

Abstract
This paper reviews the evidence that antipsychotic drugs induce neuroplasticity. We outline how the synaptic changes induced by the antipsychotic drug haloperidol may help our understanding of the mechanism of action of antipsychotic drugs in general, and how they may help to elucidate the neurobiology of schizophrenia. Studies have provided compelling evidence that haloperidol induces anatomical and molecular changes in the striatum. Anatomical changes have been documented at the level of regional brain volume, synapse morphology, and synapse number. At the molecular level, haloperidol has been shown to cause phosphorylation of proteins and to induce gene expression. The molecular responses to conventional antipsychotic drugs are predominantly observed in the striatum and nucleus accumbens, whereas atypical anti-psychotic drugs have a subtler and more widespread impact. We conclude that the ability of antipsychotic drugs to induce anatomical and molecular changes in the brain may be relevant for their antipsychotic properties. The delayed therapeutic action of antipsychotic drugs, together with their promotion of neuroplasticity suggests that modification of synaptic connections by antipsychotic drugs is important for their mode of action. The concept of schizophrenia as a disorder of synaptic organization will benefit from a better understanding of the synaptic changes induced by antipsychotic drugs.
Keywords: Haloperidol, clozapine, neuroplasticity, gene expression, schizophrenia
Introduction
Schizophrenia is a debilitating disorder that affects 1% of the world population, often at an early age (Andreasen 1995; Andreasen 1996; Carpenter and Buchanan 1994). While schizophrenic patients may present with various abnormalities of perception, thought, language, or affect, many patients develop cognitive deficits and most will show marked and long lasting impairments in social functioning (Green 1996). Despite an intense effort, the disease mechanisms and etiology of schizophrenia have remained elusive. A promising route of investigation into the causes of the disease is to study how antipsychotic drugs alter brain function. An improved understanding of how antipsychotic drugs convey their therapeutic effects during the treatment of schizophrenia can help us to unravel neural mechanisms involved in the pathogenesis of schizophrenia. Such an approach has generated a prominent model of schizophrenia, the dopamine hypothesis (Matthysse 1973).
One of the most puzzling observations about the mechanism of action of antipsychotic drugs is their delayed therapeutic effect (Hyman and Nestler 1996). This results often in prolonged treatment trials for individual patients until a particular drug has proven to be effective. In some cases, none of the existing drugs will provide remission. To improve treatment outcome, we need to understand the potential mechanisms that could explain the delayed clinical effects of antipsychotic drugs. The clinical potency of conventional antipsychotic drugs is directly correlated with their ability to inhibit dopamine D2 receptors (Creese et al 1976), but this relationship cannot explain their delayed action. Since inhibition of D2 receptors should be achieved almost instantaneously, the beneficial therapeutic effect cannot be limited to a straightforward receptor–drug interaction. Moreover, the pharmacologic profile of atypical antipsychotic drugs suggests a more complicated mechanism of action.
Which neuronal programs evolve over time? Neuroplasticity, a gradual process by which the brain adapts to changes in the environment, is a logical consideration in the delayed effects of antipsychotic drugs. Two processes contribute to neuroplasticity in the adult brain: 1) synaptic plasticity, a remodeling of synapses resulting in the rewiring and strengthening of neural circuits, and 2) neurogenesis, a creation of new neurons. A demonstration that neuroplasticity conveys some of the therapeutic effects of antipsychotic drugs could provide a valuable target for the design of novel treatment strategies.
We will focus in this article primarily on haloperidol, a conventional antipsychotic drug that has been extensively studied. A review of the literature shows that synaptic plasticity is consistently observed after treatment with haloperidol and is likely important for its effects on brain function. Molecular markers for synaptic competence such as synaptophysin are decreased in schizophrenia (Browning et al 1993; Davidsson et al 1999; Eastwood et al 1995; Eastwood et al 2000; Eastwood and Harrison 1995; Glantz and Lewis 1997; Karson et al 1999; Landen et al 1999), and increased by haloperidol (Eastwood et al 1994; Eastwood et al 1997). Synaptic reorganization by haloperidol may reverse a pathologic process of synaptic disruption in schizophrenia, as schizophrenia has been characterized as a prefrontal-temporal disconnection syndrome, with functional as well as anatomical correlates (Bogerts 1997; Friston 1998). If schizophrenia is a disconnection syndrome caused by inadequate synaptic organization (McGlashan and Hoffman 2000), neuroplasticity induced by antipsychotic drugs can achieve functional and anatomical reconnection. The time it takes to reestablish proper synaptic function could explain the delay in the therapeutic benefits of antipsychotic drugs.
Neurogenesis seems a less likely mechanism by which drugs exert their antipsychotic properties. There is agreement that neurogenesis in the mature brain is detectable only in a limited number of brain areas, most notably the hippocampus (Cameron and McKay 1998). Whereas a recent study has provided preliminary evidence that haloperidol may promote neurogenesis in the gerbil hippocampus (Dawirs et al 1998), studies in rat brain do not support this finding (Malberg et al 2000). Thus, while we cannot exclude that the creation of new neurons is important for the therapeutic properties of haloperidol, the data available are neither sufficient nor convincing to support such a conclusion.
We will therefore focus on the evidence that haloperidol mediates synaptic plasticity, and that this is an important mechanism in the therapeutic properties of many antipsychotic drugs. A major part of this review examines the role of haloperidol in striatal plasticity, because data on haloperidol and the striatum are best documented and confirmed often by independent research groups. The striatum is also a brain area where subtle changes by haloperidol should be most prominently expressed due to the high density of D2 receptors and their correlation with conventional antipsychotic drug action. While the experimental data for haloperidol and the striatum provide the foundation to formulate a sound hypothesis, we will discuss evidence that neuroplasticity as a mechanism of action is limited neither to one brain region (Figure 1), nor to a single antipsychotic drug. Neuroplasticity in brain areas other than the striatum, such as the prefrontal cortex, hippocampus and thalamus (Benes 2000; Heckers 1997), is likely as important for the treatment of schizophrenia. Antipsychotic drugs may exert some of their therapeutic properties by modulating neuroplasticity in these brain areas (Figure 1).
Figure 1.

Dopaminergic projections to brain regions implicated in schizophrenia. Substantia nigra (SN) neurons project to GABAergic neurons in the striatum (S), which receive input from the cortex (C) and send projections back to the cortex via the globus pallidus (GP) and thalamus (T). Ventral tegmental area (VTA) neurons send projections to the cortex and to limbic regions such as the hippocampus (H). Most anatomical and molecular studies of antipsychotic drugs have focused on the striatum, since it receives the densest dopaminergic projections and expresses the highest density of dopamine receptors. The effects of antipsychotic drugs on cortex and hippocampus, although more difficult to study, might be more relevant for understanding their antipsychotic effects. Two circuits that are particularly relevant in schizophrenia are highlighted: A reciprocal hippocampus-cortex connection, and the striatal-pallidal-thalamic-cortical loop. Dopaminergic connections are dashed.
We will set out with a review of the anatomic and molecular evidence that haloperidol affects synaptic plasticity, followed by a discussion of the potential mechanisms underlying these changes, and a comparison of neuroplasticity by conventional and atypical antipsychotic drugs. Finally, we will outline the implications of plastic changes induced by antipsychotic drugs for the treatment and neurobiology of schizophrenia.
Evidence That Haloperidol Affects Synaptic Plasticity
Neuroplasticity can be studied with a variety of methods. Anatomical techniques quantitatively assess macroscopic features such as regional brain volume, and microscopic features such as the morphology and number of cells, dendrites, dendritic spines, or synapses. Biochemical and molecular techniques assess the state of protein phosphorylation and the regulation of gene expression. All of these techniques have been applied to elucidate the effect of haloperidol in the striatum, resulting in convincing evidence for volumetric, ultrastructural, and molecular changes.
Volumetric and Ultrastructural Effects of Haloperidol in the Striatum
Haloperidol Increases Regional Brain Volume
Neurotrophic effects of haloperidol are supported by morphometric analyses of the caudate and putamen of treated schizophrenic patients. The initial description of striatal enlargement in patients treated with conventional antipsychotic drugs was surprising (Heckers et al 1991; Jernigan et al 1991), but subsequent studies have confirmed and extended these findings. In addition to enlarged striata, a positive correlation between medication dose and striatal volume supports a trophic effect (Bilder et al 1994; Chakos et al 1994; DeLisi et al 1991; Doraiswamy et al 1995; Gur et al 1998; Shihabuddin et al 1998; Swayze et al 1992). Time-course studies demonstrate that caudate volumes increase during treatment with conventional antipsychotic drugs and normalize after cessation of treatment or when patients are treated with atypical antipsychotics, which have a much weaker impact in the striatum (Bilder et al 1994; Chakos et al 1995; Chakos et al 1994; Keshavan et al 1994). These observations were verified in an animal model, where chronic administration of haloperidol in rats increased striatal volume (Chakos et al 1998).
Presently, the cellular processes responsible for striatal volume increase in neuroleptic-treated schizophrenic patients are not identified. Recent studies have reported that the increased striatal volume is accompanied by an overall increased number of striatal neurons (Beckmann and Lauer 1997; Lauer and Beckmann 1997). In addition, a change in the synaptic organization of the striatum, particularly the caudate nucleus, has been observed (Kung et al 1998). These initial studies of cellular changes in the striatum need to be followed up to better understand the impact of neuroleptic drugs.
The trophic effect of haloperidol and other conventional antipsychotics does not preclude neurotoxicity and loss of brain volume after long-term treatment or high dosing (Andreassen and Jorgensen 2000; Burkhardt et al 1993; Goff et al 1995). For example, caudate nucleus volumes of patients who developed tardive dyskinesia (TD) were significantly smaller in some studies (Dalgalarrondo and Gattaz 1994; Mion et al 1991), but increased in others (Brown et al 1996; Elkashef et al 1994). These inconsistent results are a reflection of the contrasting sequelae of neurotoxicity and neuroplasticity, which are simultaneously induced by haloperidol. Dependent on drug concentration, treatment time and individual sensitivity, the neurotrophic or the neurotoxic properties determine striatal volume increase or decrease respectively.
