

Abstract
Enkephalin modulates striatal function, thereby affecting motor performance and addictive behaviors. The proenkephalin gene is also used as a model to study cyclic AMP-mediated gene expression in striatal neurons. The second messenger pathway leading to proenkephalin expression demonstrates how cyclic AMP pathways are synchronized with depolarization. We show that cyclic AMP-mediated regulation of the proenkephalin gene is dependent on the activity of L-type Ca2+ channels. Inhibition of L-type Ca2+ channels blocks forskolin-mediated induction of proenkephalin. The Ca2+-activated kinase, Ca2+/calmodulin kinase, as well as the cyclic AMP-activated kinase, protein kinase A (PKA), are both necessary for the induction of the proenkephalin promoter. Similarly, both kinases are needed for the L-type Ca2+ channel-mediated induction of proenkephalin. This synchronization of second messenger pathways provides a coincidence mechanism that gates proenkephalin synthesis in striatal neurons, ensuring that levels are increased only in the presence of activated PKA and depolarization.
Keywords: Enkephalin, Protein kinase A, Ca2+/calmodulin kinase, L-type Ca2+ channels, Depolarization, Striatum
1. Introduction
The neuropeptide enkephalin plays a role in biological processes as diverse as nociception [21], stress- and immune response [7,23] and reproductive function [8]. In the central nervous system, the striatum (caudate, putamen, nucleus accumbens) is among the brain areas with the highest levels of enkephalin, which is mostly expressed in striatopallidal neurons [10,24]. Upon release, enkephalin interacts preferentially with delta and mu opiate receptors [22] and exerts a negative feedback mechanism to regulate the responsiveness of striatopallidal neurons [24]. Striatal enkephalin has been associated with motor behaviors [1,3] as well as addictive mechanisms such as reward [11,19]. Enkephalin release is precipitated by Ca2+-dependent depolarization [13] and pools are replenished via induction of proenkephalin mRNA synthesis [4,6]. Two adjacent DNA enhancer elements within 110 bp 5′ to the mRNA cap site act synergistically to activate proenkephalin mRNA synthesis [4,6]. These enhancer elements are responsive to cyclic AMP and Ca2+ stimulated second messenger pathways. Here we show that L-type Ca2+ channels mediate the induction of mRNA synthesis of the proenkephalin gene for both pathways.
2. Materials and methods
2.1. Materials
FPL 64176, forskolin and nifedipine were obtained from Sigma (St. Louis, MO, USA). KN62 (1-[N,O-bis-(5-iso-quinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine) and H89-dihydrochloride (N-[2-(( p-bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide) were obtained from Calbiochem (La Jolla, CA, USA).
2.2. Methods
E18 Sprague–Dawley rat pups were used for all experiments. Primary striatal cultures were obtained as previously described [18]. The pENKAT12, Δ80 and −772 constructs were obtained from Michael Comb. The pENKAT12 construct contains the minimal 5′ enhancer of the human proenkephalin gene, 193 bp 5′ of the CAP site. The Δ80 construct contains the 84 bp 5′ of the CAP site and is not responsive to forskolin in cell culture [4,6], and the −772 construct contains the 772 bp 5′ of the CAP site. All constructs were fused to the reporter gene chloramphenicol acetyltransferase within exon 2, and contain 1.2 kb of 3′ flanking sequence of the human proenkephalin gene [4,6]. These constructs have been used previously in primary striatal culture by our group [14]. No differences in gene regulation pattern have been observed between pENKAT12 and longer constructs, however, pENKAT12 was most responsive [14]. Enkephalin constructs were transfected into primary striatal cultures using Ca2+ transfection [25], as described previously [18]. RNA was extracted and analyzed as described previously [18].
Kinase inhibitors were added 30 min before the addition of forskolin or FPL 64176, and were present during the exposure to forskolin or FPL 64176. Exposure to forskolin or FPL 64176 lasted 6 h.
