

1. Introduction
The generation of acyl anion equivalents has been of interest to the synthetic community since the early 1960’s. Of the numerous methods available to generate acyl anions (or acyl anion equivalents), their catalytic production from aldehydes has been through the use of cyanide and N-heterocyclic carbenes (NHCs). The latter has emerged as a prominent method to catalyze the formation of acyl anion equivalents from aldehydes in an umpolung process facilitating the discovery of new transformations and the development of asymmetric variants. This rapidly growing field of organocatalysis1 has been the subject of many reviews which detail the contributions from numerous researchers in this area.2 The present review will discuss the advent and development of NHCs in the generation of acyl anion equivalents and their use in synthesis.
2. Carbenes
In 1832, Wöhler and Liebig reported the homodimerization of aldehydes in the presence of cyanide to provide benzoin products.3 In 1943, Ukai demonstrated that stoichiometric amounts of thiazolium salts in the presence of base are capable of generating acyl anion equivalents from aldehydes to yield benzoin products.4 Initially proposed by Breslow, the currently accepted mechanism for generation of the acyl anion equivalent occurs via deprotonation of the azolium thereby generating an ylide or nucleophilic carbene (Scheme 1).5
Scheme 1.
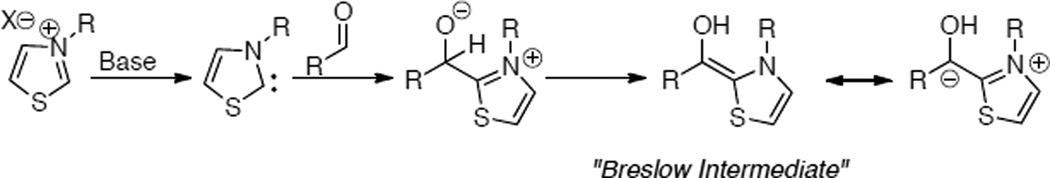
Formation of Acyl Anion Equivalent
Carbenes have long been studied, but our understanding of their stability and reactivity has dramatically improved in recent decades.6 They have typically been considered highly reactive intermediates, and only recently has it been shown that their reactivity can be harnessed and controlled upon appropriate steric and electronic manipulation. Carbenes are neutral compounds bearing a divalent carbon with six electrons in the valence shell. The two nonbonding electrons can either be spin paired (singlet) or unpaired (triplet).
Stabilizing effects in the ground state can be broken down to the π-and σ-framework.7 In the ground state, the nonbonding electrons of a singlet carbene occupy a σ-orbital leaving a vacant p-orbital available towards π-donation by proximal atoms around the divalent carbon. This π-donation results in the overall increase of s-character and aids in stabilization and nucleophilicity of the carbene. An additional stabilizing event involving a σ-withdrawing effect with a concomitant increase in s-character is also plausible. This is typically observed in crystal structures of isolable carbenes by a decrease in the X-C-X bond angle and an increase in the C-X bond length (Figure 2).8
Figure 2.

Stabilization of Singlet Carbenes
The advent of nucleophilic singlet carbenes in organic synthesis has been premised on the seminal work of Wanzlick and Arduengo. In 1962, Wanzlick reported the synthesis of bis-[1,3-diphenyl-2-imidazolidinylidene], a dimeric product obtained from the reaction of two carbenes. He also noted that the dimer has a high affinity to dissociate and react with a variety of different electrophiles and nucleophiles.9 In 1991, Arduengo’s report of an isolable NHC renewed interest in the use of such compounds as ligands and catalysts.10
3. Benzoin Reaction
The benzoin reaction became the platform for which the efficiency of novel chiral azolium carbenes would be measured. In 1966, Sheehan and Hunneman were first to make use of a chiral thiazolidine carbene (1) in facilitating benzoin formation in 22% optical purity.11 Further development of chiral thiazolidine carbenes spanning three decades all comprised of a common structural feature, in which free rotation was feasible around the chiral center, to which low enantioselectivities observed with such carbenes is attributed.12 The introduction of bicyclic thiazolium salts only led to modest improvement in enantioselectivities.13 The emergence of triazolidine carbenes, developed by Enders and Teles,14 and bicyclic triazolidine carbenes by Leeper15 resulted in acceptable enantioselectivities for the benzoin product (Chart 1).
Chart 1.

Development of Azollum Salts in the Benzoin Reaction
4. Stetter Reaction
The application of NHCs to other areas of acyl anion chemistry was pursued concurrently with the development of the benzoin reaction. In 1974, Stetter demonstrated that thiazolidine carbenes could be employed to facilitate the addition of an aldehyde via its acyl anion equivalent to activated double bonds to generate functionalized ketones (Scheme 2).16 Although Stetter had previously demonstrated this process with cyanide, the use of a thiazolidine carbene displayed improved proficiency for aliphatic aldehydes, which were not compatible under the cyanide conditions. In 1995, Ciganek illustrated an intramolecular Stetter reaction to generate chromanones.17 The asymmetric Stetter reaction did not receive considerable attention by the synthetic community for many years, and the only example prior to our work was that of Enders, who showed that triazolium precursor 6 provides the desired chromanone product in 74% ee and 42% yield (eq 1).18
Scheme 2.

Stetter Reaction
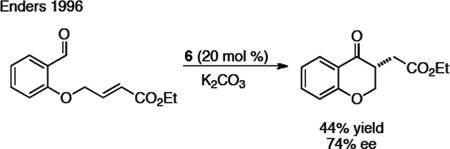 |
(eq 1) |
4.1 Catalyst Development
In 2000, our laboratory began a program to study chiral NHCs for use in organic synthesis. We envisioned accessing structurally and electronically diverse carbene precursors from readily available starting materials. The triazolium scaffold was prioritized over that of thiazolium and imidazolium precursors based on the number of sites available for structural and electronic modification and their proficiency as bicyclic carbenes in the benzoin reaction. Thiazolium precursors lead to blocking a single quadrant while corresponding triazolium-derived catalysts can effect three quadrants (Figure 3), presumably leading to improved selectivities.
Figure 3.