Haloperidol Changes Synapse Morphology and Number
In addition to volume increase, haloperidol causes ultrastructural changes in synapse morphology in the striatum. In rats treated chronically with haloperidol, an increase in the size of axon terminals has been observed (Benes et al 1985a; Kerns et al 1992; Uranova et al 1991), accompanied by an increase in the absolute number of vesicles per synapse (Benes et al 1985a), and by an increase in the size of the postsynaptic density (PSD; Figure 2), (Uranova et al 1991). Moreover, the number of synapses, mainly of the glutamatergic type, is increased in the rat striatum after chronic haloperidol treatment (Kerns et al 1992; Meshul and Casey 1989; See et al 1992; Uranova et al 1991). The number of double synapses is elevated (Kerns et al 1992), indicating synapse splitting, a process by which synapses multiply in the mature brain (Jones and Harris 1995; Kirov et al 1999; Toni et al 1999). The effects of haloperidol are reversible after cessation of treatment (Meshul and Casey 1989), and administration of clozapine does not seem to affect the number of synapses in the striatum (Meshul et al 1992). The opposing effects of haloperidol’s neurotoxic and neurotrophic mechanisms are also seen at the level of synapses. Vacuous chewing movements in rats are correlated with a significant decrease in striatal synaptic density (Roberts et al 1995).
Figure 2.
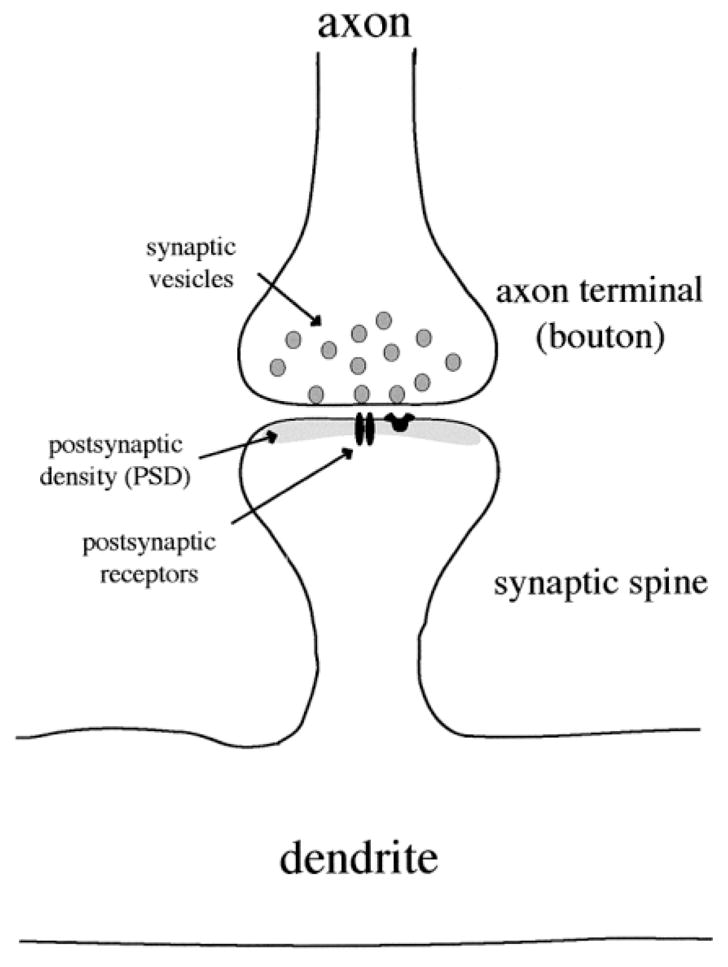
Synapse morphology in the striatum. The majority of neurons in the striatum are medium-size with dendritic spines. Synapses are formed on the dendritic spines as well as on the dendritic shafts (not shown). Synaptic vesicles in the axon terminal release neurotransmitter into the synapse. Neurotransmitters cross the synaptic cleft and interact with receptors in the postsynaptic membrane. The postsynaptic density (PSD) is an electron dense area that contains receptors, ion channels and scaffolding proteins which anchor receptors to the membrane and facilitate signal transduction.
The striatum is not the only brain area that exhibits changes in synapse morphology and synapse number in response to haloperidol treatment. Other brain areas with ultrastructural changes include the substantia nigra (Benes et al 1983) and the prefrontal cortex (Benes et al 1985b; Klintzova et al 1989). Synaptic plasticity in these affected areas may be involved in reversing the pathophysiology of schizophrenia.
Molecular Modifications During Treatment with Haloperidol
Haloperidol affects protein phosphorylation and protein synthesis. Protein phosphorylation is involved in signaling processes spanning from synaptic function to gene and protein expression (Greengard et al 1993; Schulman 1995). Protein phosphorylation and new protein synthesis are required for rapid and long-term synaptic remodeling in the developing and the mature brain. Learning and memory, the behavioral correlates of neuroplasticity, depend on protein phosphorylation (Frank and Greenberg 1994) and new protein synthesis (Davis and Squire 1984).
Haloperidol Affects Protein Phosphorylation
Conventional antipsychotic drugs influence protein phosphorylation via their ability to inhibit dopamine D2 receptors (Seeman and Lee 1975). Inhibition of D2 receptors activates adenylyl cyclase, increases levels of cyclic AMP and activates protein kinase A (PKA) (Albert et al 1990). PKA is important for haloperidol-mediated gene induction and cataleptic behavior (Adams et al 1997). PKA phosphorylates receptors and ion channels at the synapse, and modulates synaptic function (Gray et al 1998; Smart 1997; Swope et al 1999) and the activity of other protein kinases (Leveque et al 2000; Meshul and Tan 1994; Rodrigues and Dowling 1990). In addition, PKA activates transcription factors which regulate gene expression, such as the cyclic AMP and Ca2+ responsive element binding protein (CREB; Figure 3) (Gonzalez and Montminy 1989). The activation of CREB by PKA plays a critical role in neuroplasticity and memory formation in such varied species as Aplysia, Drosophila, and mouse (Frank and Greenberg 1994; Silva et al 1998). Haloperidol mediates gene expression via a signal transduction pathway that depends on PKA (Adams et al 1997) and CREB (Konradi and Heckers 1995; Konradi et al 1993), (Figure 3). The observations on the molecular mechanisms of haloperidol are important for the evaluation of the long-term and long-lasting antipsychotic effects of the drug.
Figure 3.

Gene expression and synaptogenesis by haloperidol. (A) A medium-size spiny neuron in the striatum that expresses D2 receptors contains the transcription factor CREB in the nucleus. (B) Upon interaction of haloperidol and D2 receptors, PKA is activated and proteins are phosphorylated. Phosphorylation of proteins alters their properties and activities and could be the reason for the perforation of synapses (split PSD) (Kerns et al 1992; Meshul and Casey 1989; See et al 1992). PKA also sets a signal transduction cascade in motion that translocates to the nucleus and phosphorylates the transcription factor CREB (Konradi and Heckers 1995). Phosphorylation of CREB at Ser133 (Gonzalez and Montminy 1989) enables the expression of genes and proteins that are under the control of Ca2+ and cyclic AMP second messenger pathways (Montminy 1997). (C) By incorporating newly synthesized proteins, the dendritic spine splits and yields two synapses. Compensatory mechanisms to slow down synaptogenesis will likely develop. Discontinuation of haloperidol administration reverses the process.
Haloperidol Affects Gene Expression and New Protein Synthesis
Gene expression and protein synthesis are essential for various brain functions, notably for the formation of long-term memory (Davis and Squire 1984). Haloperidol has been shown to activate levels of many different transcription factor genes in the striatum (Atkins et al 1999; Hiroi and Graybiel 1996; MacGibbon et al 1995; Nguyen et al 1992). As a consequence, haloperidol affects the regulation of many proteins. Genes of receptors (Burt et al 1977; Eastwood et al 1996; Egan et al 1994; Fitzgerald et al 1995; Florijn et al 1997; Kornhuber et al 1989; Lidow and Goldman-Rakic 1997; Riva et al 1997), neuropeptides (Konradi et al 1993; Merchant et al 1992; Tang et al 1983), and synaptic proteins (Eastwood et al 1997; Nakahara et al 1998) are regulated by haloperidol. This profile of gene regulation explains how haloperidol alters the properties of neurons and influences synaptic function. Modulation of receptor function via phosphorylation on the one hand, and via alteration of receptor protein levels on the other, changes the response properties of the affected neurons to presynaptic neurotransmitter release. The modulation of neuropeptide levels affects the amplitude of postsynaptic stimulation. The alteration in the expression of synaptic proteins is needed for synaptogenesis and the synaptic reorganization observed after chronic haloperidol treatment (Figure 3).
Mechanisms by Which Haloperidol Could Mediate Synaptic Plasticity
The various lines of evidence reviewed above provide the foundation for the hypothesis that haloperidol induces synaptic plasticity in the adult brain. But what are the mechanisms that lead to these observed changes? Because the study of synaptogenesis in the mature brain is restricted by methodological limitations, the complexity of higher brain function is principally modeled by synaptogenesis in the developing central nervous system. To discuss the relevance of synaptic plasticity for the effects of haloperidol, we will illustrate the mechanisms of synaptic plasticity established in the developing brain followed by a discussion of the data available on the mature brain.