2.3. Determination of kinase inhibitor concentration
The concentrations of KN62 and H89 needed for reliable inhibition were determined in preliminary studies using CREB phosphorylation as the model. The transcription factor CREB has been shown to be involved in forskolin-mediated induction of the proenkephalin gene [14,15]. The inhibitory potential of KN62 on CREB phosphorylation by glutamate, NMDA, KCl and FPL 64176 was determined in dose–response curves. A concentration of 30 μM KN62 inhibited CREB phosphorylation by NMDA and KCl, and significantly attenuated CREB phosphorylation by glutamate and FPL 64176, and was therefore chosen for the study. H89 was tested in dose–response curves with forskolin and dopamine, and 20 μM was chosen as the minimal concentration for reliable results.
3. Results
The proenkephalin gene is induced in primary striatal neurons after a 6-h treatment with the L-type Ca2+ channel agonist FPL 64176 (20 μM), as shown in Northern blots (Fig. 1A; the experiment was repeated four times). This induction is prevented by pretreatment for 30 min and co-treatment for 6 h with the L-type Ca2+ channel antagonist nifedipine (20 μM). Similarly, the transfected pENKAT12 construct, which contains the proenkephalin promoter linked to chloramphenicol acetyltransferase [4], was induced by FPL 64176 (20 μM) after a 6-h treatment, and inhibited by pre- and co-treatment with nifedipine (20 μM; Fig. 1B). The Δ80 construct was not responsive to FPL 64176, whereas the −772 construct yielded similar results to pENKAT12, albeit with a lower fold-induction by FPL 64176 (4.5-fold, not shown). Because the Δ80 construct was not responsive to FPL 64176, the area of activation of the proenkephalin enhancer can be narrowed to between 84 and 193 bp upstream of the transcription initiation site, to the previously described CRE1 and CRE2 sites [4,6].
Fig. 1.
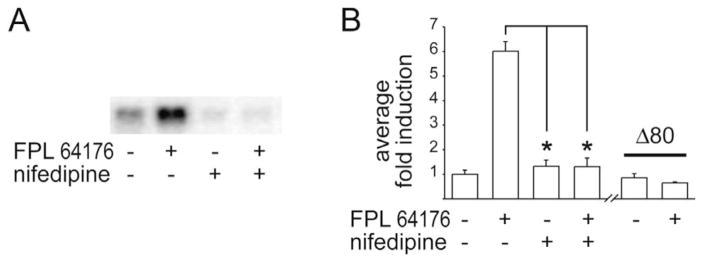
The L-type Ca2+ channel agonist FPL 64176 induces proenkephalin synthesis in primary striatal culture. (A) Proenkephalin gene levels are upregulated by the L-type Ca2+ channel agonist FPL 64176 (20 μM). This upregulation is blocked by the L-type Ca2+ channel antagonist nifedipine (20 μM). (B) The proenkephalin construct pENKAT12, transfected into primary striatal neurons, is induced by FPL 64176 (20 μM). The induction is blocked by nifedipine (20 μM). The construct with a shorter promoter, Δ80, is not induced by FPL 64176. Average fold induction of n=4–7±S.E.M. is shown. *P<0.001.
Interestingly, induction of the proenkephalin gene by the adenylate cyclase stimulating agent, forskolin (10 μM), was also inhibited by nifedipine (20 μM; Fig. 2A; the experiment was repeated four times). Comparable data were obtained with the transfected pENKAT12 construct (Fig. 2B) and the −772 construct (not shown), whereas the Δ80 construct was unresponsive. Forskolin-mediated proenkephalin expression was furthermore dependent on Ca2+/calmodulin kinase (CaMK), as the CaMK II/CaMK IV inhibitor, KN62 (30 μM), inhibited forskolin-mediated pENKAT12 induction (Fig. 2C). Similar data were observed in Northern blots with the native proenkephalin gene (not shown). Forskolin-mediated proenkephalin expression was also dependent on PKA, demonstrated by the inhibition of pENKAT12 induction by the PKA antagonist H89 (20 μM; Fig. 2D). Northern blots with native proenkephalin expression confirmed the findings (not shown). These data demonstrate that the induction of the proenkephalin gene depends on PKA as well as on CaMK.
