DA receptors and that this class of receptors is also essential for the development of dyskinesia and for the induction of ΔFosB-like proteins. Finally, we describe a broad striatal distribution of ERK1/2 activation by L-DOPA, encompassing both the dynorphinergic and the enkephalinergic striatal efferent populations.
Methods and Materials
Detailed protocols can be found in Supplement 1.
Subjects
Female Sprague–Dawley rats (225 g; Harlan, Zeist, The Netherlands) were housed under a 12-hour light–dark cycle, with ad libitum access to food and water. Animal care and treatment paradigms were approved by the Malmö-Lund Ethical Committee on Animal Research.
Dopamine Denervating Lesions
6-Hydroxydopamine (6-OHDA-HCl; Sigma-Aldrich, Stockholm, Sweden) was injected into the right-ascending DA fiber bundle, as previously described (Cenci et al. 1998). Two weeks postlesion, striatal denervation was evaluated using amphetamine-induced rotation (2.5 mg/kg D-amphetamine intraperitoneal injection [IP]). Rats showing greater than 5 full turns/min ipsilateral to the lesion, corresponding to ≥ 90% striatal dopamine depletion (Carta et al., 2006; Winkler et al. 2002), were selected for this study. The extent of the lesion was verified through immunohistochemical staining for tyrosine hydroxylase (TH) of DA axon terminals in the striatum and DA cell bodies in the substantia nigra.
Drug Treatment
Administration of L-DOPA methyl ester (10 mg/kg; Sigma-Aldrich) and the peripheral DOPA-decarboxylase inhibitor benserazide-hydrochloride (15 mg/kg; Sigma-Aldrich) was initiated 3 weeks postamphetamine screening, as described previously (Andersson et al. 2001; Cenci et al. 1999). The D1 receptor antagonist SCH23390 (.25 mg/kg or .05 mg/kg in .9% NaCl, IP; Sigma-Aldrich) or the D2 receptor antagonist raclopride (2.0 mg/kg in .9% NaCl IP; Sigma-Aldrich) were administered IP 30 min before L-DOPA.
Experimental Design
Animals with 6-OHDA lesions were randomly assigned to acute L-DOPA (12 days of saline followed by L-DOPA on Day 13), chronic L-DOPA (13 daily L-DOPA injections), or saline treatment (13 daily saline injections). Chronically treated animals were tested as previously described (Cenci et al. 1998; Cenci and Lundblad 2005; Lundblad et al. 2002) and labeled as “dyskinetic” or “nondyskinetic” (see Supplement 1). Animals defined as nondyskinetic exhibited either no or only occasional abnormal involuntary movements (AIMs) of minimal severity. The development of AIMs during chronic L-DOPA treatment in animals classified as dyskinetic or nondyskinetic is shown in Supplement 2.
Twenty-two animals (n = 6 dyskinetic, 4 nondyskinetic, 8 acute, and 4 saline control animals) were prepared for Western immunoblotting 20 min or 3 hours after the last L-DOPA injection, and 49 animals were processed for immunohistochemistry (IHC) at 20 min (n = 8 chronic and 7 acute), 60 min (n = 8 chronic and 8 acute), 120 min (n = 4 chronic and 3 acute), or 24 hours (n = 8 chronic and 3 acute) after the last L-DOPA injection.
For the studies on DA receptor subtype involvement, rats were treated with either the D1 receptor antagonist SCH23390 or the D2 receptor antagonist raclopride 30 min before an acute L-DOPA challenge (n = 8 per group). As a reference group, eight rats received saline 30 min prior to L-DOPA. Control animals received injections of saline (n = 4), SCH23990 only (n = 4), or raclopride only (n = 6). All animals were killed 30 min after the challenge injection, and the brains were processed for IHC. A group of animals (n = 8) received an acute dose of the antiakinetic drug, bromocriptine (5 mg/kg IP), which mainly acts as a D2 agonist with some D1 antagonistic properties (Bedard et al. 1986; Lieberman and Goldstein 1985). Bromocriptine-treated rats were killed 90 min (n = 4) or 180 min (n = 4) after the drug injection, which represent the beginning and the plateau phase, respectively, in the pharmacodynamic curve in rats (Lundblad et al. 2002).
In a chronic coadministration experiment, animals received SCH23390 (.25 mg/kg, n = 8; or .05 mg/kg, n = 8), 30 min before L-DOPA for 13 days. Control groups received saline 30 min before L-DOPA (n = 8) or SCH23390 (.05 mg/kg, n = 3; or .25 mg/kg, n = 3) followed by an injection of saline. All rats were killed 30 min after the last injection and the brains were processed for immunohistochemistry (IHC).
To verify the occurrence of L-DOPA–induced ERK1/2 activation in enkephalinergic neurons, three dyskinetic and three saline-treated rats were prepared for the combined detection of phospho-ERK1/2 by IHC and enkephalin mRNA by in situ hybridization.
Tissue Preparation for IHC
All animals were anesthetized with sodiumpentabarbitone (240 mg/kg body weight IP; Apoteksbolaget, Uppsala, Sweden) and transcardially perfused with 4% ice-cold, buffered (pH 7.4) paraformaldehyde (PFA; Merck, Stockholm, Sweden). Brains were rapidly extracted, postfixed for 2 hour in PFA, and transferred to 20% buffered sucrose for 24 hours; coronal sections were cut on a freezing microtome at 40-μm thickness or 16 μm for IHC in combination with in situ hybridization histochemistry (ISHH). The brains were cut between the level of nucleus accumbens (bregma +1.70) to caudal to substantia nigra (bregma −7.0; (Paxinos and Watson 1998). Free-floating sections were stored in cryoprotective solution at −20°C until further processing.
Western Immunoblotting
Striatal protein extracts were prepared from left and right striatum, and 2.3 μg protein from each rat was used for Western immunoblotting (protocol in Supplement 1).
The primary antibodies used were rabbit polyclonal antiphospho(Thr202/Tyr204) p44/42MAPK, 1:1000; and rabbit polyclonal anti-p44/42MAPK,1:2000 (both from Cell Signaling Technology, Beverley, Massachusetts). All blots were stripped and reprobed with 1:20,000 anti-βactin (Sigma).
Bright-Field Immunohistochemistry
Immunohistochemistry was performed as in Cenci et al. (1999) using a peroxidase-based detection method and 3-3′-diaminobenzidine (DAB; Sigma-Aldrich) as the chromogen. The following primary antisera were used: antiphospho(Thr202/ Tyr204) p44/42MAPK (1:250 for 36 hours; Cell Signaling Technologies), rabbit monoclonal antiphospho(Ser376) MSK-1 (1:200 for 48 hours; Epitomics, Burlingame, California), or goat anti-FosB/ΔFosB (1:10000 for 16 hours; Santa Cruz Biotechnology, Santa Cruz, California).
Two-Color Dual Antigen Fluorescence Immunohistochemistry
For dual-antigen immunofluorescence, sections were first stained for phospho-ERK1/2 using a mouse monoclonal antibody (1:100; Cell Signaling Technologies) followed by one of the following antisera: rabbit polyclonal against met-enkephalin (1:1000; BioSorin, Saluggia, Italy), rabbit polyclonal antidynorphin (1:2000, kindly provided by Dr. L. Terenius, Uppsala, Sweden), or rabbit monoclonal against phospho(Ser376) MSK-1 (1:100; protocol in Supplement 1).
Combined Immunohistochemistry and In Situ Hybridization Histochemistry
To verify the occurrence of L-DOPA–induced phospho-ERK1/2 in enkephalinergic striatal neurons, bright-field IHC staining for phospho-ERK1/2 was combined with the detection of enkephalin mRNA by radioactive in situ hybridization (protocol in Supplement 1). Sections were stained for phospho-ERK1/2 according to the standard protocol, except that all buffers used were autoclaved to minimize RNA degradation, blocking solution was made up of 3% BSA (Fraction V, Sigma-Aldrich), and blocking time was reduced to 20 min.