General Mechanisms of Synaptic Plasticity
Synaptogenesis in the Developing Brain
This chapter reviews the molecular processes that lead to synapse formation in the developing brain. Immature neurons have hairlike processes (filopodia) that explore the environment in search for other neurons to form synapses (Figures 4, 5). As the brain matures the emphasis shifts from scanning the environment for new contacts to solidifying the already existing contacts. Functional synapses are built and a mode of communication between both sides of the synapse is arranged. If proper communication cannot be established, the synapse is withdrawn. Thus, there are at least two parameters that are important for synapse density: the number of synapses that were originally established, and the effectiveness of communication between these synapses (Figure 4).
Figure 4.

Early synapse formation and developmental processes that could cause schizophrenia. Neurons extend hairlike structures (filopodia) in search for partners to form synapses. A period of extensive synapse formation is followed by synapse stabilization and use-dependent pruning. Synapses that cannot establish communication are removed. This model is supported by developmental studies in the human brain, which show a peak in synapse numbers during the early postnatal period, followed by synapse elimination (Huttenlocher 1984; Huttenlocher et al 1982). (A) Normal development. (B), (C) A consistent finding in schizophrenia is a reduction in dendrites and synapses, which is most likely based on a developmental malfunction (McGlashan and Hoffman 2000). Such a reduction can be caused by faulty synapse formation (B), or faulty synapse stabilization (C).
Figure 5.

Synaptogenesis during early neuronal development. (A–C) During early neuronal development, the number of dendritic filopodia decreases while the number of spines increases. (D) An axonal growth cone and a dentritic filopodium make contact and a molecular adhesion process ensues. (E) Prefabricated protein aggregates get trapped at the presynaptic contact zone and enable the axon to release neurotransmitter. (F) The release of neurotransmitter triggers the formation of the PSD.
Much has been accomplished in elucidating the molecular processes that are involved in the various steps from initial contact to the final synapse with synchronized pre-and postsynaptic activities. These processes are fairly complex and are described below in some detail. Of interest for schizophrenia research, glutamate receptors play an important role in synapse formation and stabilization (see below and Figure 6), thus linking two abnormalities observed in schizophrenia: the decreased neuropil and abnormal connectivity in microcircuits (Benes 2000; Selemon and Goldman-Rakic 1999), and the hypofunction of the glutamatergic system (Coyle 1996; Olney et al 1999; Tamminga 1999). The involvement of glutamate receptors in synaptogenesis suggests that reduced glutamatergic activity leads to a reduced number of synapses and the “miswiring” in schizophrenia (Figure 4).
Figure 6.

Ionotropic glutamate receptors and synapse stabilization. (A) Initially, NMDA receptors are in the postsynaptic membrane, while AMPA/kainate receptors are in the cytoplasm. (B) At glutamate release the NMDA receptor binds glutamate but does not pass ions because Mg2+ blocks the channel. The postsynaptic membrane does not depolarize. (C) A coincidence of glutamate release and depolarization opens the NMDA receptor and removes the Mg2+ block. Ions pass and AMPA/kainate receptors get recruited into the postsynaptic membrane. (D) The postsynaptic membrane contains NMDA and AMPA/kainate receptors. At glutamate release, AMPA/kainate and NMDA receptors get activated. AMPA/kainate receptors provide a local depolarization that removes the Mg2+ block from the NMDA receptor. Ions pass through the NMDA receptor.
Ontogenetic synaptogenesis consists of two consecutive processes: synapse formation and synapse stabilization (Haas 2000; Haydon and Drapeau 1995). In early neuronal development, axonal growth cones and dendritic filopodia search for partners to form synapses (Lee and Sheng 2000; Wong and Wong 2000), (Figure 5). On contact, a molecular adhesion process initiates the formation of the synapse (Hagler and Goda 1998). At the presynaptic contact area, prefabricated protein aggregates that contain molecular components for the active zone get trapped and release neurotransmitter (Ahmari et al 2000). Neurotransmitter release stimulates the postsynaptic site to attract scaffolding proteins such as SAP90/PSD-95 and neurotransmitter receptors (Friedman et al 2000), thus building the PSD and a functional synapse (Figures 2, 5). Formation of a new synapse can take as little as one to two hours after an axodendritic contact has been established (Friedman et al 2000).
A newly formed synapse can be disassembled easily and needs to be stabilized to survive. Synapse stabilization can be activity-dependent, and/or depend on molecular cues provided by adhesion molecules and neurotrophic factors (Haydon and Drapeau 1995). Activity-dependent stabilization seems to require the postsynaptic activity of NMDA receptors and of AMPA receptors (Figure 6). A synapse that contains only NMDA receptors cannot depolarize and needs a coincidence of glutamate release and synaptic depolarization to pass ions through the NMDA receptors. Opening of NMDA receptors induces an incorporation of AMPA receptors into the postsynaptic membrane on dendritic spines (Shi et al 1999; Wu et al 1996); (Figure 6A–C). Once the AMPA receptor is in the membrane, it can, upon glutamate release, provide the depolarization needed for NMDA receptor activation (Figure 6D). The consistent activation of the postsynaptic neuron by presynaptic transmitter release stabilizes the synapse (Haydon and Drapeau 1995); failure to stabilize the synapse leads to the dissolution of the spine (Jontes and Smith 2000).
Synaptic Plasticity in the Mature Brain
While synaptogenesis research deals predominantly with the construction of nervous systems during development, haloperidol is usually administered to the mature brain. The current knowledge of synaptic plasticity in the mature brain is therefore of important consideration for our understanding of haloperidol. Synaptic plasticity is achieved in the mature brain by one of three different mechanisms, discussed below: a rearrangement of existing synapses, a formation of new synapses, or a pruning of existing synapses.
Synaptic rearrangements and dendritic motility are common in the mature brain and are mediated by actin polymerization and ion concentration (Dunaevsky et al 1999; Fischer et al 1998). Rapid changes in spine shape affect synapse strength and synapse stability and are thought to be an important basis for learning and memory (Harris 1999; Matus 1999; Svoboda et al 1996). Though the spine shape is supported by the actin cytoskeleton, spines can be rearranged through swelling in response to sodium influx (Fifkova and Van Harreveld 1977; Van Harreveld and Fifkova 1975), and through actin depolymerization in response to Ca2+ influx (Matus 1999).
Novel spines can be generated in response to increased neuronal activity (Engert and Bonhoeffer 1999; Toni et al 1999), or in response to neurotrophic factors (McAllister et al 1999). Formation of new spines happens at a surprising speed in the postnatal brain, though it may take longer for the new spines to be functional. In rat hippocampal slices new spines can form within one hour of high-frequency stimulation (Engert and Bonhoeffer 1999; Maletic-Savatic et al 1999; Toni et al 1999); the number of spines in mature slices are increased by 40%–50% within one to two hours after slicing (Kirov et al 1999). While the new spines seem to form synapses, it is not precisely known how pre- and postsynaptic elements find each other in the mature brain. Unlike in early synaptogenesis, an involvement of filopodia in the formation of new spines in the mature brain is unlikely, since filopodia were replaced by synapses during maturation (Ziv and Smith 1996), (Figure 5) and are not found in mature brains (Harris 1999); however, the discovery of an activity-dependent increase in axonal boutons with multiple synapses in the mature brain is indicative of spine splitting, in which case the pre- and postsynaptic elements would be already in place (Figure 3), (Kirov et al 1999; Toni et al 1999).
A decrease in synapse number could be caused by an active mechanism such as synapse pruning, or by neuronal death. While activity strengthens synapses, cells that do not activate postsynaptic neurons have their contacts weakened and eliminated. This variant of neuroplasticity is important for learning and memory (Jontes and Smith 2000). The loss of entire neurons in the mature brain, on the other hand, is a pathologic rather than plastic phenomenon that is inevitably accompanied by a loss of synapses.
From Haloperidol-treatment to Synaptic Plasticity
Ultrastructural analyses show that treatment with haloperidol alters spine shape as well as spine density. The observed change in spine shape is indicative of haloperidol’s ability to modulate synaptic strength (Harris 1999; Matus 1999). As rapid changes in spine shape are driven by Ca2+ -dependent actin polymerization (Fischer et al 1998; Matus 1999) and by swelling secondary to the influx of ions (Fifkova and Van Harreveld 1977; Van Harreveld and Fifkova 1975), the question arises whether haloperidol can affect the influx of Ca2+ and other ions. Indeed, the signal transduction pathway activated by haloperidol can modulate NMDA receptor function, which affects Na+ and Ca2+ flux and which may be relevant for the rapid change in spine shape (Leveque et al 2000).
The increase in synapse density observed after treatment with haloperidol is accompanied by an increased synthesis of synaptic proteins (Eastwood et al 1997; Nakahara et al 1998). Synapse splitting seems to be one of the mechanisms responsible for the increase in synapse number (Kerns et al 1992) (Figure 3). Increase in synapse number, in general, can be observed after enhanced activity in neural circuits or after increased synthesis and release of neurotrophic factors. Haloperidol may employ both mechanisms. While haloperidol does not increase neuronal activity directly, it does modulate other neurotransmitter systems that promote neuronal activity. The well-documented facilitation of the glutamatergic system by haloperidol, for example, enables the indirect modulation of synaptic activity (Brene et al 1998; De Souza et al 1999; Eastwood et al 1996; Fitzgerald et al 1995; Kerns et al 1992; Leveque et al 2000; Meshul et al 1996; Meshul and Casey 1989; Moghaddam and Bunney 1993; Riva et al 1997; Schneider et al 1998; See and Chapman 1994; See et al 1992; Yamamoto and Cooperman 1994). The increase in synapse number after treatment with haloperidol could therefore be caused by an indirect enhancement of synaptic activity.