Fig. 2.

Proenkephalin induction by forskolin is inhibited by the L-type Ca2+ channel antagonist nifedipine, by the CaMK inhibitor KN62, and by the PKA inhibitor H89. (A) Forskolin (10 μM)-induced expression of the proenkephalin gene is blocked by nifedipine (20 μM). (B) The proenkephalin construct pENKAT12, transfected into primary striatal neurons, is induced by forskolin (10 μM). The induction is blocked by nifedipine (20 μM). The Δ80 construct is not induced by forskolin. (C) pENKAT12-induction by forskolin (10 μM) is blocked by KN62 (30 μM), and by H89 (20 μM), (D). Average fold induction of n=7–10±S.E.M. is shown. * P<0.001.
Proenkephalin gene expression and pENKAT12 induction by the L-type Ca2+ channel agonist FPL 64176 was also blocked by KN62 (30 μM; Fig. 3A,B). Surprisingly, FPL 64176-mediated proenkephalin expression (Fig. 3C) and pENKAT12 induction (Fig. 3D) were inhibited by H89, demonstrating a dependence of L-type Ca2+ channel-mediated proenkephalin expression in striatal neurons on PKA.
Fig. 3.

Proenkephalin induction by FPL 64176 is blocked by the CaMK inhibitor KN62 and by the PKA inhibitor H89. (A) FPL 64176 (20 μM)-mediated induction of the proenkephalin gene is blocked by KN62 (30 μM). (B) FPL 64176 (20 μM)-mediated induction of the proenkephalin construct pENKAT12 is blocked by KN62 (30 μM). (C) FPL 64176 (20 μM)-mediated induction of the proenkephalin gene is blocked by H89 (20 μM). (D) pENKAT12-induction by FPL 64176 (20 μM) is blocked by H89 (20 μM). Average fold induction of n=4–7±S.E.M. is shown. * P<0.001.
4. Discussion
In molecular models of striatal function, cyclic AMP pathways play an important role. The high density of G-protein coupled receptors, many of which are coupled to adenylyl cyclase, attracts attention to the role and function of cyclic AMP-mediated gene expression in striatal neurons. The proenkephalin gene is a perfect candidate to study cyclic AMP-mediated gene expression in the striatum, as it codes for a neuromodulator with documented functions, is located in the striatum, and is sensitive to changes in cyclic AMP levels [4,5,14,15]. Among the G-protein coupled receptors in the striatum are dopamine and adenosine receptors, both of which regulate levels of proenkephalin mRNA and enkephalin protein [12,14,15,17,20]. The most obvious increase in demand for enkephalin is during times of increased neuronal activity and, thus, increased enkephalin release, raising the question of how the cyclic AMP second messenger pathway synchronizes with neuronal activity. We show here that proenkephalin mRNA synthesis depends on L-type Ca2+ channel activity and on CaMK. Blockade of L-type Ca2+ channels, or inhibition of CaMK, disrupts PKA-stimulated proenkephalin mRNA synthesis. Thus, the cyclic AMP second messenger pathway can induce proenkephalin mRNA synthesis only in an active neuron. Because we do not add depolarizing agents with forskolin, our data suggest furthermore that the cyclic AMP pathway increases neuronal excitability to agents already present in the medium, such as KCl (5 mM) and glutamate (1–5 μM) [16]. A possible mechanism might be the phosphorylation of PKA sites on ion channels such as the L-type Ca2+ channel [2,9]. The notion that PKA contributes to neuronal excitability via modulation of ion channels is supported by our observation that inhibition of PKA prevents L-type Ca2+ channel-mediated induction of proenkephalin. The data suggest that basal phosphorylation of PKA sites is necessary for L-type Ca2+ channel-mediated proenkephalin expression, and increased phosphorylation might increase the likelihood that a neuron depolarizes. Taken together, we provide a model by which cyclic AMP-mediated gene induction is synchronized with neuronal depolarization. Increased PKA activity, as well as activation of L-type Ca2+ channels, is needed for the induction of proenkephalin gene expression. Indeed, a co-dependency of the L-type Ca2+ channel pathway with the PKA pathway provides a dual checkpoint for proenkephalin gene expression.