Image Analysis and Cell Counts
Western immunoblots were developed on autoradiographic films (Kodak, Rochester, New York), scanned, and analyzed with the Image J software (http://rsb.info.nih.gov/ij/index.html). The optical density was determined on each band of interest after the images had been calibrated using a Kodak photographic step tablet according to the software instructions. Optical density data were expressed as a percentage of saline control values within the same experiment, unless otherwise stated.
Cells immunoreactive for phospho-ERK1/2 and phospho-MSK-1 were counted by a blinded investigator using the Image J software. Sample areas (.410 × .328 mm) were visualized under a 20× objective in a Nikon Eclipse 80i microscope (Nikon, Tokyo, Japan) and digitized through a Nikon DMX 1200F video camera. Analysis was carried out in both the medial and lateral part of the caudate–putamen (CPu) in 16 sample areas per animal, distributed in two sections through the mid-rostrocaudal levels of the structure (see Figure 4E). Data are expressed as the number of immunoreactive cells per square mm.
Figure 4.
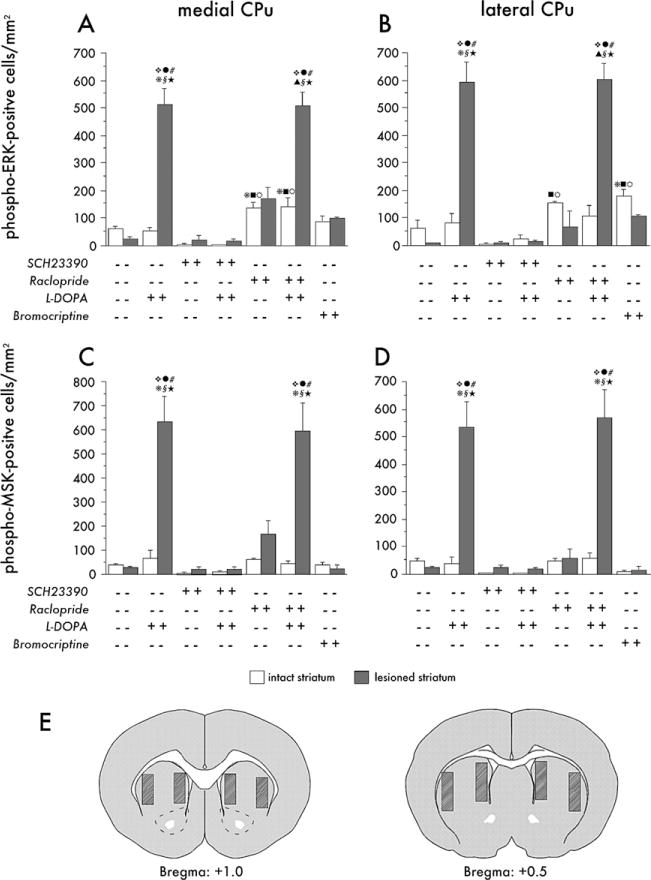
Quantitative analysis of phospho-extracellular signal-regulated kinases 1 and 2(ERK1/2) (A, B) and phospho-mitogen- and stress-activated protein kinase-1 (MSK-1) (C, D) immunoreactivity in the medial (A, C) and the lateral part (B, D) of the caudate-putamen (CPu). Animals were euthanized 30 min after an acute injection of 3,4-dihydroxyphenyl-L-alanine (L-DOPA), alone or combined with the D1-antagonist, SCH23390, or the D2-antagonist, raclopride. The pair of bars farthest to the right in each diagram illustrate the effects of bromocriptine. Grey bars represent the lesioned hemisphere and white bars the intact hemisphere in each group. Data are expressed as number of immunoreactive cells per square millimeter. Cell counting was carried out at the two rostrocaudal levels represented by the drawings (E), where hatched areas indicate the medial and lateral regions sampled. p < .05 versus the lesioned side of, § vehicle treatment (no drug); # SCH23390 only; ϖ SCH23390 + L-DOPA; λ raclopride only, ★ Bromocriptine.p < .05 versus the intact side of * L-DOPA only; σ Raclopride + L-DOPA; v SCH23390 only;
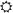 SCH23390 + L-DOPA; υ Bromocriptine (Honestly Significant Difference test after two-factor analysis of variance, where p< .001 for the effects of group, side, and the group-side interaction in each A, B, C, and D).
SCH23390 + L-DOPA; υ Bromocriptine (Honestly Significant Difference test after two-factor analysis of variance, where p< .001 for the effects of group, side, and the group-side interaction in each A, B, C, and D).
Antigen colocalization on immunofluorescent sections were analyzed using a Zeiss Confocal laser scanning microscope and the LSM 5 PASCAL software (Carl Zeiss, Heidelberg, Germany). Sampling was done on two sections per animal (480 μm apart) from mid-rostrocaudal CPu. Sample areas were selected using a 40× objective and magnified threefold before scanning. The sample areas were systematically moved along the dorsoventral axis of the section at two mediolateral coordinates (approximately 1.25 and 2.25 mm lateral to the midline, respectively), randomly covering both striosomal and matrix compartments. Cells were evaluated in the z dimension with a minimum of five consecutive optical sections. One hundred cells were sampled from the medial part and the lateral part of the striatum. Sections immunostained for phospho-ERK1/2 and subsequently hybridized for PPE mRNA were examined under bright-field optics.
Statistical Analyses
Counts of phospho-ERK1/2 and phospho-MSK1 immunoreactive cells in the intact and lesioned striatum were analyzed by two-factor analysis of variance (ANOVA; group × side), followed by Tukey’s Honestly Significant Difference (HSD) test. Western blots and counts of immunoreactive cells on the DA-denervated side of the striatum were analyzed by one-factor ANOVA followed by post hoc Newman–Keuls test. Global AIMs scores were analyzed by repeated-measures ANOVA, and relevant differences within or between groups were analyzed pairwise by Tukey’s HSD test. The null hypothesis was rejected when p < .05.
Results
Striatal ERK1/2 Activation after Acute or Chronic L-DOPA Treatment
Levels of phosphorylated (Figure 1A) and total ERK1/2 (Figure 1B) were assessed in tissue lysates from the DA-denervated striatum from rats treated with L-DOPA either chronically or acutely. Both antibodies revealed two bands with the expected molecular weights of ERK1 (44 kDa) and ERK2 (42 kDa), with ERK2 as the prominent isoform (Mazzucchelli et al. 2002).
Figure 1.
Western immunoblots and bar histograms of phospho-extracellular signal-regulated kinases 1 and 2 (ERK1/2), total ERK1/2 and β-actin levels in dopamine-denervated striata after treatment with 3,4-dihydroxyphenyl-L-alanine (L-DOPA) or saline. (A) Phospho-ERK1 (thin band) and phospho-ERK2 (thicker band) were upregulated in chronically L-DOPA-treated dyskinetic animals (d) compared with nondyskinetic cases (n) and saline-injected controls (s). Chronically L-DOPA-treated rats were killed 20 min after the last injection. After acute L-DOPA treatment, ERK1/2 phosphorylation was significantly elevated at 20 min (group a), but not 3 hours postinjection (group A). Data are expressed as a percentage of the optical density (O.D.) levels of saline control animals. * p < .05 versus saline-treated control group, # p < .05 versus chronic, nondyskinetic L-DOPA group, §p < .05 versus acute L-DOPA group (Newman–Keuls test after one-factor analysis of variance, where group effect = .002). (B and C) The total levels of ERK1/2 and β-actin did not differ between any of the experimental groups. (D) Simple regression of phospho-ERK1/2 O.D. levels on the cumulative axial, limb, and orolingual AIM scores recorded from chronically L-DOPA treated rats (groups d and n) shows that ERK1/2 phosphorylation was positively correlated with dyskinesia severity. Pearson’s correlation coefficient (R) and the probability value of the regression (p) are given in the bottom right corner.