It has recently been shown that an upregulation of the neurotrophin brain derived neurotrophic factor (BDNF) plays a role in the clinical effects of antidepressants (Altar 1999). It is therefore of interest to examine the interaction of antipsychotic drugs with neurotrophic factors. Because BDNF is not synthesized in striatal neurons (Altar et al 1997), and neither haloperidol nor clozapine increase BDNF levels in cortex or hippocampus (Angelucci et al 2000; Linden et al 2000; Nibuya et al 1995), BDNF itself does not seem to play much of a role in the action of antipsychotic drugs; however, levels of another neurotrophin, nerve growth factor, are increased in the striatum after treatment with haloperidol (Ozaki 2000; Ozaki et al 1999). No data are available on the role of nerve growth factor in antipsychotic properties. Taken together, a contribution of neurotrophic factors to synaptogenesis by antipsychotic drugs does not seem likely but needs further examination before it should be ruled out.
While the decrease in synapse number reported in some studies (Roberts et al 1995) seems to be predominantly a consequence of neurotoxicity, it is also possible that haloperidol may reduce the synaptic activity of some connections, causing destabilization and withdrawal of synapses. Even though there is substantial evidence for neurotoxic effects of haloperidol in the striatum (Harrison 1999), decreased synapse number is also compatible with a pruning of synapses in response to suppressed activity in specific neuronal circuits.
Does Haloperidol Affect Neurogenesis?
Adult neurogenesis, the creation of new neurons in the mature brain, has recently gained renewed attention (Barinaga 1998; Gross 2000). While the vast majority of neurons are generated during early brain development, limited neurogenesis occurs in the mature brain with regional specificity. The addition of new neurons to an adult brain can, in principle, improve brain function and acquisition of memory. Hippocampal neurons and olfactory bulb interneurons can be generated in the adult mammalian nervous system (Cameron and McKay 1998). In the adult human brain, mitotically competent neuronal precursors have been found in the hippocampus (Eriksson et al 1998; Roy et al 2000). The authenticity of neurogenesis reported in other brain areas (e.g., cortex) is presently a matter of debate (Cameron and McKay 1998; Gould et al 1999; Nowakowski and Hayes 2000). A resolution of this discussion will be helpful for any investigation of neuroplasticity by antipsychotic drugs, particularly since a recent report of increased neurogenesis in the adult rat hippocampus after chronic treatment with antidepressant drugs (Malberg et al 2000) raised the question whether antipsychotic drugs have similar effects. Paradoxically, haloperidol seems to cause neurogenesis in the gerbil hippocampus (Dawirs et al 1998), but not in the rat hippocampus (Malberg et al 2000). Its effect on neurogenesis in the human brain is not known. Because of the potential species specificity and the lack of data in the human brain, neurogenesis by antipsychotic drugs cannot, at this point, be critically evaluated.
What About Atypical Antipsychotic Drugs?
The striatum is the brain area best suited to study neuroplasticity by conventional antipsychotic drugs, although it may not be the principal locus for the cognitive deficits that characterize schizophrenia. Since D2 receptors, the main target for conventional antipsychotic drugs, are abundantly expressed in the striatum, the striatum is the brain area where responses to antipsychotic drug exposure are most noticeable. Importantly, the effects of haloperidol are not limited to the striatum. Other brain areas implicated in schizophrenia such as the prefrontal cortex, hippocampus and thalamus, are affected as well, albeit to a lesser extent (Figure 1). The action of haloperidol in these brain areas may be more relevant for its antipsychotic properties, but studies from these brain areas are sparse and more ambiguous. Atypical antipsychotic drugs such as clozapine or olanzapine may have their biggest impact in these brain areas, and they provide valuable information for our understanding of the restorative processes induced by treatment.
Atypical antipsychotic drugs affect the dopaminergic system to a lesser extent than conventional antipsychotic drugs and, in addition, inhibit serotonergic, muscarinic, histaminergic and α1 adrenergic receptors (Bymaster et al 1996; Bymaster et al 1999; Farde et al 1992; Kapur et al 1999; Nordstrom et al 1995; Pilowsky et al 1992; Seeman et al 1997; Stockmeier et al 1993; Trichard et al 1998). Atypical antipsychotic drugs have a more widespread yet temperate impact across neurotransmitter systems and brain regions. They are less potent in the striatum, and anatomical changes are generally more subtle and difficult to uncover. Atypical antipsychotic drugs unquestionably affect neuroplasticity, as they induce gene expression in many brain areas (Deutch and Duman 1996; Lidow and Goldman-Rakic 1997; MacGibbon et al 1994; Merchant et al 1994; Nguyen et al 1992; Robertson and Fibiger 1992). C-Fos expression is induced by atypical antipsychotic drugs with a different regional expression profile and intensity than conventional antipsychotic drugs (Deutch and Duman 1996; Nguyen et al 1992; Robertson and Fibiger 1992). In the striatum, atypical antipsychotic drugs induce gene expression to a lesser extent and with a somewhat different distribution than conventional antipsychotic drugs (Leveque et al 2000; MacGibbon et al 1994; Robertson and Fibiger 1992; Robertson and Fibiger 1996; Robertson et al 1994). More importantly, in agreement with their pharmacological profile, atypical antipsychotic drugs induce genes in brain areas that are implicated in the pathophysiology of schizophrenia, such as the prefrontal cortex (Robertson and Fibiger 1992; Robertson and Fibiger 1996; Robertson et al 1994; Sebens et al 1995; Vahid-Ansari et al 1996; Wan et al 1995). Thus, atypical antipsychotic drugs can further facilitate our understanding of the anatomical and cellular deficiencies in schizophrenia. Moreover, they support the notion that modulation of neuroplasticity in specific brain areas is of critical consideration for the treatment of schizophrenia.
Implications for the Treatment and Neuropathology of Schizophrenia
The delayed therapeutic action of antipsychotic drugs, together with their promotion of neuroplasticity, suggests that the ultrastructural modulation of neuronal circuits is an important contributor to antipsychotic properties. Despite our focus on the striatum, subtler changes in other brain areas may be responsible for, or contribute to, the therapeutic effect of antipsychotic drugs. Such brain areas include limbic structures and the prefrontal cortex (Figure 1), which are implicated in schizophrenia, and which show synaptic rearrangements and altered gene expression in response to antipsychotic drug treatment (Benes et al 1985b; Eastwood et al 1997; Fitzgerald et al 1995; Klintzova et al 1989; Robertson and Fibiger 1992). Neuroplasticity in extrastriatal structures such as the prefrontal cortex, nucleus accumbens and other brain areas (Damask et al 1996; Fitzgerald et al 1995; Robertson and Fibiger 1992; Vahid-Ansari et al 1996; Vincent et al 1991) could explain some of the differences in the pharmacological profile and the therapeutic effects of typical and atypical antipsychotic drugs. Thus, while the striatum was the best place to initiate studies on the effects of antipsychotic drugs on neuroplasticity, a thorough examination of extrastriatal areas is now needed.
So far, the importance of neuroplasticity for the beneficial action of antipsychotic drugs has not been sufficiently explored. If neuroplasticity is a crucial mechanism for antipsychotic properties, it could lead to the development of a new class of antipsychotic drugs. The data on haloperidol and neuroplasticity suggest that there may be a novel therapeutic avenue. Data on atypical antipsychotic drugs support that notion and help to identify the relevant brain areas. The side effects of drugs currently available, and the high percentage of patients who do not respond to treatment, are fueling the quest for more therapeutic options.
Should antipsychotic drugs indeed ‘rewire’ the brain, such a realization could provide us with new insights into the pathology of schizophrenia. Recent neuropathology and neuroimaging studies of schizophrenia have provided results that are compatible with the hypothesis that successful treatment of schizophrenia is accomplished by modification of synaptic connectivity. For example, anatomical changes in the prefrontal cortex in schizophrenia include decreased neuropil (Selemon and Goldman-Rakic 1999), selective decrease of GABAergic terminals (Woo et al 1998), and altered expression of a family of genes that control presynaptic function (Mirnics et al 2000). These abnormalities are potentially amenable to treatment with compounds that improve synaptic connectivity. The abnormality of the functional integration of the prefrontal cortex and temporal lobe structures in schizophrenia (Fletcher 1998; Heckers et al 1998), and the prefrontal-temporal disconnection syndrome (Bogerts 1997; Friston 1998) also suggest a malfunction of neuroplasticity. Furthermore, schizophrenia has been conceptualized as a neurodevelopmental rather than neurodegenerative disorder (Woods 1998), with abnormal neuronal migration and an incomplete establishment of cortico-cortical and cortico-subcortical projections (Akbarian et al 1993). Treatment that targets synaptic remodeling of such abnormalities could potentially be beneficial, particularly at early age.
Taken together, the hypothesis that antipsychotic drug treatment targets synaptic plasticity is consistent with recent evidence of abnormal synaptic function in schizophrenia. Modulation and facilitation of synaptic plasticity should be considered in future drug development.
Conclusion
There is ample evidence that the typical antipsychotic drug haloperidol affects neuroplasticity in the mature brain. The effect of haloperidol on synapse formation and rearrangement may be important for its antipsychotic properties, and may provide insight into the brain abnormalities leading to schizophrenia. A further analysis of the role of neuroplasticity in the cause and treatment of schizophrenia could provide us with a novel treatment approach.
Acknowledgments
This work was supported by the National Alliance for Research on Schizophrenia and Depression (CK, SH), and a National Institute of Drug Abuse grant DA07134 (CK).