Acknowledgments
We thank Dr Michael Comb for the enkephalin constructs. This work was supported by DA07134.
Abbreviations
- PKA
protein kinase A
- CaMK
Ca2+/calmodulin kinase
References
- 1.Brotchie JM, Henry B, Hille CJ, Crossman AR. Opioid peptide precursor expression in animal models of dystonia secondary to dopamine-replacement therapy in Parkinson’s disease. Adv Neurol. 1998;78:41–52. [PubMed] [Google Scholar]
- 2.Bünemann M, Gerhardstein BL, Gao T, Hosey MM. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the beta(2) subunit. J Biol Chem. 1999;274:33851–33854. doi: 10.1074/jbc.274.48.33851. [DOI] [PubMed] [Google Scholar]
- 3.Calon F, Di Paolo T. Levodopa response motor complications: GABA receptors and preproenkephalin expression in human brain. Parkinsonism Relat Disord. 2002;8:449–454. doi: 10.1016/s1353-8020(02)00029-9. [DOI] [PubMed] [Google Scholar]
- 4.Comb M, Birnberg NC, Seasholtz A, Herbert E, Goodman HM. A cyclic AMP- and phorbol ester-inducible DNA element. Nature. 1986;323:353–356. doi: 10.1038/323353a0. [DOI] [PubMed] [Google Scholar]
- 5.Comb M, Hyman SE, Kobierski L, Chu HM, Van Nguyen T. Mechanisms underlying synaptic regulation of proenkephalin transcription. NIDA Res Monogr. 1991;111:149–170. [PubMed] [Google Scholar]
- 6.Comb M, Mermod N, Hyman SE, Pearlberg J, Ross ME, Goodman HM. Proteins bound at adjacent DNA elements act synergistically to regulate human proenkephalin cAMP inducible transcription. EMBO J. 1988;7:3793–3805. doi: 10.1002/j.1460-2075.1988.tb03264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drolet G, Dumont EC, Gosselin I, Kinkead R, Laforest S, Trottier JF. Role of endogenous opioid system in the regulation of the stress response. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:729–741. doi: 10.1016/s0278-5846(01)00161-0. [DOI] [PubMed] [Google Scholar]
- 8.Fabbri A, Jannini EA, Gnessi L, Ulisse S, Moretti C, Isidori A. Neuroendocrine control of male reproductive function. The opioid system as a model of control at multiple sites. J Steroid Biochem. 1989;32:145–150. doi: 10.1016/0022-4731(89)90155-6. [DOI] [PubMed] [Google Scholar]
- 9.Gao T, Yatani A, Dell’Acqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–196. doi: 10.1016/s0896-6273(00)80358-x. [DOI] [PubMed] [Google Scholar]
- 10.Gerfen CR, Young WS., III Distribution of striatonigral and striatopallidal peptidergic neurons in both patch and matrix compartments: an in situ hybridization histochemistry and fluorescent retrograde tracing study. Brain Res. 1988;460:161–167. doi: 10.1016/0006-8993(88)91217-6. [DOI] [PubMed] [Google Scholar]
- 11.Hayward MD, Pintar JE, Low MJ. Selective reward deficit in mice lacking beta-endorphin and enkephalin. J Neurosci. 2002;22:8251–8258. doi: 10.1523/JNEUROSCI.22-18-08251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hong JS, Yang HY, Gillin JC, Di Giulio AM, Fratta W, Costa E. Chronic treatment with haloperidol accelerates the biosynthesis of enkephalins in rat striatum. Brain Res. 1979;160:192–195. doi: 10.1016/0006-8993(79)90618-8. [DOI] [PubMed] [Google Scholar]