Acute administration of L-DOPA significantly increased ERK1/2 phosphorylation in the lesioned striatum 20 min, but not 3 hours postinjection (Figure 1A). Among chronically L-DOPA treated rats (killed 20 min after the last drug injection), dyskinetic animals had 50% higher levels of ERK1/2 phosphorylation than non-dyskinetic animals (Figure 1A). No significant differences in ERK1/2 protein levels (Figure 1B) or β-actin levels (Figure 1C) were found. Accordingly, a similar pattern of group differences was found when phospho-ERK1/2 levels were expressed as a ratio over β-actin (170 ± 10 for dyskinetic rats; 124 ± 15 for nondyskinetic rats; 138 ± 19 for acute 20 min; 117 ± 12 for acute 3 hours; and 101 ± 8 for saline-treated control rats; p = .002, one-factor ANOVA).
A positive linear relationship was observed between the severity of dyskinesia and the amount of phospho-ERK1/2 in the lesioned striatum in chronically L-DOPA treated rats (Figure 1D). In the intact hemisphere, striatal levels of phospho-ERK1/2, total ERK1/2, and β-actin were unaffected by acute or chronic L-DOPA treatment and/or by the occurrence of dyskinesia (data not shown).
Time Course and Distribution of L-DOPA–Induced ERK1/2 Phosphorylation
The distribution and amount of phospho-ERK1/2 expression were evaluated semiquantitatively at the onset (20 min), the peak (60 min), and the decline phase (120 min) of the L-DOPA dosing cycle in rats treated with the drug either acutely or chronically and compared with the pattern in animals killed 24 hours after the last (or only) injection of L-DOPA.
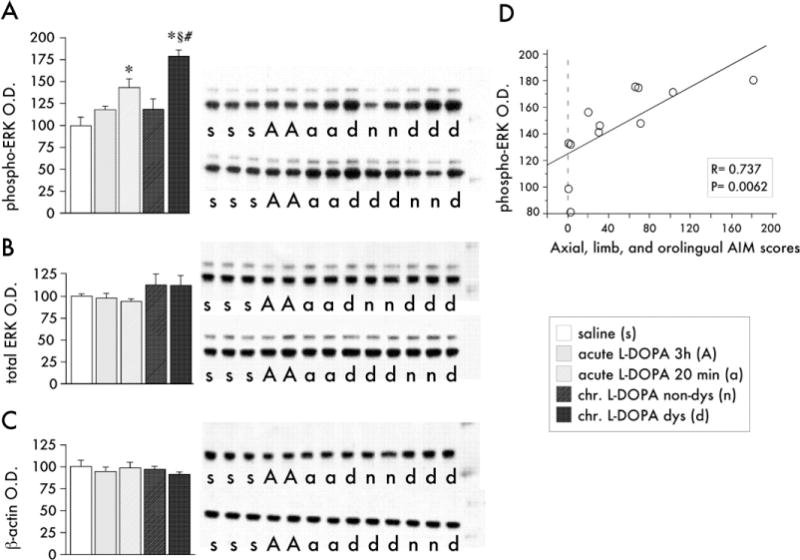
The distribution of phospho-ERK1/2 did not show any consistent difference between the medial and the lateral part of the striatum after either acute or chronic L-DOPA administration (Figure 2; see also Figures 3 and 4). In rats treated acutely with L-DOPA, phospho-ERK1/2 immunoreactivity was induced at 20 and 60 min post– drug injection (Figure 2 A, 2A′, 2B, and 2B′), and decreased visibly by 120 min (Figure 2C and 2C′). In the chronically L-DOPA–treated group, levels of phospho-ERK1/2 immunoreactivity were upregulated 20 and 60 min after the injection (Figure 2E–2F′) and remained elevated also at the 120 min time point (Figure 2G and 2G′). One animal per time point in the chronic L-DOPA group did not develop dyskinesia, and these rats displayed much fewer phospho-ERK1/2 immunoreactive cells. Twenty-four hours after the final L-DOPA injection, phospho-ERK1/2 was no longer detectable in either the acute or the chronic L-DOPA groups (Figure 2D and 2D′ and 2H and 2H′).
Figure 2.
Photomicrographs of phospho-extracellular signal-regulated kinases 1 and 2 (ERK1/2) immunoreactive cells in the medial and lateral part of the caudate-putamen at 20, 60, and 120 min, and 24 hours after acute (A–D and A′–D′) or chronic 3,4-dihydroxyphenyl-L-alanine (L-DOPA) treatment with dyskinesia (E–H and E′–H′). Photos were taken on the side of the striatum ipsilateral to the 6-hydroxydopamine lesions. Scale =100 μm.
Figure 3.
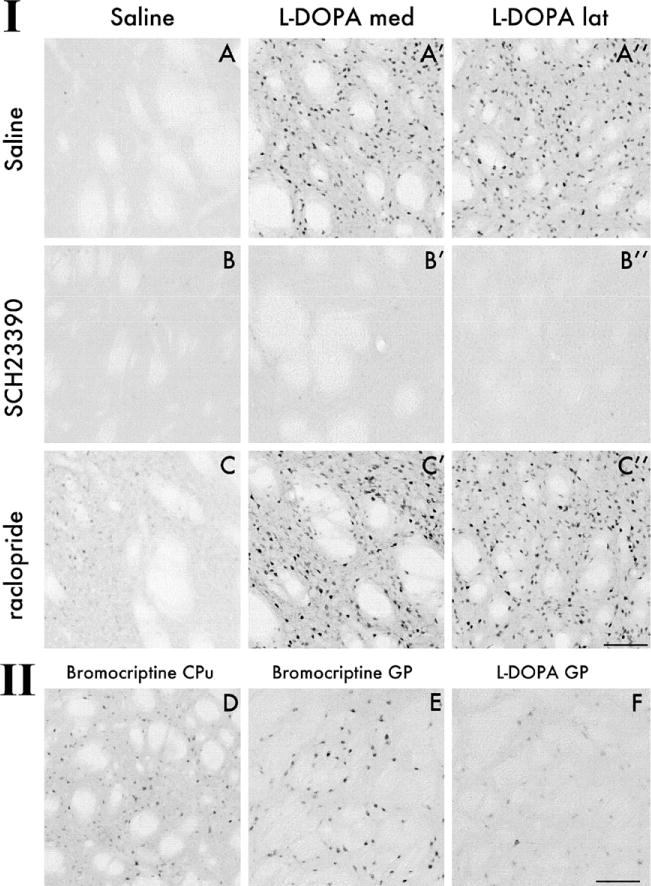
Photomicrographs illustrating the effects of D1-and D2-receptor antagonist pretreatment on extracellular signal-regulated kinases 1 and 2 (ERK1/2) phosphorylation as assessed 30 min after an acute challenge injection of 3,4-dihydroxyphenyl-L-alanine (L-DOPA) (Panel I). Each animal was given two injections in sequence, which are specified by the captions on left-hand side (first injection) and on the top of the panel (second injection). In both the lateral and the medial striatum, administration of SCH23390 30 min before L-DOPA completely inhibited phospho-ERK1/2 immunoreactivity (B′ and B″), whereas raclopride administration had no effect (C′ and C″, cf. with L-DOPA alone in A′ and A″). Photomicrographs in Panel II illustrate the effects of bromocriptine, an antiparkinsonian drug that does not induce dyskinesia. Although having only a weak stimulatory effect on phospho-ERK1/2 in the striatum (D), bromocriptine induced strong neuronal immunoreactivity for phospho-ERK1/2 in the globus pallidus (GP) (E), a structure where L-DOPA did not have any effect (F). The bromocriptine-treated animal illustrated in this picture was euthanized 90 min postinjection, a time point at which the motor effects of the drug in this rat model start to become appreciable. All pictures were taken on the side of the striatum ipsilateral to the 6-hydroxydopamine lesion. Scale = 100 μm.