References
- Adams MR, Brandon EP, Chartoff EH, Idzerda RL, Dorsa DM, McKnight GS. Loss of haloperidol induced gene expression and catalepsy in protein kinase A-deficient mice. Proc Natl Acad Sci USA. 1997;94:12157–12161. doi: 10.1073/pnas.94.22.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nature Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Bunney WE, Jr, Potkin SG, et al. Altered distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase cells in frontal lobe of schizophrenics implies disturbances of cortical development. Arch Gen Psychiatry. 1993;50:169–177. doi: 10.1001/archpsyc.1993.01820150007001. [DOI] [PubMed] [Google Scholar]
- Albert PR, Neve KA, Bunzow JR, Civelli O. Coupling of a cloned rat dopamine-D2 receptor to inhibition of adenylyl cyclase and prolactin secretion. J Biol Chem. 1990;265:2098–2104. [PubMed] [Google Scholar]
- Altar CA. Neurotrophins and depression. Trends Pharmacol Sci. 1999;20:59–61. doi: 10.1016/s0165-6147(99)01309-7. [DOI] [PubMed] [Google Scholar]
- Altar CA, Cai N, Bliven T, et al. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Andreasen NC. Symptoms, signs, and diagnosis of schizophrenia. Lancet. 1995;346:477–481. doi: 10.1016/s0140-6736(95)91325-4. [DOI] [PubMed] [Google Scholar]
- Andreasen NC. Pieces of the schizophrenia puzzle fall into place. Neuron. 1996;16:697–700. doi: 10.1016/s0896-6273(00)80090-2. [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Jorgensen HA. Neurotoxicity associated with neuroleptic-induced oral dyskinesias in rats. Implications for tardive dyskinesia? Prog Neurobiol. 2000;61:525–541. doi: 10.1016/s0301-0082(99)00064-7. [DOI] [PubMed] [Google Scholar]
- Angelucci F, Mathe AA, Aloe L. Brain-derived neurotrophic factor and tyrosine kinase receptor TrkB in rat brain are significantly altered after haloperidol and risperidone administration. J Neurosci Res. 2000;60:783–794. doi: 10.1002/1097-4547(20000615)60:6<783::AID-JNR11>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Atkins JB, Chlan-Fourney J, Nye HE, Hiroi N, Carlezon WA, Jr, Nestler EJ. Region-specific induction of deltaFosB by repeated administration of typical versus atypical antipsychotic drugs. Synapse. 1999;33:118–128. doi: 10.1002/(SICI)1098-2396(199908)33:2<118::AID-SYN2>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Barinaga M. No-new-neurons dogma loses ground. Science. 1998;279:2041–2042. doi: 10.1126/science.279.5359.2041. [DOI] [PubMed] [Google Scholar]
- Beckmann H, Lauer M. The human striatum in schizophrenia. II. Increased number of striatal neurons in schizophrenics. Psychiatry Res. 1997;68:99–109. doi: 10.1016/s0925-4927(96)02947-2. [DOI] [PubMed] [Google Scholar]
- Benes FM. Emerging principles of altered neural circuitry in schizophrenia. Brain Res Brain Res Rev. 2000;31:251–269. doi: 10.1016/s0165-0173(99)00041-7. [DOI] [PubMed] [Google Scholar]
- Benes FM, Paskevich PA, Davidson J, Domesick VB. The effects of haloperidol on synaptic patterns in the rat striatum. Brain Res. 1985a;329:265–273. doi: 10.1016/0006-8993(85)90532-3. [DOI] [PubMed] [Google Scholar]
- Benes FM, Paskevich PA, Davidson J, Domesick VB. Synaptic rearrangements in medial prefrontal cortex of haloperidol-treated rats. Brain Res. 1985b;348:15–20. doi: 10.1016/0006-8993(85)90353-1. [DOI] [PubMed] [Google Scholar]
- Benes FM, Paskevich PA, Domesick VB. Haloperidol-induced plasticity of axon terminals in rat substantia nigra. Science. 1983;221:969–971. doi: 10.1126/science.6879197. [DOI] [PubMed] [Google Scholar]
- Bilder RM, Wu H, Chakos MH, et al. Cerebral morphometry and clozapine treatment in schizophrenia. J Clin Psychiatry. 1994;55:53–56. [PubMed] [Google Scholar]
- Bogerts B. The temporolimbic system theory of positive schizophrenic symptoms. Schizophr Bull. 1997;23:423–435. doi: 10.1093/schbul/23.3.423. [DOI] [PubMed] [Google Scholar]
- Brene S, Messer C, Nestler EJ. Expression of messenger RNAs encoding ionotropic glutamate receptors in rat brain: Regulation by haloperidol. Neuroscience. 1998;84:813–823. doi: 10.1016/s0306-4522(97)00490-9. [DOI] [PubMed] [Google Scholar]
- Brown KW, White T, Wardlaw JM, Walker N, Foley D. Caudate nucleus morphology in tardive dyskinesia. Br J Psychiatry. 1996;169:631–636. doi: 10.1192/bjp.169.5.631. [DOI] [PubMed] [Google Scholar]
- Browning MD, Dudek EM, Rapier JL, Leonard S, Freedman R. Significant reductions in synapsin but not synaptophysin specific activity in the brains of some schizophrenics. Biol Psychiatry. 1993;34:529–535. doi: 10.1016/0006-3223(93)90195-j. [DOI] [PubMed] [Google Scholar]
- Burkhardt C, Kelly JP, Lim YH, Filley CM, Parker WD., Jr Neuroleptic medications inhibit complex I of the electron transport chain. Ann Neurol. 1993;33:512–517. doi: 10.1002/ana.410330516. [DOI] [PubMed] [Google Scholar]
- Burt DR, Creese I, Snyder SH. Antischizophrenic drugs: Chronic treatment elevates dopamine receptor binding in brain. Science. 1977;196:326–328. doi: 10.1126/science.847477. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Hemrick-Luecke SK, Perry KW, Fuller RW. Neurochemical evidence for antagonism by olanzapine of dopamine, serotonin, alpha 1-adrenergic and muscarinic receptors in vivo in rats. Psychopharmacology. 1996;124:87–94. doi: 10.1007/BF02245608. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Nelson DL, DeLapp NW, et al. Antagonism by olanzapine of dopamine D1, serotonin2, muscarinic, histamine H1 and alpha 1-adrenergic receptors in vitro. Schizophr Res. 1999;37:107–122. doi: 10.1016/s0920-9964(98)00146-7. [DOI] [PubMed] [Google Scholar]
- Cameron HA, McKay R. Stem cells and neurogenesis in the adult brain. Curr Opin Neurobiol. 1998;8:677–680. doi: 10.1016/s0959-4388(98)80099-8. [DOI] [PubMed] [Google Scholar]
- Carpenter WT, Jr, Buchanan RW. Schizophrenia. N Engl J Med. 1994;330:681–690. doi: 10.1056/NEJM199403103301006. [DOI] [PubMed] [Google Scholar]
- Chakos MH, Lieberman JA, Alvir J, Bilder R, Ashtari M. Caudate nuclei volumes in schizophrenic patients treated with typical antipsychotics or clozapine. Lancet. 1995;345:456–457. doi: 10.1016/s0140-6736(95)90441-7. [DOI] [PubMed] [Google Scholar]
- Chakos MH, Lieberman JA, Bilder RM, et al. Increase in caudate nuclei volumes of first-episode schizophrenic patients taking antipsychotic drugs. Am J Psychiatry. 1994;151:1430–1436. doi: 10.1176/ajp.151.10.1430. [DOI] [PubMed] [Google Scholar]
- Chakos MH, Shirakawa O, Lieberman J, Lee H, Bilder R, Tamminga CA. Striatal enlargement in rats chronically treated with neuroleptic. Biol Psychiatry. 1998;44:675–684. doi: 10.1016/s0006-3223(98)00029-8. [DOI] [PubMed] [Google Scholar]
- Coyle JT. The glutamatergic dysfunction hypothesis for schizophrenia. Harvard Rev Psychiatry. 1996;3:241–253. doi: 10.3109/10673229609017192. [DOI] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- Dalgalarrondo P, Gattaz WF. Basal ganglia abnormalities in tardive dyskinesia. Possible relationship with duration of neuroleptic treatment. Eur Arch Psychiatry Clin Neurosci. 1994;244:272–277. doi: 10.1007/BF02190380. [DOI] [PubMed] [Google Scholar]
- Damask SP, Bovenkerk KA, de la Pena G, et al. Differential effects of clozapine and haloperidol on dopamine receptor mRNA expression in rat striatum and cortex. Brain Res Mol Brain Res. 1996;41:241–249. doi: 10.1016/0169-328x(96)00101-5. [DOI] [PubMed] [Google Scholar]
- Davidsson P, Gottfries J, Bogdanovic N, et al. The synaptic-vesicle-specific proteins rab3a and synaptophysin are reduced in thalamus and related cortical brain regions in schizophrenic brains. Schizophr Res. 1999;40:23–29. doi: 10.1016/s0920-9964(99)00037-7. [DOI] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: A review. Psychological Bull. 1984;96:518–559. [PubMed] [Google Scholar]
- Dawirs RR, Hildebrandt K, Teuchert-Noodt G. Adult treatment with haloperidol increases dentate granule cell proliferation in the gerbil hippocampus. J Neural Transm (Budapest) 1998;105:317–327. doi: 10.1007/s007020050061. [DOI] [PubMed] [Google Scholar]
- De Souza IE, McBean GJ, Meredith GE. Chronic haloperidol treatment impairs glutamate transport in the rat striatum. Eur J Pharm. 1999;382:139–142. doi: 10.1016/s0014-2999(99)00589-0. [DOI] [PubMed] [Google Scholar]
- DeLisi LE, Hoff AL, Schwartz JE, et al. Brain morphology in first-episode schizophrenic-like psychotic patients: A quantitative magnetic resonance imaging study. Biol Psychiatry. 1991;29:159–175. doi: 10.1016/0006-3223(91)90044-m. [DOI] [PubMed] [Google Scholar]
- Deutch AY, Duman RS. The effects of antipsychotic drugs on Fos protein expression in the prefrontal cortex: Cellular localization and pharmacological characterization. Neuroscience. 1996;70:377–389. doi: 10.1016/0306-4522(95)00357-6. [DOI] [PubMed] [Google Scholar]
- Doraiswamy PM, Tupler LA, Krishnan KR. Neuroleptic treatment and caudate plasticity. Lancet. 1995;345:734–735. [PubMed] [Google Scholar]