Mapping Steric Space About Carbene
We conceived that the chiral backbone could be readily introduced from amino alcohols (morpholine series) or amino acids (pyrrolidine series) and would provide steric blocking of quadrant II (Figure 3). We also envisaged that substitution in quadrants I and IV could arise from the hydrazine component which would enable an additional modulation of electronic and steric parameters (Figure 4). The use of substituted aryl hydrazines would give rise to differences in reactivity given its proximity to the carbene carbon.
Figure 4.

Catalyst Design
The intramolecular Stetter reaction was the platform wherein the effectiveness of our catalysts would be measured. We quickly identified amino indanol as the optimal chiral amino alcohol in the morpholine series, which could be coupled with a variety of structurally and electronically diverse hydrazines to furnish bench stable triazolium salts (Chart 2).19 In the pyrrolidine series, a variety of sidechains (ultimately derived from amino acids such as phenyl alanine and valine) were identified as efficient chiral scaffolds which could also incorporate a variety of hydrazines to furnish bench stable triazolium salts (Chart 2).
Chart 2.

Triazolium Salts
4.2 Asymmetric Intramolecular Stetter Reaction
Our work on the intramolecular Stetter reaction has been reviewed extensively and therefore will not be the focus of this review.20 A variety of salicylaldehyde derived substrates and aliphatic aldehydes bearing a range of linkers, including ether, thioether, sulfone, and protected amines, are effective in this process. An electron-withdrawing group on the prochiral alkene is still a necessary requirement21 but encompasses esters, thioesters, amides, ketones, aldehydes, nitriles, phosphine oxides, and phosphonates (eq 2).22
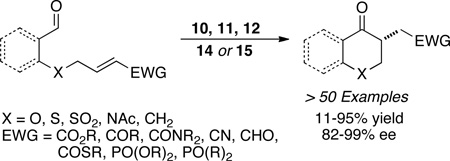 |
(eq 2) |
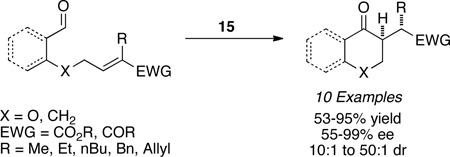
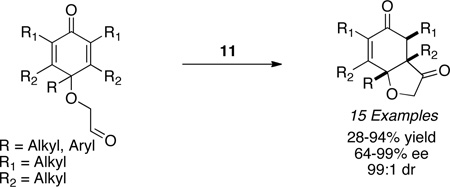
Michael acceptors bearing a second β-substituent also participate in the reaction in the presence of precatalyst 11 to furnish quaternary stereocenters (eq 3).23 Contiguous stereocenters could also be generated by using precatalyst 15 with a variety of salicylaldehyde substrates to yield a highly enantioselective and diastereoselective process. The reaction is not limited to salicylaldehyde derived substrates as aliphatic aldehydes also partake in the reaction with high levels of enantio- and diastereoselectivity, the latter being attributed to an intramolecular proton transfer event (eq 4).24 In advancing the scope of the Stetter reaction, we next focused on the synthesis of hydrobenzofuranones arising from a Stetter reaction on cyclohexadienones. These substrates are readily accessible in two steps and when subjected to optimized reaction conditions with triazolium salt 10 provide hydrobenzofuranones in excellent enantio- and diastereoselectivity (eq 5).25 With these successes in the intramolecular Stetter reaction in hand, we turned our attention to the intermolecular version.
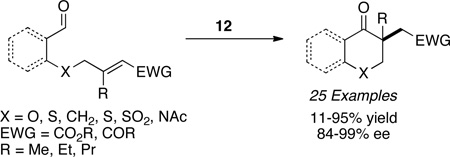 |
(eq 3) |
 |
(eq 4) |
 |
(eq 5) |
4.3 Asymmetric Intermolecular Stetter Reaction
The intermolecular addition of acyl anion equivalents into activated double bonds had been thoroughly studied by Stetter,24 but the asymmetric variant of this reaction has been nearly non-existent. The use of chiral NHCs in this process has not been fruitful as benzoin products dominate. One solution to the benzoin product is the use of acyl silanes to generate acyl anion equivalents via Brook rearrangement. Johnson has shown that in the presence of a chiral metallophosphite, acyl silane, and an activated prochiral olefin, one can obtain sila-Stetter products.26 Scheidt has also shown the addition of acyl silanes into chalcones which give rise to racemic products in the presence of a NHC.27 In 2006, this process was rendered enantioselective by generation of the acyl anion in the presence of stoichiometric carbene and chiral thiourea.28 While the use of acyl silanes is an elegant solution, it only maneuvers around the problem at hand.
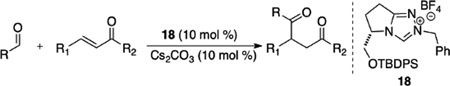
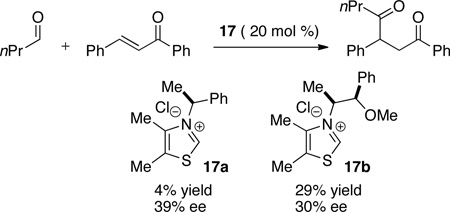
Prior to our work in this area, only three examples were known of an asymmetric intermolecular Stetter reaction, all reported by Enders. The reaction of butanal and chalcone in the presence of precatalysts 17 provides the 1,4-dicarbonyl product in low yield and poor ee (eq 6).29 Enders introduced an improved triazolium catalyst for the Stetter reaction of aromatic aldehydes and chalcones (Table 1).30 The benzyl substituent on the triazolium was found to be key to providing observable yields.
Table 1.