Effects of SCH23390 and Raclopride
The effect of selective antagonists for D1- or D2-type receptors was examined with SCH23390 and raclopride, respectively. As a marker of signaling pathway activation downstream of ERK1/2, we studied phospho(Ser376) MSK-1 (Deak et al. 1998), a histone H3 kinase that plays an essential role in ERK1/2-dependent chromatin remodeling and transcriptional changes (Dunn et al. 2005). Although MSK-1 can also be phosphorylated by p38MAPK kinase (Dunn et al. 2005), this pathway is not recruited by L-DOPA under our experimental conditions (data not shown). Confocal microscopy showed that phospho-ERK1/2 and phospho-MSK-1 were colocalized (see Figure 7H–7H″).
Figure 7.
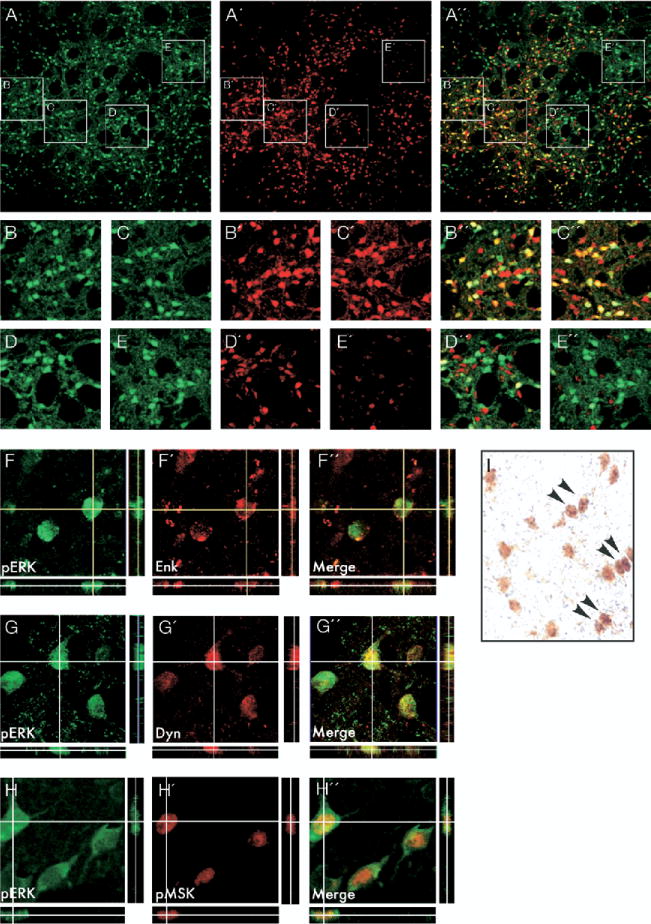
Distribution of phospho-extracellular signal-regulated kinases 1 and 2 (ERK1/2) immunoreactive neurons in a dyskinetic rat relative to the striosome and matrix compartment of the striatum. The matrix is identified by high levels of calbindin expression. Photomicrograph of a striatal section double-stained for phospho-ERK1/2 (A), and calbindin (A′). The merged image is shown in A″. Boxed insets (B–E; B′–E′, and B″–E″) are shown at higher magnification below. Colocalization of phospho-ERK1/2 with the peptide markers enkephalin and dynorphin is illustrated by confocal three-dimensional pictures in F–F′ and G–G″, respectively. Colocalization of phospho-ERK1/2 with preproenkephalin (PPE) mRNA is shown in the bright-field photomicrograph in I. Positive labeling for PPE mRNA appears as a dense cluster of black (silver) grains, and arrowheads show examples of double-labeled cells. Confocal photomicrographs in H–H″ show cells colabeled for phospho-MSK-1 and phospho-ERK1/2 under the confocal microscope. All phospho-MSK-1 immunoreactive cells stained positively also for phospho-ERK1/2.
The dose of .25 mg/kg SCH23390 was used to yield a maximal D1 receptor occupancy without losing selectivity (Bischoff et al. 1986), and 2.0 mg/kg raclopride was chosen because it has been proven sufficient to block D2 receptor-mediated cellular responses in the striatum (Paul et al. 1992). The behavioral effects of the selected doses were evaluated in a preliminary experiment (see Supplement 1).
In all the experimental animals, the antagonists were given 30 min before an acute injection of L-DOPA, and the rats were killed 30 min later to study striatal ERK1/2 and MSK-1 activation by IHC. Counts of phospho-ERK1/2 and phospho-MSK-1 immunoreactive cells were carried out in the medial and the lateral part of the CPu on both sides of the brain. When administered alone, L-DOPA induced a highly significant 10- to 20-fold increase in the number of phospho-ERK1/2 and MSK-1 immunoreactive cells in all aspects of the DA-denervated CPu (Figure 3A–A″; phospho-MSK-1 is shown in photomicrographs in Supplement 3, panel IA–A″) whereas the intact side was not affected at all (Figure 4A–4D). The induction of phospho-ERK1/2 and phospho-MSK-1 by L-DOPA was completely inhibited in animals pretreated with SCH23390 (Figures 3 and Supplement 3, panels B′ and B″), whereas raclopride had no effect (Figures 3 and Supplement 3, panels C′ and C″). Either alone or combined with L-DOPA, raclopride induced a significant increase in the number of phospho-ERK1/2 immunoreactive cells in the intact but not the denervated striatum (Figure 4A and 4B), which is in agreement with the report by Gerfen and coworkers (2002) that D2 receptor antagonists require an intact nigrostriatal system to activate ERK1/2 in striatal neurons. Raclopride alone did not however, produce a significant change in the number of phospho-MSK-1 positive cells on either the intact or the DA-denervated side of the striatum (Figure 4C and 4D). SCH23390 alone had no effect on the number of phospho-ERK1/2 or phospho-MSK-1 positive cells (Figure 4A and 4D).
Striatal ERK1/2 and MSK-1 phosphorylation was also examined after treatment with bromocriptine, an antiparkinsonian drug that produces intense motor activation without dyskinesia (Lundblad et al. 2002). A high dose of bromocriptine (5 mg/kg; Lundblad et al. 2002) induced some weak cellular staining for phospho-ERK1/2 (Figure 3D) in both the intact and the denervated striatum, and the effect reached significance in the lateral CPu on the intact side (Figure 4B). Interestingly, bromocriptine produced strong ERK1/2 activation in neurons in the globus pallidus (GP; Figure 3E), a structure where neither L-DOPA (Figure 3F) nor any of the other treatments tested had induced phospho-ERK1/2. Bromocriptine did not raise the number of phospho-MSK-1 immunoreactive cells above control levels in either the CPu (Figures 4C and 4D, and Supplement 3) or the GP (Supplement 3).
Chronic Treatment with SCH23390 in Combination with L-DOPA Inhibited the Development of Dyskinesia
To examine the effects of D1-antagonist treatment on dyskinesia, animals were injected with either of two doses of SCH23390 (.05 or .25 mg/kg/day) or with saline, 30 min before their daily L-DOPA injection over 13 days. Ratings of AIMs were carried out daily. The D1 antagonist suppressed the development of dyskinesia in a dose-dependent manner (Figure 5A). The reduction in global AIMs per session by SCH23390 was due to decreased severity of dyskinesia (lower AIMs score per observation point) as well as to a shortening of dyskinesia duration (Figure 5B). The effect of SCH23390 cannot be ascribed to general motor suppression, which lasted for only 1 hour in our preliminary experiments (cf. Supplement 1). Moreover, the antagonist was injected 50 min before the first observation point in the session and could thus have affected only the initial score.