- Dunaevsky A, Tashiro A, Majewska A, Mason C, Yuste R. Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci USA. 1999;96:13438–13443. doi: 10.1073/pnas.96.23.13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood SL, Burnet PW, Harrison PJ. Striatal synaptophysin expression and haloperidol-induced synaptic plasticity. Neuroreport. 1994;5:677–680. doi: 10.1097/00001756-199402000-00004. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Burnet PW, Harrison PJ. Altered synaptophysin expression as a marker of synaptic pathology in schizophrenia. Neuroscience. 1995;66:309–319. doi: 10.1016/0306-4522(94)00586-t. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Cairns NJ, Harrison PJ. Synaptophysin gene expression in schizophrenia. Investigation of synaptic pathology in the cerebral cortex. Br J Psychiatry. 2000;176:236–242. doi: 10.1192/bjp.176.3.236. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Harrison PJ. Decreased synaptophysin in the medial temporal lobe in schizophrenia demonstrated using immunoautoradiography. Neuroscience. 1995;69:339–343. doi: 10.1016/0306-4522(95)00324-c. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Heffernan J, Harrison PJ. Chronic haloperidol treatment differentially affects the expression of synaptic and neuronal plasticity-associated genes. Mol Psychiatry. 1997;2:322–329. doi: 10.1038/sj.mp.4000238. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Porter RH, Harrison PJ. The effect of chronic haloperidol treatment on glutamate receptor subunit (GluR1, GluR2, KA1, KA2, NR1) mRNAs and glutamate binding protein mRNA in rat forebrain. Neurosci Lett. 1996;212:163–166. doi: 10.1016/0304-3940(96)12801-9. [DOI] [PubMed] [Google Scholar]
- Egan MF, Hurd Y, Hyde TM, Weinberger DR, Wyatt RJ, Kleinman JE. Alterations in mRNA levels of D2 receptors and neuropeptides in striatonigral and striatopallidal neurons of rats with neuroleptic-induced dyskinesias. Synapse. 1994;18:178–189. doi: 10.1002/syn.890180303. [DOI] [PubMed] [Google Scholar]
- Elkashef AM, Buchanan RW, Gellad F, Munson RC, Breier A. Basal ganglia pathology in schizophrenia and tardive dyskinesia: An MRI quantitative study. Am J Psychiatry. 1994;151:752–755. doi: 10.1176/ajp.151.5.752. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, et al. Neurogenesis in the adult human hippocampus. Nature Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Farde L, Nordstrom AL, Wiesel FA, Pauli S, Halldin C, Sedvall G. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Arch Gen Psychiatry. 1992;49:538–544. doi: 10.1001/archpsyc.1992.01820070032005. [DOI] [PubMed] [Google Scholar]
- Fifkova E, Van Harreveld A. Long-lasting morphological changes in dendritic spines of dentate granular cells following stimulation of the entorhinal area. J Neurocytol. 1977;6:211–230. doi: 10.1007/BF01261506. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Knutti D, Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. doi: 10.1016/s0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- Fitzgerald LW, Deutch AY, Gasic G, Heinemann SF, Nestler EJ. Regulation of cortical and subcortical glutamate receptor subunit expression by antipsychotic drugs. J Neurosci. 1995;15:2453–2461. doi: 10.1523/JNEUROSCI.15-03-02453.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher P. The missing link: A failure of fronto-hippocampal integration in schizophrenia. Nature Neurosci. 1998;1:266–267. doi: 10.1038/1078. [DOI] [PubMed] [Google Scholar]
- Florijn WJ, Tarazi FI, Creese I. Dopamine receptor subtypes: Differential regulation after 8 months treatment with antipsychotic drugs. J Pharmacol Exp Ther. 1997;280:561–569. [PubMed] [Google Scholar]
- Frank DA, Greenberg ME. CREB: A mediator of long-term memory from mollusks to mammals. Cell. 1994;79:5–8. doi: 10.1016/0092-8674(94)90394-8. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: Time course and temporal order of synaptic molecular recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Friston KJ. The disconnection hypothesis. Schizophr Res. 1998;30:115–125. doi: 10.1016/s0920-9964(97)00140-0. [DOI] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA. Reduction of synaptophysin immunoreactivity in the prefrontal cortex of subjects with schizophrenia. Regional and diagnostic specificity. Arch Gen Psychiatry. 1997;54:660–669. doi: 10.1001/archpsyc.1997.01830190088009. [DOI] [PubMed] [Google Scholar]
- Goff DC, Tsai G, Beal MF, Coyle JT. Tardive dyskinesia and substrates of energy metabolism in CSF. Am J Psychiatry. 1995;152:1730–1736. doi: 10.1176/ajp.152.12.1730. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Gould E, Reeves AJ, Fallah M, Tanapat P, Gross CG, Fuchs E. Hippocampal neurogenesis in adult Old World primates. Pro Natl Aca Sci USA. 1999;96:5263–5267. doi: 10.1073/pnas.96.9.5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray PC, Scott JD, Catterall WA. Regulation of ion channels by cAMP-dependent protein kinase and A-kinase anchoring proteins. Curr Opin Neurobiol. 1998;8:330–334. doi: 10.1016/s0959-4388(98)80057-3. [DOI] [PubMed] [Google Scholar]
- Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry. 1996;153:321–330. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- Gross CG. Neurogenesis in the adult brain: Death of a dogma. Nature Rev Neurosci. 2000;1:67–73. doi: 10.1038/35036235. [DOI] [PubMed] [Google Scholar]
- Gur RE, Maany V, Mozley PD, Swanson C, Bilker W, Gur RC. Subcortical MRI volumes in neuroleptic-naive and treated patients with schizophrenia. Am J Psychiatry. 1998;155:1711–1717. doi: 10.1176/ajp.155.12.1711. [DOI] [PubMed] [Google Scholar]
- Haas K. Prefabrication provides synapses on demand. Trends Neurosci. 2000;23:449. doi: 10.1016/s0166-2236(00)01678-7. [DOI] [PubMed] [Google Scholar]
- Hagler DJ, Jr, Goda Y. Synaptic adhesion: The building blocks of memory? Neuron. 1998;20:1059–1062. doi: 10.1016/s0896-6273(00)80486-9. [DOI] [PubMed] [Google Scholar]
- Harris KM. Structure, development, and plasticity of dendritic spines. Curr Opin Neurobiol. 1999;9:343–348. doi: 10.1016/s0959-4388(99)80050-6. [DOI] [PubMed] [Google Scholar]
- Harrison PJ. The neuropathological effects of antipsychotic drugs. Schizophr Res. 1999;40:87–99. doi: 10.1016/s0920-9964(99)00065-1. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Drapeau P. From contact to connection: Early events during synaptogenesis. Trends Neurosci. 1995;18:196–201. doi: 10.1016/0166-2236(95)93901-9. [DOI] [PubMed] [Google Scholar]
- Heckers S. Neuropathology of schizophrenia: Cortex, thalamus, basal ganglia, and neurotransmitter-specific projection systems. Schizophr Bull. 1997;23:403–421. doi: 10.1093/schbul/23.3.403. [DOI] [PubMed] [Google Scholar]
- Heckers S, Heinsen H, Heinsen Y, Beckmann H. Cortex, white matter, and basal ganglia in schizophrenia: A volumetric postmortem study. Biol Psychiatry. 1991;29:556–566. doi: 10.1016/0006-3223(91)90091-y. [DOI] [PubMed] [Google Scholar]
- Heckers S, Rauch SL, Goff D, et al. Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Nature Neurosci. 1998;1:318–323. doi: 10.1038/1137. [DOI] [PubMed] [Google Scholar]
- Hiroi N, Graybiel AM. Atypical and typical neuroleptic treatments induce distinct programs of transcription factor expression in the striatum. J Comp Neurol. 1996;374:70–83. doi: 10.1002/(SICI)1096-9861(19961007)374:1<70::AID-CNE5>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR. Synapse elimination and plasticity in developing human cerebral cortex. Am J Mental Deficiency. 1984;88:488–496. [PubMed] [Google Scholar]
- Huttenlocher PR, de Courten C, Garey LJ, Van der Loos H. Synaptogenesis in human visual cortex–evidence for synapse elimination during normal development. Neurosci Lett. 1982;33:247–252. doi: 10.1016/0304-3940(82)90379-2. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Nestler EJ. Initiation and adaptation: A paradigm for understanding psychotropic drug action. Am J Psychiatry. 1996;153:151–162. doi: 10.1176/ajp.153.2.151. [DOI] [PubMed] [Google Scholar]
- Jernigan TL, Zisook S, Heaton RK, Moranville JT, Hesselink JR, Braff DL. Magnetic resonance imaging abnormalities in lenticular nuclei and cerebral cortex in schizophrenia. Arch Gen Psychiatry. 1991;48:881–890. doi: 10.1001/archpsyc.1991.01810340013002. [DOI] [PubMed] [Google Scholar]
- Jones DG, Harris RJ. An analysis of contemporary morphological concepts of synaptic remodelling in the CNS: Perforated synapses revisited. Rev Neurosci. 1995;6:177–219. doi: 10.1515/revneuro.1995.6.3.177. [DOI] [PubMed] [Google Scholar]
- Jontes JD, Smith SJ. Filopodia, spines, and the generation of synaptic diversity. Neuron. 2000;27:11–14. doi: 10.1016/s0896-6273(00)00003-9. [DOI] [PubMed] [Google Scholar]