Intermolecular stetter
 | ||||
|---|---|---|---|---|
| R | R1 | R2 | yield (%) | ee (%) |
| Ph | Ph | Ph | 89 | 66 |
| 4-MeC6H4 | Ph | Ph | 85 | 78 |
| 3-MeC6H4 | Ph | Ph | 73 | 70 |
| 4-ClC6H4 | Ph | Ph | 89 | 67 |
| 4-BrC6H4 | Ph | Ph | 72 | 56 |
| 2-Naphthyl | Ph | Ph | 87 | 70 |
| 2-Furyl | Ph | Ph | 82 | 56 |
| Ph | 4-MeC6H4 | Ph | 83 | 64 |
| Ph | 4-ClC6H4 | Ph | 85 | 56 |
 |
(eq 6) |
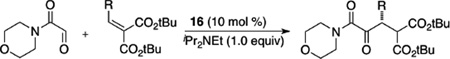
Following our successes in the intramolecular asymmetric Stetter reaction, we turned our attention to relaying the lessons learned there to the problem of the intermolecular reaction. Based on Stetter’s extensive work,31 we identified glyoxamides as competent aldehyde partners and β-substituted alkylidene malonates as reactive electrophiles. During catalyst optimization it was noted that while precatalyst 16 produces the desired product in 50% yield and 51% ee, the phenyl analogue 14 does not provide any product. These results again stress the significance of the N-aryl substituent in impacting carbene catalyzed reactions.32 A morpholine-derived glyoxamide results in superior reactivity with a variety of substituents tolerated at the β-position of the alkylidene malonate to provide α-ketoamides (Table 2). Under the reaction conditions, the glyoxamide is rapidly consumed to generate the benzoin product, which simply serves as the reservoir for the aldehyde via a retro-benzoin, and then participates in the desired Stetter process.33
Table 2.
Intermolecular Glyoxal Stetter Reaction
 | ||
|---|---|---|
| R | yield (%) | ee (%) |
| Et | 84 | 90 |
| Me | 68 | 87 |
| Pr | 83 | 90 |
| Bu | 70 | 90 |
| CH2CH2Ph | 81 | 88 |
| iBu | 51 | 91 |
| CH2CH2OBn | 91 | 80 |
| CH2CH2CH2Cl | 84 | 81 |
| CH2CH2CH(SCH2CH2CH2S) | 88 | 84 |
| CH2CH2CHCH2 | 97 | 89 |
This reaction was further developed to generate contiguous stereocenters in a highly enantioselective and diastereoselective manner. We hypothesized that a highly diastereoselective protonation event should result in a second stereocenter if the two activating carbonyls are different. The resultant stereocenter would be difficult to maintain under the basic reaction conditions unless one of the activating carbonyls was a tertiary amide which would insulate the stereocenter due to A1,3 strain. The use of alkylidine ketoamides with glyoxamide under the mediation of precatalyst 16 leads to the desired Stetter product in 68–97% yield and 81–97% ee and high diastereoselectivity (Table 3).34
Table 3.
Contiguous Stereocenters in the Glyoxamide Stetter Reaction
 | ||||
|---|---|---|---|---|
| R | R' | yield (%) | ee (%) | dr |
| Et | Et | 84 | 90 | 12:1 |
| Et | Me | 68 | 87 | 7:1 |
| Et | Pr | 83 | 90 | 6:1 |
| Et | Bu | 70 | 90 | 12:1 |
| Et | iBu | 81 | 88 | 11:1 |
| Et | CH2CH2Ph | 51 | 91 | 19:1 |
| Et | CH2CH2OBn | 91 | 80 | 11:1 |
| Et | CH2CH2CH2Cl | 84 | 81 | 10:1 |
| Et | CH2CH2CHCH2 | 88 | 84 | 14:1 |
| Et | CH2CH2CCH | 97 | 89 | 4:1 |
| Et | CH2CH2C(SCH2CH2CH2S) | 97 | 89 | 9:1 |
| Pr | Et | 97 | 89 | 11:1 |
| Pr | CH2CH2Ar | 97 | 89 | 5:1 |
| CH2CH2CHCH2 | Et | 97 | 97 | 9:1 |
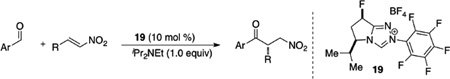
The above studies, although inherently limited to glyoxamide as the sole nucleophile, taught us much about necessary reactivity for our azolium catalysts in the asymmetric intermolecular Stetter reaction. To wit, electron-deficient aldehydes and more activated Michael acceptors provide higher yields with the triazolium catalysts. We thus focused our efforts at using nitroalkenes as electrophiles and heteroaryl aldehydes as nucleophiles. We found that triazolium salts derived from the pyrrolidine series bearing the electron deficient pentafluorophenyl group are necessary to obtain reactivity and a sterically demanding substituent such as an iso-propyl group is necessary in quadrant II (Figure 3) to obtain moderate enantioselectivity. Further modification of the pyrrolidine core with a fluorine provides optimal reactivity and selectivity. Upon synthesis of both diastereomers, it was identified that the cis-diastereomer provides the β-nitro ketone in 95% yield and 95% ee while the trans only provides the product in 22% yield and 88% ee. The difference is selectivity between diastereomers is currently hypothesized to arise form a conformational pucker (exo/endo) induced via a stereoelectronic effect. The scope of the reaction is restricted to heteroaromatic aldehydes while a variety of aliphatic substituents can be tolerated on the nitroalkene (Table 4).35
Table 4.
N-heterocyclic Carbene Catalyzed Nitro Ketone Synthesis
 | ||||
|---|---|---|---|---|
| Ar | R | Product | Yield (%) | ee (%) |
| 2-pyridyl | cyclohexyl | 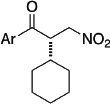 |
95 | 95 |
| 2-pyrazyl | cyclohexyl | 99 | 96 | |
| 3-pyridazyl | cyclohexyl | 88 | 94 | |
| 2-thiazolyl | cyclohexyl | 70 | 96 | |
| 2-furyl | cyclohexyl | 75 | 87 | |
| 4-oxazolyl | cyclohexyl | 76 | 86 | |
| 2-pyridyl | cyclopentyl | 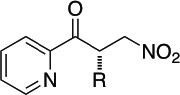 |
98 | 90 |
| 2-pyridyl | cyclopropyl | 72 | 87 | |
| 2-pyridyl | iso-propyl | 85 | 95 | |
| 2-pyridyl | propyl | 97 | 89 | |
| 2-pyridyl | propyl | 97 | 89 | |
| 2-pyridyl | sec-butyl | 97 | 89 | |
| 2-pyridyl | cyclohexyl | 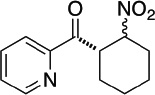 |
97 | 89 |
5. Redox Reaction
During our efforts in the intramolecular Stetter reaction, we inadvertently discovered a unique reactivity of acyl anion equivalents which was reminiscent of the observation by Wallach where chloral generates dichloroacetic acid when treated with aqueous cyanide.36 The mechanism of the Wallach method was long debated and the currently accepted mechanism is that proposed by Nowak (Scheme 3).37 The net process is an internal redox wherein one functionality gets reduced while a second is oxidized. An acylation using these intermediates thus constitutes an example of redox economy.38
Scheme 3.