Figure 5.
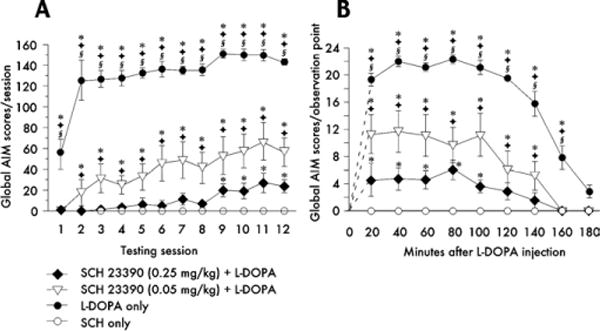
Effects of SCH23390 coadministration on 3,4-dihydroxyphenyl-L-alanine (L-DOPA)-induced abnormal involuntary movements (AIMs). The evolution of AIM scores during 12 days of L-DOPA treatment is shown in A. Animals receiving saline instead of SCH23390 before the daily L-DOPA dose showed a rapid development of dyskinesia (filled circles). Pretreatment with .05 mg/kg SCH23390 partially inhibited the development of L-DOPA-induced AIMs, whereas .25 mg/kg SCH23390 completely suppressed dyskinesia during the first 8 days of treatment. The time plot in B shows the AIM scores recorded at each observation point (20–180 min) during test session 11. SCH23390 dose-dependently reduced both the severity of the AIMs at each observation point and their duration. p < .05 versus *SCH23390 only, Φ .25 mg SCH23390 + L-DOPA, § .05 mg/kg SCH23390 + L-DOPA (Honestly Significant Difference test after repeated-measures analysis of variance, in which p < .001 for the effects of group, time, and the group–time interaction in both A and B).
SCH23390 Inhibited ERK1/2 and MSK-1 Phosphorylation and the Induction of FosB/ΔFosB by Chronic L-DOPA
Striatal levels of phospho-ERK1/2, phospho-MSK-1, and FosB/ΔFosB were analyzed by IHC in the animals pretreated with SCH23390 during chronic L-DOPA administration, which were euthanized 30 min after the final drug injection. SCH23390 suppressed the activation of ERK1/2 and MSK-1 by L-DOPA in a dose-dependent manner in both the medial and the lateral part of the CPu (photomicrographs in Supplement 4; quantification in Figure 6A–D). In animals cotreated with L-DOPA and the higher dose of the antagonist, levels of phospho-ERK1/2 and phospho-MSK-1 did not differ from control values (Figure 6A–6D). The lower dose of SCH23390 could not fully inhibit L-DOPA–induced activation of phospho-ERK1/2 in the striatum (Supplement 4 and Figures 6A and 6B). No correlation was found between this residual number of phospho-ERK1/2 immunoreactive cells and the severity of dyskinesia (p = .38, regression analysis of AIMs score and number of phospho-ERK1/2 immunoreactive cells in the low-dose SCH23390 group).
Figure 6.
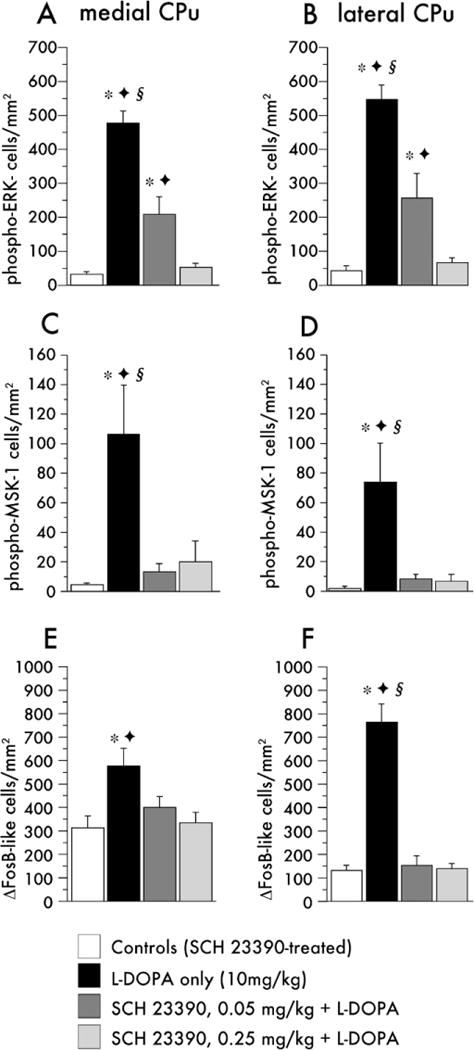
Quantitative analysis of phospho-extracellular signal-regulated kinases 1 and 2 (ERK1/2) (A, B), phospho-mitogen- and stress-activated protein kinase-1 (MSK-1) (C, D) and FosB/ΔFosB immunoreactivity (E, F) in the DA-denervated caudate-putamen (CPu) after chronic treatment with 3,4-dihydroxyphenyl-L-alanine (L-DOPA) and SCH23390. Note that the scaling on the y axes differs among markers to match their different absolute levels of expression. p < .05 versus *SCH23390 only, Φ .25mg SCH23390 + L-DOPA, § .05 mg/kg SCH23390 + L-DOPA (Newman–Keuls test after one-factor analysis of variance, where p ≤ .01 for the effect of group in each comparison).
FosB/ΔFosB immunoreactivity was increased in dyskinetic animals in the patchy striosomes of the medial striatum and along the rim of the lateral CPu, as reported earlier (Andersson et al. 1999; Cenci et al. 1999). The distribution of FosB/ΔFosB immunoreactivity thus was different from phospho-ERK1/2 and phospho-MSK-1, which were rather uniform across striatal compartments and regions (Supplement 4, compare B and B′, F and F′, and J and J′). Interestingly, the induction of FosB/ΔFosB by chronic L-DOPA treatment was completely inhibited by both doses of SCH23390 (Supplement 4; Figure 6E and 6F).
In Dyskinetic Rats, Phospho-ERK1/2 Was Present in Dynorphinergic and Enkephalinergic Neurons, in Striosomes, and in the Matrix
L-DOPA–induced dyskinesia has been suggested to require a preferential activation of the striosome versus the matrix compartment (Graybiel et al. 2000) and of the dynorphinergic versus the enkephalinergic striatal efferent pathway (Andersson et al. 1999; Sgambato-Faure et al. 2005; St-Hilaire et al. 2005). We thus examined the compartmental distribution and cellular phenotype of striatal ERK1/2 activation in four chronically L-DOPA–treated dyskinetic rats. Dual-antigen immunofluorescence for phospho-ERK1/2 and the matrix marker, Calbindin D28 (Kincaid and Wilson 1996; Moratalla et al. 1996), revealed no difference in either the density or the staining intensity of phospho-ERK1/2 immunoreactive cells between calbindin-rich (matrix) and calbindin-poor (striosomes) areas (Figure 7A–E″).
Dual-antigen immunofluorence for phospho-ERK1/2 and either dynorphin or met-enkephalin was used to determine the distribution of activated ERK1/2 in the two main efferent populations of striatal neurons (Gerfen 1992). In agreement with previous observations (Cenci et al. 1999), the distribution of dynorphin immunoreactivity in the DA-denervated striatum of dyskinetic animals was patchy in the medial striatum but exceeded the striosomal borders in the lateral part of the CPu. The distribution of enkephalin immunoreactivity was even in all striatal subregions (data not shown). Analysis of antigen colocalization was carried out by random selection of 200 phospho-ERK immunoreactive cells per peptide marker per rat along the dorsoventral coordinate in both the medial and the lateral parts of the CPu. Of the phospho-ERK1/2 positive cells sampled in this way, similar numbers stained positively for enkephalin (43.2% ± 5.3%; Figure 7F–F″) and for dynorphin (52.0% ± 5.5%; Figure 7G–G″).