- 13.Iversen LL, Lee CM, Gilbert RF, Hunt S, Emson PC. Regulation of neuropeptide release. Proc R Soc Lond B. 1980;210:91–111. doi: 10.1098/rspb.1980.0121. [DOI] [PubMed] [Google Scholar]
- 14.Konradi C, Cole RL, Green D, Senatus P, Leveque JC, Pollack A, Grossbard SJ, Hyman SE. Analysis of the proenkephalin second messenger-inducible enhancer in rat striatal cultures. J Neurochem. 1995;65:1007–1015. doi: 10.1046/j.1471-4159.1995.65031007.x. [DOI] [PubMed] [Google Scholar]
- 15.Konradi C, Kobierski LA, Nguyen TV, Heckers S, Hyman SE. The cAMP-response-element-binding protein interacts, but Fos protein does not interact, with the proenkephalin enhancer in rat striatum. Proc Natl Acad Sci USA. 1993;90:7005–7009. doi: 10.1073/pnas.90.15.7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konradi C, Leveque JC, Hyman SE. Amphetamine and dopamine-induced immediate early gene expression in striatal neurons depends on postsynaptic NMDA receptors and calcium. J Neurosci. 1996;16:4231–4239. doi: 10.1523/JNEUROSCI.16-13-04231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kowalski C, Giraud P. Dopamine decreases striatal enkephalin turnover and proenkephalin messenger RNA abundance via D2 receptor activation in primary striatal cell cultures. Neuroscience. 1993;53:665–672. doi: 10.1016/0306-4522(93)90614-l. [DOI] [PubMed] [Google Scholar]
- 18.Leveque JC, Macías W, Rajadhyaksha A, Carlson RR, Barczak A, Kang S, Li XM, Coyle JT, Huganir RL, Heckers S, Konradi C. Intracellular modulation of NMDA receptor function by antipsychotic drugs. J Neurosci. 2000;20:4011–4020. doi: 10.1523/JNEUROSCI.20-11-04011.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McBride WJ, Murphy JM, Ikemoto S. Localization of brain reinforcement mechanisms: intracranial self-administration and intracranial place-conditioning studies. Behav Brain Res. 1999;101:129–152. doi: 10.1016/s0166-4328(99)00022-4. [DOI] [PubMed] [Google Scholar]
- 20.Pollack AE, Wooten GF. Differential regulation of striatal pre-proenkephalin mRNA by D1 and D2 dopamine receptors. Mol Brain Res. 1992;12:111–119. doi: 10.1016/0169-328x(92)90074-l. [DOI] [PubMed] [Google Scholar]
- 21.Przewlocki R, Przewlocka B. Opioids in chronic pain. Eur J Pharmacol. 2001;429:79–91. doi: 10.1016/s0014-2999(01)01308-5. [DOI] [PubMed] [Google Scholar]
- 22.Raynor K, Kong H, Law S, Heerding J, Tallent M, Livingston F, Hines J, Reisine T. Molecular biology of opioid receptors. NIDA Res Monogr. 1996;161:83–103. [PubMed] [Google Scholar]
- 23.Salzet M, Vieau D, Day R. Crosstalk between nervous and immune systems through the animal kingdom: focus on opioids. Trends Neurosci. 2000;23:550–555. doi: 10.1016/s0166-2236(00)01642-8. [DOI] [PubMed] [Google Scholar]
- 24.Steiner H, Gerfen CR. Role of dynorphin and enkephalin in the regulation of striatal output pathways and behavior. Exp Brain Res. 1998;123:60–76. doi: 10.1007/s002210050545. [DOI] [PubMed] [Google Scholar]
- 25.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]