Redox Mechanism
5.1 Redox Esterification
We saw this as an opportunity to implement this methodology as a mild acylating process using α-reducible aldehydes and NHCs. A resurgence in the development of α-halogenation of aldehydes39 led to their use as substrates with alcohols as nucleophiles. Investigation of the leaving group reveals that bromide is more facile to eliminate than chloride with a variety of alcohols participating in the acylation process (60–99%). Enantioenriched ethyl lactate may also be used with acylation proceeding with minor epimerization, while the use of racemic lactate with chiral carbene occurs with enantioenrichment suggesting acylation occurs on the acyl azolium (Table 5).40
Table 5.
α-Halo Aldehyde Redox Esterification
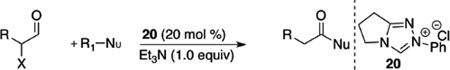 | |||
|---|---|---|---|
| R | X | R1 | yield (%) |
| H | Br | BnOH | 60 |
| Bn | Br | BnOH | 80 |
| Bn | Cl | BnOH | 65 |
| Cy | Br | BnOH | 99 |
| Bn | Br | MeOH | 78 |
| Bn | Br | EtOH | 78 |
| Bn | Br | PhCH2CH2CH2CH2OH | 78 |
| Bn | Br | i-PrOH | 66 |
| Bn | Br | CyOH | 66 |
| Bn | Br | PhOH | 55 |
| Bn | Br | 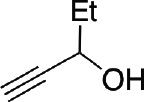 |
65 |
| Bn | Br | 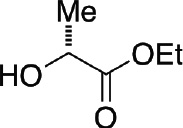 |
56 |
| Bn | Br | PhNH2 | 91 |
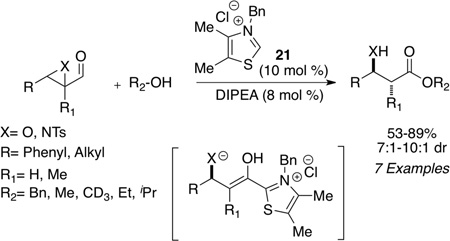
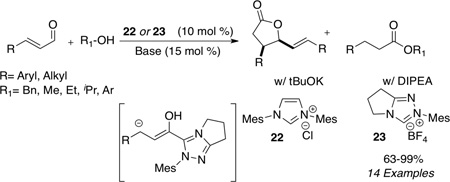
Independently and concurrently, Bode showed that epoxy aldehydes are viable substrates in the NHC redox reaction with a variety of alcohols indentified as competent nucleophiles (eq 7) to furnish β-hydroxy esters.41 Since the two initial reports by Bode and ourselves, the carbene catalyzed redox process has been developed to include other α-reducible aldehydes which participate in the esterification process. Bode42 and Scheidt43 have independently shown that enals in the presence of imidazolium salt 22 or triazolium salt 23 can either generate a lactone dimer or saturated ester upon choice of reaction conditions. The use of a strong base such as t-butoxide leads to generation of the lactone dimer (eq 8), presumably due to an inefficient proton source (pKa of t-butanol = 29.4 in DMSO); however, when a base whose conjugate acid is sufficiently acidic is employed, one obtains the protio-acylation product (pKa of iPr2EtNH+= 13 in THF). A variety of alcohols are tolerated in the reaction to produce the saturated esters in 63–99% yield (eq 8).
 |
(eq 7) |
 |
(eq 8) |
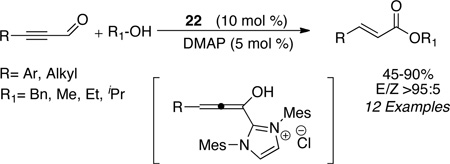
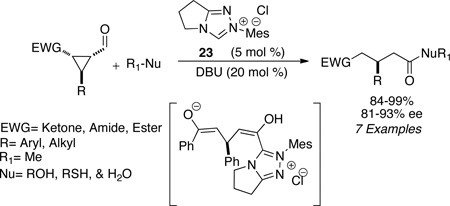
The synthesis of (E)-α,β-unsaturated esters can also be accomplished via redox esterification of propargylic aldehydes. Zeitler had identified imidazolium salt 22 to be efficient in this process in the presence of DMAP as the base to furnish high levels of E:Z selectivity (typically >95:5) (eq 9). A variety of alcohols participate in the reaction and aromatic, heteroaromatic, and aliphatic substituents are tolerated on the propargylic aldehyde (45–90%) (eq 9).44 Bode has also found that formylcyclopropanes are competent in the N-heterocyclic redox esterification with a variety of nucleophiles including alcohols, thiols, and water (eq 10). Subjection of the chiral substrates under optimized reaction conditions with precatalyst 23 proceeds with minor epimerization to furnish the 1,5 dicarbonyl adducts in good yields (84–93%) (eq 10).45
 |
(eq 9) |
 |
(eq 10) |
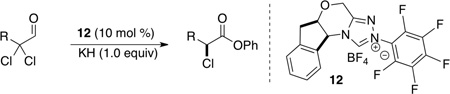
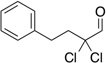
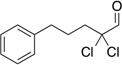
Treatment of α,α-dichloro aldehydes with phenols in the presence of NHC precursor 12 allows access to enantioenriched α-chloro aryl esters, via asymmetric protonation46 of the enol.47 Generating the potassium phenoxide with KH and 18-crown-6 provides the desired product in 75% yield and 81% ee. The additional use of 2,6-dibromo-4-methyl phenol provides a reservoir for the base, minimizing epimerization, leading to the product in 79% yield and 92% ee. A variety of α,α-dichloro aldehydes participate in the reaction to provide the respective esters in 65–79% yield and 84–93% ee (Table 6), with the current limitation being β-branching on the aldehyde. A variety of substituted phenols work in the reaction which provides distinct advantages and complementarity to other methods generating enantioenriched α-chloro aryl esters (Table 7).48
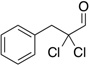
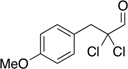
Table 6.