The occurrence of phospho-ERK1/2 in enkephalinergic neurons was verified in a group of 4 dyskinetic animals in which phospho-ERK1/2 was detected by IHC and preproenkephalin (PPE) mRNA by radioactive in situ hybridization. Positive immunostaining for phospho-ERK1/2 occurred in a large number of neurons that contained dense clusters of 35S-generated silver grains (> 30 grains per cell), indicating labeling by the PPE probe (Figure 7I).
Discussion
Phosphorylation of ERK1/2 was induced in DA-denervated but not intact striatal neurons after both acute and chronic treatment with L-DOPA. Phosphorylated ERK1/2 was detectable for as long as 2 hours but not at 24 hours after a drug dose. This induction kinetics rules out a possible accumulation of phospho-ERK1/2 in striatal neurons during the course of chronic L-DOPA treatment and implies de novo phosphorylation after each single drug injection. However, the temporal pattern of L-DOPA–induced ERK1/2 activation was prolonged compared with DA agonist-induced phospho-ERK1/2 in the intact striatum, which peaks after 5–15 min, declines at 30 min, and returns to baseline by 60 min (Gerfen et al. 2002; Sgambato et al. 1998a; Sgambato et al. 1998b; Valjent et al. 2000). The sustained activation of ERK1/2 by L-DOPA likely reflects the supersensitivity of DA receptor-dependent signaling that is produced by severe DA-denervating lesions.
In chronically L-DOPA treated rats, the extent of striatal ERK1/2 phosphorylation produced by the last drug dose was positively and strongly correlated with the AIMs scores recorded during the treatment period. Levels of ERK1/2 activation in chronically L-DOPA treated, nondyskinetic animals did not differ significantly from baseline values. Thus, the pronounced activation of ERK1/2 in DA-denervated striatal neurons provided a molecular counterpart to the induction of AIMs by L-DOPA. Because an increased phosphorylation of ERK1/2 was also produced by acute L-DOPA treatment (which does not elicit significant AIMs), our data suggest that the core signaling alteration associated with dyskinesia consists in an inability to desensitize the phospho-ERK1/2 response with repeated exposure to L-DOPA.
Both D1 and D2 receptor agonists have been reported to stimulate ERK1/2 phosphorylation (Cai et al. 2000; Gerfen et al. 2002; Zhen et al. 2002). In our study, the D2 receptor antagonist raclopride did not have an effect on L-DOPA–induced ERK1/2 activation. Similarly, L-DOPA–mediated induction of phospho(Ser376) MSK-1, a histone kinase that plays an essential role in the transcriptional changes brought about by ERK1/2 (Deak et al. 1998; Dunn et al. 2005), was not affected by raclopride. In contrast, the D1 receptor antagonist SCH 23390 completely blocked phosphorylation of both kinases. We further showed that SCH 23390 was able to inhibit ERK1/2 and MSK-1 activation after a chronic course of L-DOPA treatment, which leads to internalization and possible desensitization of D1 receptors (Fiorentini et al. 2006). Bromocriptine, an antiparkinsonian drug with strong motor-stimulating properties but no dyskinesiogenic potential (Lundblad et al. 2002), exerting agonistic activity at D2 but not D1 receptors (Bedard et al. 1993; De Keyser et al. 1995), did not produce a significant activation of ERK1/2 and MSK-1 in the DA-denervated striatum. Taken together, these data indicate that D2 receptors are not implicated in the induction of ERK1/2-dependent signaling by L-DOPA, and support an association between ERK1/2 activation and the supersensitivity of D1 receptor-mediated signaling that follows DA denervation (Gerfen et al. 2002). Increased signaling through D1 receptors is implicated in the abnormal regulation of molecular and synaptic responses in striatal neurons at the heart of L-DOPA–induced dyskinesia (Aubert et al. 2005; Konradi et al. 2004; Picconi et al. 2003). Our data suggest an involvement of ERK1/2 in these abnormal striatal responses. Indeed, while blocking ERK1/2 and MSK-1 phosphorylation, SCH 23390 produced a similar dose-dependent suppression of L-DOPA–induced AIMs and prevented the striatal upregulation of FosB/ΔFosB proteins, which is causally linked with dyskinesia development in this rat model (Andersson et al. 1999, 2001).
Unexpectedly, the striatal pattern of phospho-ERK1/2 in dyskinetic rats differed greatly from the distribution of FosB/ΔFosB immunoreactivity. Moreover, although FosB/ΔFosB is selectively expressed in dynorphinergic neurons (Andersson et al. 1999; Cenci et al. 1999), the activation of phospho-ERK1/2 by L-DOPA in dyskinetic rats did not show selectivity for dynorphinversus enkephaline-positive cells. The great efficacy of the D1 antagonist in blocking all ERK1/2 activation is somewhat difficult to explain when considering that enkephalinergic cells express predominantly D2 receptors (Gerfen et al. 1992). Previous studies have shown a positive modulation of gene expression in striatal enkephalin neurons by muscarinic receptor activation (Wang and McGinty 1996). However, it is highly unlikely that treatment with L-DOPA would lead to an increased activation of striatal muscarinic receptors in our model (Jackson et al. 1993). The most likely explanation for the phospho-ERK1/2 response in enkephalin neurons implicates an activation of polysynaptic, corticobasal ganglionic circuits. L-DOPA–induced dyskinesia is indeed associated with an excessive activation of motor and premotor cortical areas (Rascol et al. 1998), and cortical activation induces immediate-early gene expression (Berretta et al. 1997) and ERK1/2 phosphorylation (Gerfen et al. 2002) predominantly in “indirect pathway” neurons. The importance of cortical afferents in activating these neurons has been further demonstrated by Ferguson and Robinson (2004). In their study, treatment of rats with amphetamine induced c-fos both in enkephalin-positive and in enkephalin-negative neurons within the striatum, but only the response evoked in the enkephalin-positive population was dependent on the integrity of the corticostriatal pathway. Interestingly, this response was also dependent on the activation of ERK1/2 (Ferguson and Robinson 2004). Supporting a facilitatory role of striatal D1 receptors on cortical function, Steiner and Kitai (2000) have shown that local intrastriatal antagonism of D1 receptors results in blockade of DA-dependent immediate-early gene expression both in the striatum and in the cerebral cortex.
The molecular adaptations produced by chronic L-DOPA treatment in indirect pathway neurons are poorly understood. Some recent studies performed on rats with 6-OHDA lesions have specifically addressed the relative localization of L-DOPA–induced changes in striatal gene expression to dynorphinergic or enkephalinergic neurons and have consistently found a selective upregulation of transcription factors and plasticity genes in the former population (Carta et al. 2005; Sgambato-Faure et al. 2005; St-Hilaire et al. 2005). Although it activates the transcription of plasticity genes in direct pathway neurons, chronic treatment with L-DOPA can also normalize denervation-induced changes in gene expression in enkephalin-positive neurons (Aubert et al. 2006), and it is tempting to propose involvement of ERK1/2 in these effects. Alternatively, ERK1/2 activation in enkephalin-positive neurons may not result in alterations of gene expression. Studies in several model systems have clearly shown that cellular activation of ERK1/2 is not in itself sufficient to cause changes at the nuclear level, but that the duration of and the signaling context in which this response occurs are crucial in determining the gene transcription outcome (Caunt et al. 2006; Impey et al. 1998). After treatment with L-DOPA, a simultaneous activation of ERK1/2 and other positive modulators of nuclear signaling, such as the PKA-DARPP 32 pathway (Greengard et al. 1998), occurs in striatal neurons that bear D1 but not D2 receptors. This may explain the occurrence of large gene expression changes in one striatal efferent population but not in the other one, despite similar levels of ERK1/2 activation.