- Kapur S, Zipursky RB, Remington G. Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiatry. 1999;156:286–293. doi: 10.1176/ajp.156.2.286. [DOI] [PubMed] [Google Scholar]
- Karson CN, Mrak RE, Schluterman KO, Sturner WQ, Sheng JG, Griffin WS. Alterations in synaptic proteins and their encoding mRNAs in prefrontal cortex in schizophrenia: A possible neurochemical basis for ‘hypofrontality’. Mol Psychiatry. 1999;4:39–45. doi: 10.1038/sj.mp.4000459. [DOI] [PubMed] [Google Scholar]
- Kerns JM, Sierens DK, Kao LC, Klawans HL, Carvey PM. Synaptic plasticity in the rat striatum following chronic haloperidol treatment. Clin Neuropharmacol. 1992;15:488–500. doi: 10.1097/00002826-199212000-00006. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Bagwell WW, Haas GL, Sweeney JA, Schooler NR, Pettegrew JW. Changes in caudate volume with neuroleptic treatment. Lancet. 1994;344:1434. doi: 10.1016/s0140-6736(94)90599-1. [DOI] [PubMed] [Google Scholar]
- Kirov SA, Sorra KE, Harris KM. Slices have more synapses than perfusion-fixed hippocampus from both young and mature rats. J Neurosci. 1999;19:2876–2886. doi: 10.1523/JNEUROSCI.19-08-02876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klintzova AJ, Haselhorst U, Uranova NA, Schenk H, Istomin VV. The effects of haloperidol on synaptic plasticity in rat’s medial prefrontal cortex. J Hirnforsch. 1989;30:51–57. [PubMed] [Google Scholar]
- Konradi C, Heckers S. Haloperidol-induced Fos expression in striatum is dependent upon transcription factor cyclic AMP response element binding protein. Neuroscience. 1995;65:1051–1061. doi: 10.1016/0306-4522(94)00546-h. [DOI] [PubMed] [Google Scholar]
- Konradi C, Kobierski LA, Nguyen TV, Heckers S, Hyman SE. The cAMP-response-element-binding protein interacts, but Fos protein does not interact, with the proenkephalin enhancer in rat striatum. Proc Natl Acad Sci USA. 1993;90:7005–7009. doi: 10.1073/pnas.90.15.7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhuber J, Riederer P, Reynolds GP, Beckmann H, Jellinger K, Gabriel E. 3H-spiperone binding sites in postmortem brains from schizophrenic patients: Relationship to neuroleptic drug treatment, abnormal movements, and positive symptoms. J Neural Transm. 1989;75:1–10. doi: 10.1007/BF01250639. [DOI] [PubMed] [Google Scholar]
- Kung L, Conley R, Chute DJ, Smialek J, Roberts RC. Synaptic changes in the striatum of schizophrenic cases: A controlled postmortem ultrastructural study. Synapse. 1998;28:125–139. doi: 10.1002/(SICI)1098-2396(199802)28:2<125::AID-SYN3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Landen M, Davidsson P, Gottfries CG, Grenfeldt B, Stridsberg M, Blennow K. Reduction of the small synaptic vesicle protein synaptophysin but not the large dense core chromogranins in the left thalamus of subjects with schizophrenia. Biol Psychiatry. 1999;46:1698–1702. doi: 10.1016/s0006-3223(99)00160-2. [DOI] [PubMed] [Google Scholar]
- Lauer M, Beckmann H. The human striatum in schizophrenia. I. Increase in overall relative striatal volume in schizophrenics. Psychiatry Res. 1997;68:87–98. doi: 10.1016/s0925-4927(96)02946-0. [DOI] [PubMed] [Google Scholar]
- Lee SH, Sheng M. Development of neuron-neuron synapses. Curr Opin Neurobiol. 2000;10:125–131. doi: 10.1016/s0959-4388(99)00046-x. [DOI] [PubMed] [Google Scholar]
- Leveque J-C, Macías W, Rajadhyaksha A, et al. Intracellular modulation of NMDA receptor function by antipsychotic drugs. J Neurosci. 2000;20:4011–4020. doi: 10.1523/JNEUROSCI.20-11-04011.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidow MS, Goldman-Rakic PS. Differential regulation of D2 and D4 dopamine receptor mRNAs in the primate cerebral cortex vs. neostriatum: Effects of chronic treatment with typical and atypical antipsychotic drugs. J Pharmacol Exp Ther. 1997;283:939–946. [PubMed] [Google Scholar]
- Linden AM, Vaisanen J, Lakso M, Nawa H, Wong G, Castren E. Expression of neurotrophins BDNF and NT-3, and their receptors in rat brain after administration of antipsychotic and psychotrophic agents. J Mol Neurosci. 2000;14:27–37. doi: 10.1385/JMN:14:1-2:027. [DOI] [PubMed] [Google Scholar]
- MacGibbon GA, Lawlor PA, Bravo R, Dragunow M. Clozapine and haloperidol produce a differential pattern of immediate early gene expression in rat caudate-putamen, nucleus accumbens, lateral septum and islands of Calleja. Brain Res Mol Brain Res. 1994;23:21–32. doi: 10.1016/0169-328x(94)90207-0. [DOI] [PubMed] [Google Scholar]
- MacGibbon GA, Lawlor PA, Hughes P, Young D, Dragunow M. Differential expression of inducible transcription factors in basal ganglia neurons. Brain Res Mol Brain Res. 1995;34:294–302. doi: 10.1016/0169-328x(95)00184-t. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Matthysse S. Antipsychotic drug actions: A clue to the neuropathology of schizophrenia? Federation Proc. 1973;32:200–205. [PubMed] [Google Scholar]
- Matus A. Postsynaptic actin and neuronal plasticity. Curr Opin Neurobiol. 1999;9:561–565. doi: 10.1016/S0959-4388(99)00018-5. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Ann Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- McGlashan TH, Hoffman RE. Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch Gen Psychiatry. 2000;57:637–648. doi: 10.1001/archpsyc.57.7.637. [DOI] [PubMed] [Google Scholar]
- Merchant KM, Dobie DJ, Dorsa DM. Expression of the proneurotensin gene in the rat brain and its regulation by antipsychotic drugs. Ann NY Acad Sci. 1992;668:54–69. doi: 10.1111/j.1749-6632.1992.tb27339.x. [DOI] [PubMed] [Google Scholar]
- Merchant KM, Dobie DJ, Filloux FM, Totzke M, Aravagiri M, Dorsa DM. Effects of chronic haloperidol and clozapine treatment on neurotensin and c-fos mRNA in rat neostriatal subregions. J Pharmacol Exp Ther. 1994;271:460–471. [PubMed] [Google Scholar]
- Meshul CK, Andreassen OA, Allen C, Jorgensen HA. Correlation of vacuous chewing movements with morphological changes in rats following 1-year treatment with haloperidol. Psychopharmacology. 1996;125:238–247. doi: 10.1007/BF02247334. [DOI] [PubMed] [Google Scholar]
- Meshul CK, Casey DE. Regional, reversible ultrastructural changes in rat brain with chronic neuroleptic treatment. Brain Res. 1989;489:338–346. doi: 10.1016/0006-8993(89)90867-6. [DOI] [PubMed] [Google Scholar]
- Meshul CK, Janowsky A, Casey DE, Stallbaumer RK, Taylor B. Effect of haloperidol and clozapine on the density of “perforated” synapses in caudate, nucleus accumbens, and medial prefrontal cortex. Psychopharmacology. 1992;106:45–52. doi: 10.1007/BF02253587. [DOI] [PubMed] [Google Scholar]
- Meshul CK, Tan SE. Haloperidol-induced morphological alterations are associated with changes in calcium/calmodulin kinase II activity and glutamate immunoreactivity. Synapse. 1994;18:205–217. doi: 10.1002/syn.890180306. [DOI] [PubMed] [Google Scholar]
- Mion CC, Andreasen NC, Arndt S, Swayze VWd, Cohen GA. MRI abnormalities in tardive dyskinesia. Psychiatry Res. 1991;40:157–166. doi: 10.1016/0925-4927(91)90007-d. [DOI] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28:53–67. doi: 10.1016/s0896-6273(00)00085-4. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Bunney BS. Depolarization inactivation of dopamine neurons: Terminal release characteristics. Synapse. 1993;14:195–200. doi: 10.1002/syn.890140302. [DOI] [PubMed] [Google Scholar]
- Montminy M. Transcriptional regulation by cyclic AMP. Ann Rev Biochem. 1997;66:807–822. doi: 10.1146/annurev.biochem.66.1.807. [DOI] [PubMed] [Google Scholar]
- Nakahara T, Nakamura K, Tsutsumi T, et al. Effect of chronic haloperidol treatment on synaptic protein mRNAs in the rat brain. Brain Res Mol Brain Res. 1998;61:238–242. doi: 10.1016/s0169-328x(98)00230-7. [DOI] [PubMed] [Google Scholar]
- Nguyen TV, Kosofsky BE, Birnbaum R, Cohen BM, Hyman SE. Differential expression of c-fos and zif268 in rat striatum after haloperidol, clozapine, and amphetamine. Proc Natl Acad Sci USA. 1992;89:4270–4274. doi: 10.1073/pnas.89.10.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordstrom AL, Farde L, Nyberg S, Karlsson P, Halldin C, Sedvall G. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: A PET study of schizophrenic patients. Am J Psychiatry. 1995;152:1444–1449. doi: 10.1176/ajp.152.10.1444. [DOI] [PubMed] [Google Scholar]
- Nowakowski RS, Hayes NL. New neurons Extraordinary evidence or extraordinary conclusion? Science. 2000;288:771. doi: 10.1126/science.288.5467.771a. [DOI] [PubMed] [Google Scholar]
- Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatric Res. 1999;33:523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- Ozaki T. Comparative effects of dopamine D(1) and D(2) receptor antagonists on nerve growth factor protein induction. Eur J Pharmacol. 2000;402:39–44. doi: 10.1016/s0014-2999(00)00493-3. [DOI] [PubMed] [Google Scholar]