Aldehyde Scope for Synthesis of Enantioenriched α-Chloro Aryl Esters
 | |||||
|---|---|---|---|---|---|
| Substrate | yield (%) | ee (%) | Substrate | yield (%) | ee (%) |
 |
79 | 93 | 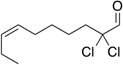 |
71 | 88 |
 |
76 | 90 | 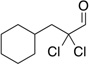 |
65 | 93 |
 |
73 | 85 | 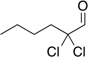 |
65 | 89 |
 |
68 | 89 | 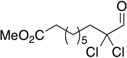 |
75 | 84 |
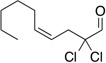 |
74 | 90 | 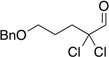 |
71 | 84 |
Table 7.
Scope of Phenols
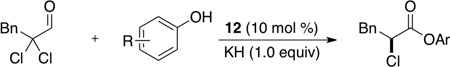 | ||
|---|---|---|
| Substituted Phenols | yield (%) | ee (%) |
| 4-methyl | 71 | 89 |
| 4-methoxy | 71 | 91 |
| 4-chloro | 75 | 83 |
| 2-methyl | 62 | 90 |
| 2-chloro | 75 | 91 |
| 2,6-dichloro | 65 | 82 |
| 2,6-dibromo-4-methyl | 85 | 76 |
| 2,4,6-trimethyl | 0 | NA |
| 3,4-dimethyl | 80 | 89 |
5.2 Redox Amidation
Expanding the scope of this mild acylation process to incorporate amine nucleophiles would afford amides. Amines have been previously studied as coupling partners with acyl azoliums derived from thiazolium and imidazolium carbenes and were determined to be less competent when compared to alcohols and hydroxide.49 We had initially shown that aniline was an effective nucleophile with α-bromo aldehyde and carbene 20 to furnish the desired amide in excellent yield (Table 4); however, other amines failed in the reaction or only provided the amide in low yields. Others working in this area expressed similar sentiments50 and further investigation into this process revealed that two byproducts of the reaction are imine formation and hydration product arising from water incorporation in the redox reaction.
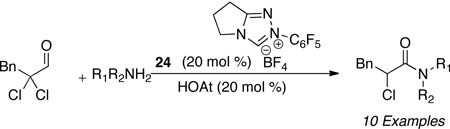
This problem was solved by addition of a nucleophilic additive which acts as an acyl transfer agent thus liberating the carbene 24 from the acyl azolium.51 The acylated additive then undergoes the desired N-acylation to yield the respective amide. Common peptide coupling reagents were identified to participate in this process with HOAt displaying optimal reactivity.52 Exclusion of water was key to prevent hydration. A variety of primary and secondary amines participate as nucleophiles in the acylation process as well as the respective hydrochloride salts (Table 8). The scope of the reducible aldehyde includes α,α-dichloro aldehydes, epoxy aldehydes, and enals (Table 9).53
Table 8.
Amine Scope of NHC-Amidation
 | ||
|---|---|---|
| R1 | R2 | yield (%) |
| Et | H | 89 |
| Cy | H | 85 |
| t-Bu | H | 73 |
| Et | Et | 89 |
| Me | OMe | 72 |
| Ph | H | 87 |
| 3-ClC6H4 | H | 82 |
| 4-OMeC6H4 | H | 83 |
| Alanine | H | 85 |
Table 9.
Substrate Scope of NHC-Amidation
 | |||
|---|---|---|---|
| X | R1 | yield (%) | dr |
| O | Bn | 86 | >19:1 |
| O | Alanine | 75 | 15:1 |
| NTs | Bn | 72 | >19:1 |
Concurrent to our work, Bode and coworkers reported a similar concept in facilitating N-acylation in the redox reaction using stoichiometric imidazole and triazolium carbene 23. Imine formation is suppressed by stoichiometric generation of the acyl imidazole followed by sequential addition of the amine to generate the amide. Primary and secondary amines are tolerated, however, amine hydrochloride salts are not compatible. The reducible aldehyde substrate scope entails formylcyclopropanes, α-chloro aldehydes, and α,β-unsaturated aldehydes to generate the respective amide (Scheme 4).54 Recently, Bode and coworkers have extended the substrate scope to include α’-hydroxyenones as surrogates for α,β-unsaturated aldehydes with 1,2,4-triazole used in substoichiometric quantities to facilitate amidation (Scheme 4).55
Scheme 4.

NHC-Amidation
5.3 Redox Azidation
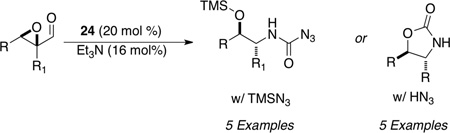
We also sought to generate carbamoyl azides and oxazolidinones from epoxy aldehydes in the presence of NHCs and azide as the nucleophile. Using triazolium salt 24 we were able to attenuate product selectivity based on reaction conditions. Using a combination of azidotrimethylsilane and sodium azide in a 2.5:1 ratio provides the carbamoyl azide selectively in 20–84% yields and diastereoselectivities of 2.6:1 to 6.5:1. The use of pseudo HN3 conditions (which require a 1:1:1 ratio of azidotrimethylsilane to ethanol to sodium azide) affords the oxazolidinone products in 53–83% yields and moderate diastereoselectivities (1:1 to 6.5:1). The varying levels of diastereoselectivities are attributed to epimerization occurring at the acyl azide, as the α-proton is significantly acidic (Table 10).56
Table 10.