In conclusion, our results show that treatment with L-DOPA in dyskinetic animals produces a pronounced, sustained, and broad striatal activation of ERK1/2 signaling. This response is dependent on D1 receptors but occurs in both dynorphinergic (D1-receptor-rich) and enkephalinergic (D2-receptor-rich) striatal neurons. Activation of ERK1/2 in dynorphinergic neurons is likely implicated in the abnormal molecular changes associated with LID (Andersson et al. 1999, 2001; Cenci et al. 1998). The significance of ERK1/2 activation in enkephalinergic neurons is currently unknown, but our findings provide the first evidence of modulation of intracellular signaling processes by L-DOPA in this cellular population in an animal model of PD.
Supplementary Material
Acknowledgments
This work was supported by grants from the Michael J. Fox Foundation for Parkinson’s Research, the Elsa and Thorsten Segerfalk Foundation, the Johan and Greta Kock Foundations, the King Gustaf V and Queen Victoria Foundation, the Swedish Foundation for Parkinson’s Research, the Swedish National Research Council (MAC), and NIH Grant No. NS048235 (CK). We thank the Gaemzeus Foundation and the Swedish Society for Medical Research for their financial support to JEW, the Marie Curie Host Fellowship Training Program for its support to LV, and the Parkinson Society Canada for its support to EMS. We thank Inga-Lill Bertilsson and Ulla Jarl for their assistance in the histologic part of the study.
Footnotes
Supplementary material cited in this article is available online.
References
- Andersson M, Hilbertson A, Cenci MA. Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson’s disease. Neurobiol Dis. 1999;6:461–474. doi: 10.1006/nbdi.1999.0259. [DOI] [PubMed] [Google Scholar]
- Andersson M, Konradi C, Cenci MA. cAMP response element-binding protein is required for dopamine-dependent gene expression in the intact but not the dopamine-denervated striatum. J Neurosci. 2001;21:9930–9943. doi: 10.1523/JNEUROSCI.21-24-09930.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Aubert I, Guigoni C, Hakansson K, Li Q, Dovero S, Barthe N, et al. Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann Neurol. 2005;57:17–26. doi: 10.1002/ana.20296. [DOI] [PubMed] [Google Scholar]
- Aubert I, Guigoni C, Li Q, Dovero S, Bioulac BH, Gross CE, et al. Enhanced preproenkephalin-b-derived opioid transmission in striatum and subthalamic nucleus converges upon globus pallidus internalis in L-3,4-dihydroxyphenylalanine-induced dyskinesia. Biol Psychiatry. 2006 doi: 10.1016/j.biopsych.2006.06.038. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Bedard PJ, Di Paolo T, Falardeau P, Boucher R. Chronic treatment with L-DOPA, but not bromocriptine induces dyskinesia in MPTP-parkinsonian monkeys. Correlation with [3H]spiperone binding. Brain Res. 1986;379:294–299. doi: 10.1016/0006-8993(86)90783-3. [DOI] [PubMed] [Google Scholar]
- Bedard PJ, Gomez-Mancilla B, Blanchette P, Gagnon C, Falardeau P, DiPaolo T. Role of selective D1 and D2 agonists in inducing dyskinesia in drug-naive MPTP monkeys. Adv Neurol. 1993;60:113–118. [PubMed] [Google Scholar]
- Berretta S, Parthasarathy HB, Graybiel AM. Local release of GABAergic inhibition in the motor cortex induces immediate-early gene expression in indirect pathway neurons of the striatum. J Neurosci. 1997;17:4752–4763. doi: 10.1523/JNEUROSCI.17-12-04752.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff S, Heinrich M, Sonntag JM, Krauss J. The D-1 dopamine receptor antagonist SCH 23390 also interacts potently with brain serotonin (5-HT2) receptors. Eur J Pharmacol. 1986;129:367–370. doi: 10.1016/0014-2999(86)90449-8. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Gnesutta N, Minichiello L, White G, Roylance AJ, Herron CE, et al. Arole for the Ras signalling pathway in synaptic transmission and long-term memory. Nature. 1997;390:281–286. doi: 10.1038/36849. [DOI] [PubMed] [Google Scholar]
- Brotchie JM, Henry B, Hille CJ, Crossman AR. Opioid peptide precursor expression in animal models of dystonia secondary to dopamine-replacement therapy in Parkinson’s disease. Adv Neurol. 1998;78:41–52. [PubMed] [Google Scholar]
- Cai G, Zhen X, Uryu K, Friedman E. Activation of extracellular signalregulated protein kinases is associated with a sensitized locomotor response to D(2) dopamine receptor stimulation in unilateral 6-hydroxydopamine-lesioned rats. J Neurosci. 2000;20:1849–1857. doi: 10.1523/JNEUROSCI.20-05-01849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta AR, Tronci E, Pinna A, Morelli M. Different responsiveness of striatonigral and striatopallidal neurons to L-DOPA after a subchronic intermittent L-DOPA treatment. Eur J Neurosci. 2005;21:1196–1204. doi: 10.1111/j.1460-9568.2005.03944.x. [DOI] [PubMed] [Google Scholar]
- Carta M, Lindgren HS, Lundblad M, Stancampiano R, Fadda F, Cenci MA. Role of striatal L-DOPA in the production of dyskinesia in 6-hydroxydopamine lesioned rats. J Neurochem. 2006;96:1718–1727. doi: 10.1111/j.1471-4159.2006.03696.x. [DOI] [PubMed] [Google Scholar]
- Caunt CJ, Finch AR, Sedgley KR, McArdle CA. Seven-transmembrane receptor signalling and ERK compartmentalization. Trends Endocrinol Metab. 2006;17:276–283. doi: 10.1016/j.tem.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Lee CS, Bjorklund A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin-and glutamic acid decarboxylase mRNA. Eur J Neurosci. 1998;10:2694–2706. [PubMed] [Google Scholar]
- Cenci MA, Lundblad M. Utility of 6-hydroxydopamine lesioned rats in the preclinical screening of novel treatments for Parkinson disease. In: Le-Doux M, editor. Animal Models of Movement Disorders. San Diego: Elsevier Academic Press; 2005. pp. 193–208. [Google Scholar]
- Cenci MA, Tranberg A, Andersson M, Hilbertson A. Changes in the regional and compartmental distribution of FosB- and JunB-like immunoreactivity induced in the dopamine-denervated rat striatum by acute or chronic L-dopa treatment. Neuroscience. 1999;94:515–527. doi: 10.1016/s0306-4522(99)00294-8. [DOI] [PubMed] [Google Scholar]
- Crow T, Xue-Bian JJ, Siddiqi V, Kang Y, Neary JT. Phosphorylation of mitogen-activated protein kinase by one-trial and multi-trial classical conditioning. J Neurosci. 1998;18:3480–3487. doi: 10.1523/JNEUROSCI.18-09-03480.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keyser J, De Backer JP, Wilczak N, Herroelen L. Dopamine agonists used in the treatment of Parkinson’s disease and their selectivity for the D1, D2, and D3 dopamine receptors in human striatum. Prog Neuropsychopharmacol Biol Psychiatry. 1995;19:1147–1154. doi: 10.1016/0278-5846(95)00232-4. [DOI] [PubMed] [Google Scholar]
- Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen-and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. Embo J. 1998;17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet JP, Nakabeppu Y, Bedard PJ, Hope BT, Nestler EJ, Jasmin BJ, et al. Chronic alterations in dopaminergic neurotransmission produce a persistent elevation of deltaFosB-like protein(s) in both the rodent and primate striatum. Eur J Neurosci. 1996;8:365–381. doi: 10.1111/j.1460-9568.1996.tb01220.x. [DOI] [PubMed] [Google Scholar]