- Ozaki T, Mui K, Yamagami S. Comparison of the effects of dopamine D1 and D2 receptor antagonists on nerve growth factor mRNA expression. Eur J Pharmacol. 1999;369:133–143. doi: 10.1016/s0014-2999(99)00059-x. [DOI] [PubMed] [Google Scholar]
- Pilowsky LS, Costa DC, Ell PJ, Murray RM, Verhoeff NP, Kerwin RW. Clozapine, single photon emission tomography, and the D2 dopamine receptor blockade hypothesis of schizophrenia. Lancet. 1992;340:199–202. doi: 10.1016/0140-6736(92)90467-h. [DOI] [PubMed] [Google Scholar]
- Riva MA, Tascedda F, Lovati E, Racagni G. Regulation of NMDA receptor subunit messenger RNA levels in the rat brain following acute and chronic exposure to antipsychotic drugs. Brain Res Mol Brain Res. 1997;50:136–142. doi: 10.1016/s0169-328x(97)00175-7. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Gaither LA, Gao XM, Kashyap SM, Tamminga CA. Ultrastructural correlates of haloperidol-induced oral dyskinesias in rat striatum. Synapse. 1995;20:234–243. doi: 10.1002/syn.890200307. [DOI] [PubMed] [Google Scholar]
- Robertson GS, Fibiger HC. Neuroleptics increase c-fos expression in the forebrain: Contrasting effects of haloperidol and clozapine. Neuroscience. 1992;46:315–328. doi: 10.1016/0306-4522(92)90054-6. [DOI] [PubMed] [Google Scholar]
- Robertson GS, Fibiger HC. Effects of olanzapine on regional C-Fos expression in rat forebrain. Neuropsychopharmacology. 1996;14:105–110. doi: 10.1016/0893-133X(95)00196-K. [DOI] [PubMed] [Google Scholar]
- Robertson GS, Matsumura H, Fibiger HC. Induction patterns of Fos-like immunoreactivity in the forebrain as predictors of atypical antipsychotic activity. J Pharmacol Exp Ther. 1994;271:1058–1066. [PubMed] [Google Scholar]
- Rodrigues PdS, Dowling JE. Dopamine induces neurite retraction in retinal horizontal cells via diacylglycerol and protein kinase C. Proc Natl Acad Sci USA. 1990;87:9693–9697. doi: 10.1073/pnas.87.24.9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy NS, Wang S, Jiang L, et al. In vitro neurogenesis by progenitor cells isolated from the adult human hippocampus. Nature Med. 2000;6:271–277. doi: 10.1038/73119. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Wade T, Lidsky TI. Chronic neuroleptic treatment alters expression of glial glutamate transporter GLT-1 mRNA in the striatum. Neuroreport. 1998;9:133–136. doi: 10.1097/00001756-199801050-00026. [DOI] [PubMed] [Google Scholar]
- Schulman H. Protein phosphorylation in neuronal plasticity and gene expression. Curr Opin Neurobiol. 1995;5:375–381. doi: 10.1016/0959-4388(95)80051-4. [DOI] [PubMed] [Google Scholar]
- Sebens JB, Koch T, Ter Horst GJ, Korf J. Differential Fos-protein induction in rat forebrain regions after acute and long-term haloperidol and clozapine treatment. Eur J Pharmacol. 1995;273:175–182. doi: 10.1016/0014-2999(94)00692-z. [DOI] [PubMed] [Google Scholar]
- See RE, Chapman MA. Chronic haloperidol, but not clozapine, produces altered oral movements and increased extracellular glutamate in rats. Eur J Pharmacol. 1994;263:269–276. doi: 10.1016/0014-2999(94)90722-6. [DOI] [PubMed] [Google Scholar]
- See RE, Chapman MA, Meshul CK. Comparison of chronic intermittent haloperidol and raclopride effects on striatal dopamine release and synaptic ultrastructure in rats. Synapse. 1992;12:147–154. doi: 10.1002/syn.890120208. [DOI] [PubMed] [Google Scholar]
- Seeman P, Corbett R, Van Tol HH. Atypical neuroleptics have low affinity for dopamine D2 receptors or are selective for D4 receptors. Neuropsychopharmacology. 1997;16:93–110. doi: 10.1016/S0893-133X(96)00187-X. [DOI] [PubMed] [Google Scholar]
- Seeman P, Lee T. Antipsychotic drugs: Direct correlation between clinical potency and presynaptic action on dopamine neurons. Science. 1975;188:1217–1219. doi: 10.1126/science.1145194. [DOI] [PubMed] [Google Scholar]
- Selemon L, Goldman-Rakic P. The reduced neuropil hypothesis: A circuit based model of schizophrenia. Biol Psychiatry. 1999;45:17–25. doi: 10.1016/s0006-3223(98)00281-9. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, et al. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Shihabuddin L, Buchsbaum MS, Hazlett EA, et al. Dorsal striatal size, shape, and metabolic rate in never-medicated and previously medicated schizophrenics performing a verbal learning task. Arch Gen Psychiatry. 1998;55:235–243. doi: 10.1001/archpsyc.55.3.235. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Ann Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Smart TG. Regulation of excitatory and inhibitory neurotransmitter-gated ion channels by protein phosphorylation. Curr Opin Neurobiol. 1997;7:358–367. doi: 10.1016/s0959-4388(97)80063-3. [DOI] [PubMed] [Google Scholar]
- Stockmeier CA, DiCarlo JJ, Zhang Y, Thompson P, Meltzer HY. Characterization of typical and atypical antipsychotic drugs based on in vivo occupancy of serotonin2 and dopamine2 receptors. J Pharmacol Exp Ther. 1993;266:1374–1384. [PubMed] [Google Scholar]
- Svoboda K, Tank DW, Denk W. Direct measurement of coupling between dendritic spines and shafts. Science. 1996;272:716–719. doi: 10.1126/science.272.5262.716. [DOI] [PubMed] [Google Scholar]
- Swayze VWd, Andreasen NC, Alliger RJ, Yuh WT, Ehrhardt JC. Subcortical and temporal structures in affective disorder and schizophrenia: A magnetic resonance imaging study. Biol Psychiatry. 1992;31:221–240. doi: 10.1016/0006-3223(92)90046-3. [DOI] [PubMed] [Google Scholar]
- Swope SL, Moss SI, Raymond LA, Huganir RL. Regulation of ligand-gated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res. 1999;33:49–78. doi: 10.1016/s1040-7952(99)80005-6. [DOI] [PubMed] [Google Scholar]
- Tamminga C. Glutamatergic aspects of schizophrenia. Br J Psychiatry Suppl. 1999;(37):12–15. [PubMed] [Google Scholar]
- Tang F, Costa E, Schwartz JP. Increase of proenkephalin mRNA and enkephalin content of rat striatum after daily injection of haloperidol for 2 to 3 weeks. Proc Natl Acad Sci USA. 1983;80:3841–3844. doi: 10.1073/pnas.80.12.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni N, Buchs P-A, Nikonenko I, Bron CR, Muller D. LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature. 1999;402:421–425. doi: 10.1038/46574. [DOI] [PubMed] [Google Scholar]
- Trichard C, Paillere-Martinot ML, Attar-Levy D, Recassens C, Monnet F, Martinot JL. Binding of antipsychotic drugs to cortical 5-HT2A receptors: A PET study of chlorpromazine, clozapine, and amisulpride in schizophrenic patients. Am J Psychiatry. 1998;155:505–508. doi: 10.1176/ajp.155.4.505. [DOI] [PubMed] [Google Scholar]
- Uranova NA, Orlovskaya DD, Apel K, Klintsova AJ, Haselhorst U, Schenk H. Morphometric study of synaptic patterns in the rat caudate nucleus and hippocampus under haloperidol treatment. Synapse. 1991;7:253–259. doi: 10.1002/syn.890070402. [DOI] [PubMed] [Google Scholar]
- Vahid-Ansari F, Nakabeppu Y, Robertson GS. Contrasting effects of chronic clozapine, Seroquel(TM) (ICI 204,636) and haloperidol administration of deltaFosB-like immunoreactivity in the rodent forebrain. Eur J Neurosci. 1996;8:927–936. doi: 10.1111/j.1460-9568.1996.tb01579.x. [DOI] [PubMed] [Google Scholar]
- Van Harreveld A, Fifkova E. Swelling of dendritic spines in the fascia dentata after stimulation of the perforant fibers as a mechanism of post-tetanic potentiation. Exp Neurol. 1975;49:736–749. doi: 10.1016/0014-4886(75)90055-2. [DOI] [PubMed] [Google Scholar]
- Vincent SL, McSparren J, Wang RY, Benes FM. Evidence for ultrastructural changes in cortical axodendritic synapses following long-term treatment with haloperidol or clozapine. Neuropsychopharmacology. 1991;5:147–155. [PubMed] [Google Scholar]
- Wan W, Ennulat DJ, Cohen BM. Acute administration of typical and atypical antipsychotic drugs induces distinctive patterns of Fos expression in the rat forebrain. Brain Res. 1995;688:95–104. doi: 10.1016/0006-8993(95)00544-z. [DOI] [PubMed] [Google Scholar]
- Wong WT, Wong RO. Rapid dendritic movements during synapse formation and rearrangement. Curr Opin Neurobiol. 2000;10:118–124. doi: 10.1016/s0959-4388(99)00059-8. [DOI] [PubMed] [Google Scholar]
- Woo TU, Whitehead RE, Melchitzky DS, Lewis DA. A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci USA. 1998;95:5341–5346. doi: 10.1073/pnas.95.9.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods BT. Is schizophrenia a progressive neurodevelopmental disorder? Toward a unitary pathogenetic mechanism. Am J Psychiatry. 1998;155:1661–1670. doi: 10.1176/ajp.155.12.1661. [DOI] [PubMed] [Google Scholar]
- Wu G, Malinow R, Cline HT. Maturation of a central glutamatergic synapse. Science. 1996;274:972–976. doi: 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Cooperman MA. Differential effects of chronic antipsychotic drug treatment on extracellular glutamate and dopamine concentrations. J Neurosci. 1994;14:4159–4166. doi: 10.1523/JNEUROSCI.14-07-04159.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]