Azidation of Aldehydes
 | |||
|---|---|---|---|
| R | R1 | w/ TMSN3 5 Examples yield (%)a (dr)c |
w/ HN3 5 Examples yield (%)b (dr)c |
| Ph | Me | 75 (4:1) | 75 (4:1) |
| Ph | C5H11 | 83 (6.5:1) | 83 (6.5:1) |
| Ph | Ph | 84 (2.6:1) | 47 (1:1) |
| 4-ClC6H4 | Me | 79 (3.6:1) | 53 (3.5:1) |
| 2-Naphthyl | Me | 52 (5.6:1) | 53 (3:1) |
| 1-Naphthyl | Me | 20 (2.6:1) | 60 (5:1) |
| Cy | Me | 78 (2.7:1) | 78 (3:1) |
5.4 Redox Hydration
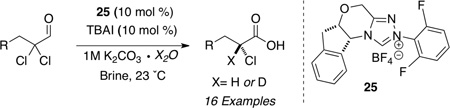
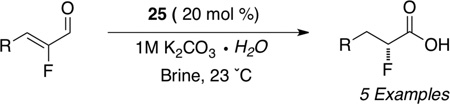
In recent years, the generation of α-halo acyl derivatives has received considerable attention. To the best of our knowledge, however, no catalytic asymmetric method is available to generate α-halo acids directly.57 We identified chiral NHC precatalyst 2558 in promoting the redox hydration of α,α-dichloro aldehydes with 1M potassium carbonate in water. The reaction can also generate α-deuterio labeled chloro acids by simply using D2O (Table 11). The hydration process has been extended to α-fluoro enals to generate enantioenriched fluoro carboxylic acids (Table 12). The α-deuterio, α-fluoro acids can also be obtained from the use of α-fluoro, α-bromo aldehydes as the reducible aldehyde.59
Table 11.
Synthesis of Enatioenriched α-Chloro Acids
 | |||
|---|---|---|---|
| R | X | yield (%) | ee (%) |
| Ph | H/D | 89/ 95 | 88/ 88 |
| 2-OMeC6H4 | H/D | 80/ 83 | 94/ 95 |
| 4-BocNHC6H4 | H/D | 78/ 75 | 78/ 79 |
| Cyclopentyl | H/D | 89/ 90 | 94/ 95 |
| Cyclohexyl | H/D | 85/ 88 | 94/ 95 |
| CH2OBn | H/D | 78/ 75 | 88/ 89 |
| CH2CH2Ph | H/D | 86/ 78 | 88/ 89 |
| CH2CH2CH2CO2Et | H/D | 75/ 77 | 90/ 91 |
Table 12.
Synthesis of Enatioenriched α-Fluoro Acids
 | |||
|---|---|---|---|
| R | X | yield(%) | ee(%) |
| 4-OMeC6H4 | H | 74 | 93 |
| 2-OMeC6H4 | H | 80 | 96 |
| 2-Naphthyl | H | 77 | 94 |
| 2-Thiazolyl | H | 70 | 90 |
| Cyclohexyl | H | 65 | 96 |
6. Cascade Catalysis
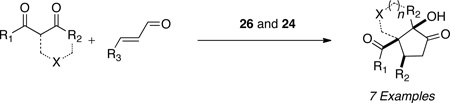
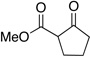
Having shown cooperative catalysis to be fruitful is resolving our amidation chemistry, we turned our attention to coupling other branches of organocatalysis with carbene catalysis.60 The generation of substituted cyclopentanones was achieved using secondary amine catalysis coupled with carbene catalysis with readily available starting materials in a Michael/Benzoin cascade reaction. Substitution is tolerated on the enal and the 1,3-dicarbonyl to provide the respective cyclopentanones in excellent enantioselectivities and modest diastereoselectivties (eq 11 and Table 13). Importantly, both catalysts are present throughout the course of the reaction and the protocol involves immediate charging of all reactants and reagents with no slow addition necessary.61
Table 13.
Michael/Benzoin Cascade
 | ||||
|---|---|---|---|---|
| R1 | R2 | R3 | yield(%) (dr) |
ee(%) |
| OMe | Me | Me | 90 (64:33:3:<1) | 91 |
| OEt | Me | n-Pr | 80 (60:30:8:2) | 93 |
| Ot-Bu | Me | Ph | 86(60:35:5:<1) | 97 |
| OBn | Me | Me | 90 (58:39:2<1) | 82 |
| Ets | Me | n-Pr | 56 (69:22:6:3) | 81 |
 |
Me | 79 (80:20:<1:<1) | 94 | |
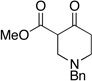 |
Me | 76 (85:15:<1:<1) | 90 | |
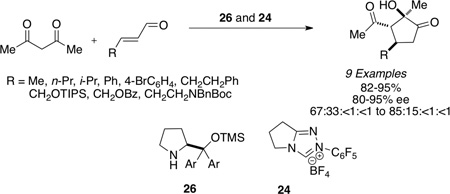 |
(eq 11) |
Our laboratory has also extended the Michael/benzoin cascade to a Michael/Stetter cascade reaction with salicylaldehydes and activated acetylenes to provide hydrobenzofuranones. The reaction scope is tolerant of substitution on the salicylaldehyde with dimethylacetylene dicarboxylate (DMAD) as the Michael acceptor to provide the respective benzofuranones in good yields (62–80%) and enantioselectivities (85–94%). The Michael acceptor is currently limited to acetylene dicarboxylates and activated allenes (eq 12).62
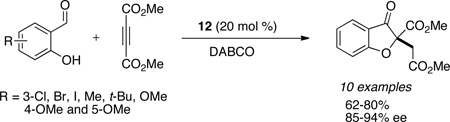 |
(eq 12) |
Córdova63 and Jørgensen64 have independently shown secondary amine catalysis coupled with carbene catalysis in the realm of redox reactivity to furnish β-hydroxy and β-amino esters. The optimized reaction conditions rely on the consumption of the enal with secondary amine catalysis to furnish the epoxy or aziridinyl aldehyde prior to the addition of azolium salt and alcohol to furnish the respective esters.
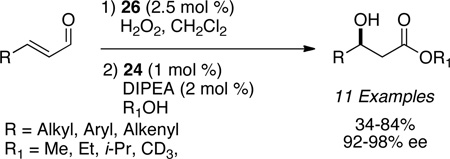 |
(eq 13) |
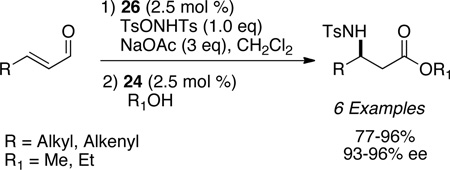 |
(eq 14) |
7. Conclusion
This review has presented an overview of NHC catalyzed formation of acyl anion equivalents from aldehydes and their reactivity in the Stetter, α-redox reaction and cascade catalysis. Although this field is still in its infancy compared to other areas of organocatalysis, the further development of novel carbene precursors will pave the way to new reactivity and its utilization in natural product synthesis, and industrial processes.