- Dunn KL, Espino PS, Drobic B, He S, Davie JR. The Ras-MAPK signal transduction pathway, cancer and chromatin remodeling. Biochem Cell Biol. 2005;83:1–14. doi: 10.1139/o04-121. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Robinson TE. Amphetamine-evoked gene expression in striatopallidal neurons: Regulation by corticostriatal afferents and the ERK/MAPK signaling cascade. J Neurochem. 2004;91:337–348. doi: 10.1111/j.1471-4159.2004.02712.x. [DOI] [PubMed] [Google Scholar]
- Fiorentini C, Rizzetti MC, Busi C, Bontempi S, Collo G, Spano P, Missale C. Loss of synaptic D1 dopamine/N-methyl-D-aspartate glutamate receptor complexes in L-DOPA-induced dyskinesia in the rat. Mol Pharmacol. 2006;69:805–812. doi: 10.1124/mol.105.016667. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;23:S71–77. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Miyachi S, Paletzki R, Brown P. D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J Neurosci. 2002;22:5042–5054. doi: 10.1523/JNEUROSCI.22-12-05042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Canales JJ, Capper-Loup C. Levodopa-induced dyskinesias and dopamine-dependent stereotypies: A new hypothesis. Trends Neurosci. 2000;23:S71–77. doi: 10.1016/s1471-1931(00)00027-6. [DOI] [PubMed] [Google Scholar]
- Greengard P, Nairn AC, Girault JA, Ouimet CC, Snyder GL, Fisone G, et al. The DARPP-32/protein phosphatase-1 cascade: A model for signal integration. Brain Res Brain Res Rev. 1998;26:274–284. doi: 10.1016/s0165-0173(97)00057-x. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, et al. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Jackson D, Abercrombie ED, Zigmond MJ. Impact of L-dopa on striatal acetylcholine release: Effects of 6-hydroxydopamine. J Pharmacol Exp Ther. 1993;267:912–918. [PubMed] [Google Scholar]
- Keyse SM. Protein phosphatases and the regulation of mitogenactivated protein kinase signalling. Curr Opin Cell Biol. 2000;12:186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- Kincaid AE, Wilson CJ. Corticostriatal innervation of the patch and matrix in the rat neostriatum. J Comp Neurol. 1996;374:578–592. doi: 10.1002/(SICI)1096-9861(19961028)374:4<578::AID-CNE7>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Konradi C, Westin JE, Carta M, Eaton ME, Kuter K, Dekundy A, et al. Transcriptome analysis in a rat model of L-DOPA-induced dyskinesia. Neurobiol Dis. 2004;17:219–236. doi: 10.1016/j.nbd.2004.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazo PS, Dorfman K, Noguchi T, Mattei MG, Bravo R. Structure and mapping of the fosB gene. FosB downregulates the activity of the fosB promoter. Nucleic Acids Res. 1992;20:343–350. doi: 10.1093/nar/20.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman AN, Goldstein M. Bromocriptine in Parkinson disease. Pharmacol Rev. 1985;37:217–227. [PubMed] [Google Scholar]
- Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson’s disease. Eur J Neurosci. 2002;15:120–132. doi: 10.1046/j.0953-816x.2001.01843.x. [DOI] [PubMed] [Google Scholar]
- Marsden CD. Problems with long-term levodopa therapy for Parkinson’s disease. Clin Neuropharmacol. 1994;17(suppl 2):S32–44. [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, et al. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/s0896-6273(02)00716-x. [DOI] [PubMed] [Google Scholar]
- Moratalla R, Elibol B, Vallejo M, Graybiel AM. Network-level changes in expression of inducible Fos-Jun proteins in the striatum during chronic cocaine treatment and withdrawal. Neuron. 1996;17:147–156. doi: 10.1016/s0896-6273(00)80288-3. [DOI] [PubMed] [Google Scholar]
- Nutt JG. Levodopa-induced dyskinesia: Review, observations, and speculations. Neurology. 1990;40:340–345. doi: 10.1212/wnl.40.2.340. [DOI] [PubMed] [Google Scholar]
- Paul ML, Graybiel AM, David JC, Robertson HA. D1-like and D2-like dopamine receptors synergistically activate rotation and c-fos expression in the dopamine-depleted striatum in a rat model of Parkinson’s disease. J Neurosci. 1992;12:3729–3742. doi: 10.1523/JNEUROSCI.12-10-03729.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic Press; 1998. [DOI] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Hakansson K, Bernardi G, Greengard P, Fisone G, et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA-in-duced dyskinesia. Nat Neurosci. 2003;6:501–506. doi: 10.1038/nn1040. [DOI] [PubMed] [Google Scholar]
- Rascol O, Sabatini U, Brefel C, Fabre N, Rai S, Senard JM, et al. Cortical motor overactivation in parkinsonian patients with L-dopa-induced peak-dose dyskinesia. Brain. 1998;121(pt 3):527–533. doi: 10.1093/brain/121.3.527. [DOI] [PubMed] [Google Scholar]
- Sgambato V, Pages C, Rogard M, Besson MJ, Caboche J. Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J Neurosci. 1998a;18:8814–8825. doi: 10.1523/JNEUROSCI.18-21-08814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgambato V, Vanhoutte P, Pages C, Rogard M, Hipskind R, Besson MJ, Caboche J. In vivo expression and regulation of Elk-1, a target of the extracellular-regulated kinase signaling pathway, in the adult rat brain. J Neurosci. 1998b;18:214–226. doi: 10.1523/JNEUROSCI.18-01-00214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgambato-Faure V, Buggia V, Gilbert F, Levesque D, Benabid AL, Berger F. Coordinated and spatial upregulation of arc in striatonigral neurons correlates with L-dopa-induced behavioral sensitization in dyskinetic rats. J Neuropathol Exp Neurol. 2005;64:936–947. doi: 10.1097/01.jnen.0000186922.42592.b7. [DOI] [PubMed] [Google Scholar]
- St-Hilaire M, Landry E, Levesque D, Rouillard C. Denervation and repeated L-DOPA induce complex regulatory changes in neurochemical phenotypes of striatal neurons: Implication of a dopamine D1-dependent mechanism. Neurobiol Dis. 2005;20:450–460. doi: 10.1016/j.nbd.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Steiner H, Kitai ST. Regulation of rat cortex function by D1 dopamine receptors in the striatum. J Neurosci. 2000;20:5449–5460. doi: 10.1523/JNEUROSCI.20-14-05449.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J. Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci. 2000;20:8701–8709. doi: 10.1523/JNEUROSCI.20-23-08701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JQ, McGinty JF. Muscarinic receptors regulate striatal neuropeptide gene expression in normal and amphetamine-treated rats. Neuroscience. 1996;75:43–56. doi: 10.1016/0306-4522(96)00277-1. [DOI] [PubMed] [Google Scholar]
- Westin JE, Andersson M, Lundblad M, Cenci MA. Persistent changes in striatal gene expression induced by long-term L-DOPA treatment in a rat model of Parkinson’s disease. Eur J Neurosci. 2001;14:1171–1176. doi: 10.1046/j.0953-816x.2001.01743.x. [DOI] [PubMed] [Google Scholar]
- Winkler C, Kirik D, Bjorklund A, Cenci MA. L-DOPA-induced dyskinesia in the intrastriatal 6-hydroxydopamine model of Parkinson’s disease: Relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis. 2002;10:165–186. doi: 10.1006/nbdi.2002.0499. [DOI] [PubMed] [Google Scholar]
- Zhen X, Torres C, Cai G, Friedman E. Inhibition of protein tyrosine/mitogen-activated protein kinase phosphatase activity is associated with D2 dopamine receptor supersensitivity in a rat model of Parkinson’s disease. Mol Pharmacol. 2002;62:1356–1363. doi: 10.1124/mol.62.6.1356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.