Acknowledgments
We gratefully acknowledge our many coworkers without whose intellectual and experimental contributions none of this would have been possible. Special thanks go to Mark Kerr and Javier Read de Alaniz who initiated the early work with these catalysts. We thank NIGMS (GM72586) for support. H.U.V. thanks Lilly for a graduate fellowship. T.R. thanks Amgen and Roche for support. We thank Donald Gauthier (Merck) for a generous gift of aminoindanol.
References
- 1.(a) Bertelsen S, Jørgensen KA. Chem. Soc. Rev. 2009;38:2178. doi: 10.1039/b903816g. [DOI] [PubMed] [Google Scholar]; (b) Gaunt MJ, Johansson CCC, McNally A, Vo NT. Drug Discovery Today. 2007;12:8. doi: 10.1016/j.drudis.2006.11.004. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 2.(a) Enders D, Niemeier O, Henseler A. Chem Rev. 2007;107:5606. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; (b) Marion N, Díez-González S, Nolan SP. Angew. Chem. Int. Ed. 2007;46:2988. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]; (C) Nair V, Vellath S, Babu BP. Chem. Soc. Rev. 2008;37:2691. doi: 10.1039/b719083m. [DOI] [PubMed] [Google Scholar]; (d) Moore JL, Rovis T. Top. Curr. Chem. 2009;291:8. doi: 10.1007/978-3-642-02815-1_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wöhler F, Liebig J. Annalen der Pharmacie. 1832:249. [Google Scholar]
- 4.Ukai T, Tanaka R, Dokawa T. J. Pharm. Soc. Jpn. 1943;63:296. [Google Scholar]
- 5.(a) Breslow R. J. Am. Chem. Soc. 1958;80:3719. [Google Scholar]; (b) Lapworth A. J. Chem. Soc. 1903;83:995. [Google Scholar]
- 6.Bertrand G, editor. Carbene Chemistry. 270 Madison Ave/New York/NY 10016/USA: Marcel Dekker; 2002. [Google Scholar]
- 7.(a) Dixon DA, Arduengo AJ., III J. Phys. Chem. 1991;95:4180. [Google Scholar]; (b) Arduengo AJ, III, Dias HVR, Harlow RL, Kline M. J. Am. Chem. Soc. 1992;114:5530. [Google Scholar]; (c) Arduengo AJ., III Acc. Chem. Res. 1999;32:913. [Google Scholar]
- 8.Bauschlicher CW, Schaefer HF, III, Bagus PS. J. Am. Chem. Soc. 1977:7106. [Google Scholar]
- 9.Wanzlick H-W. Angew. Chem. Int. Ed. Engl. 1962;1:75. [Google Scholar]
- 10.Arduengo AJ, III, Harlow RL, Kline M. J. Am. Chem. Soc. 1991;113:361. [Google Scholar]
- 11.Sheehan JC, Hunneman DH. J. Am. Chem. Soc. 1966;88:3666. [Google Scholar]
- 12.(a) Tagaki W, Tamura Y, Yano Y. Bull. Chem. Soc. Jpn. 1980;53:478. [Google Scholar]; (b) Marti J, Castells J, Lopez-Calahora F. Tetrahedron Lett. 1994;35:699. [Google Scholar]
- 13.(a) Knight RL, Leeper FJ. Tetrahedron Lett. 1997;38:3611. [Google Scholar]; (b) Gerhard AU, Leeper FJ. Tetrahedron Lett. 1997;38:3615. [Google Scholar]; (c) Dvorak CA, Rawal VH. Tetrahedron Lett. 1998;39:2925. [Google Scholar]
- 14.(a) Enders D, Breuer K, Raabe G, Runsink J, Teles JH, Melder J-P, Ebel K, Brode S. Angew. Chem. Int. Ed. Engl. 1995;34:1021. [Google Scholar]; (b) Enders D, Breuer K. Helv. Chim. Acta. 1996;79:1217. [Google Scholar]; (c) Enders D, Kallfass U. Angew. Chem. Int. Ed. 2002;41:1743. doi: 10.1002/1521-3773(20020517)41:10<1743::aid-anie1743>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 15.Knight RJ, Leeper FJ. J. Chem. Soc. Perkin Trans. 1998:1891. [Google Scholar]
- 16.Stetter H. Angew. Chem. Int. Ed. Engl. 1976;15:639. [Google Scholar]
- 17. Ciganek E. Synthesis. 1995:1311. Trost had previously demonstrated an intramolecular Stetter reaction en route to hirsutic acid see: Trost BM, Shuey CD, DiNinno F., Jr J. Am. Chem. Soc. 1979;101:1284.
- 18.Enders D, Breuer K, Runsink J, Teles JH. Helv. Chim. Acta. 1996;79:1899. [Google Scholar]
- 19.(a) Kerr MS, Read de Alaniz J, Rovis T. J. Org. Chem. 2005;70:5725. doi: 10.1021/jo050645n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vora HU, Lathrop SP, Reynolds NT, Kerr MS, Read de Alaniz J, Rovis T. Org. Synth. 2010;87:350. [Google Scholar]
- 20. Read de Alaniz J, Rovis T. Synlett. 2009:1189. doi: 10.1055/s-0029-1216654. Also see references 3a and 3b.
- 21.Note, however, that a Stetter-like process has been described for aldehydes bearing unactivated terminal alkenes; see: He J, Zheng J, Liu J, She X, Pan X. Org. Lett. 2006;8:4637. doi: 10.1021/ol061924f. He J, Tang S, Liu J, Siu Y, Pan X, She X. Tetrahedron. 2008;64:8797. Hirano K, Biju AT, Piel I, Glorius F. J. Am. Chem. Soc. 2009;131:14190. doi: 10.1021/ja906361g. Biju AT, Wurz NE, Glorius F. J. Am. Chem. Soc. 2010;132:5970. doi: 10.1021/ja102130s.
- 22.(a) Kerr MS, Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2002;124:10298. doi: 10.1021/ja027411v. [DOI] [PubMed] [Google Scholar]; (b) Kerr MS, Rovis T. Synlett. 2003;12:1934. [Google Scholar]; (c) Reynolds NT, Rovis T. Tetrahedron. 2005;61:6368. [Google Scholar]; (d) Read de Alaniz J, Kerr MS, Moore JL, Rovis T. J. Org. Chem. 2008;73:2033. doi: 10.1021/jo702313f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Cullen SC, Rovis T. Org. Lett. 2008;10:3141. doi: 10.1021/ol801047k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Kerr MS, Rovis T. J. Am. Chem. Soc. 2004;126:8876. doi: 10.1021/ja047644h. [DOI] [PubMed] [Google Scholar]; (b) Moore JL, Kerr MS, Rovis T. Tetrahedron. 2006;62:11477. [Google Scholar]
- 24.Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2005;127:6284. doi: 10.1021/ja0425132. [DOI] [PubMed] [Google Scholar]
- 25.(a) Liu Q, Rovis T. J. Am. Chem. Soc. 2006;128:2552. doi: 10.1021/ja058337u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu Q, Rovis T. Org. Process Res. Dev. 2007;11:598. doi: 10.1021/op600278f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stetter H, Kuhlmann H. Org. React. 1991;40:407. [Google Scholar]
- 26.(a) Nahm MR, Lighu X, Potnick JR, Yates CM, White PS, Johnson JS. Angew. Chem. Int. Ed. 2005;44:2377. doi: 10.1002/anie.200462795. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nahm MR, Potnick JR, White PS, Johnson JS. J. Am. Chem. Soc. 2006;128:2751. doi: 10.1021/ja056018x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mattson AE, Bharadwaj AR, Zuhl AM, Scheidt KA. J. Org. Chem. 2006;71:5715. doi: 10.1021/jo060699c. [DOI] [PubMed] [Google Scholar]
- 28.Mattson AE, Zuhl AM, Reynolds TE, Scheidt KA. J. Am. Chem. Soc. 2006;128:4932. doi: 10.1021/ja056565i. [DOI] [PubMed] [Google Scholar]
- 29.(a) Enders D, Breuer K. Addition of Acyl Carbanion Equivalents to Carbonyl Groups and Enones, In Comprehensive Asymmetric Catalysis I–III. Vol. 3 [Google Scholar]; Jacobsen EN, Pfaltz A, Yamamoto H, editors. Berlin: Springer-Verlag; 1999. p. 1093. [Google Scholar]; (b) Enders D, Balensiefer T. Acc. Chem. Res. 2004;37:534. doi: 10.1021/ar030050j. [DOI] [PubMed] [Google Scholar]
- 30.Enders D, Han J, Henseler A. Chem. Commun. 2008:3989. doi: 10.1039/b809913h. [DOI] [PubMed] [Google Scholar]
- 31. Stetter H, Kuhlmann H. Org. React. 1991;40:407. This chapter lists >500 examples of the Stetter reaction involving 93 different aldehydes. In terms of the Michael acceptor, the vast majority are unsubstituted at the beta-position, or are quite activated (maleates, fumarates, chalcones, etc.). Five examples contain simple alkyl substitution at the beta-position of the Michael acceptor (cyclopentenone [30% yield], pentenone [34% yield], and ethylidine acetoacetate [3 examples, 32–49% yield]).
- 32.Rovis T. Chem. Lett. 2008;37:2. [Google Scholar]
- 33.Liu Q, Perreault S, Rovis T. J. Am. Chem. Soc. 2008;130:14066. doi: 10.1021/ja805680z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Q, Rovis T. Org. Lett. 2009;11:2856. doi: 10.1021/ol901081a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DiRocco DA, Oberg KM, Dalton DM, Rovis T. J. Am. Chem. Soc. 2009;131:10872. doi: 10.1021/ja904375q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wallach O. Ber. Dtsch. Chem. Ges. 1873;6:114. [Google Scholar]
- 37.Nowak RM. J. Org. Chem. 1963;28:1182. [Google Scholar]
- 38.Burns NZ, Baran PS, Hoffmann RW. Angew. Chem. Int. Ed. 2009;48:2854. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- 39.Amatore M, Beeson TD, Brown SP, MacMillan DWC. Angew. Chem. Int. Ed. 2009;48:5121. doi: 10.1002/anie.200901855. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reynolds NT, Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2004;126:9518. doi: 10.1021/ja046991o. [DOI] [PubMed] [Google Scholar]
- 41.Chow KY-K, Bode JW. J. Am. Chem. Soc. 2004;126:8126. doi: 10.1021/ja047407e. [DOI] [PubMed] [Google Scholar]
- 42.Sohn SS, Bode JW. Org. Lett. 2005;7:3873. doi: 10.1021/ol051269w. [DOI] [PubMed] [Google Scholar]
- 43.Chan A, Scheidt KA. Org. Lett. 2005;7:905. doi: 10.1021/ol050100f. [DOI] [PubMed] [Google Scholar]
- 44.Zeitler K. Org. Lett. 2006;8:637. doi: 10.1021/ol052826h. [DOI] [PubMed] [Google Scholar]
- 45.Sohn SS, Bode JW. Angew. Chem. Int. Ed. 2006;45:6021. doi: 10.1002/anie.200601919. [DOI] [PubMed] [Google Scholar]
- 46.Mohr JT, Hong AY, Stoltz BM. Nature Chem. 2009;1:359. doi: 10.1038/nchem.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reynolds NT, Rovis T. J. Am. Chem. Soc. 2005;127:16406. doi: 10.1021/ja055918a. [DOI] [PubMed] [Google Scholar]
- 48.(a) Wack H, Taggi AE, Hafez AM, Drury WJ, III, Lectka T. J. Am. Chem. Soc. 2001;123:1531. doi: 10.1021/ja005791j. [DOI] [PubMed] [Google Scholar]; (b) France S, Wack H, Taggi AE, Hafez AM, Waggerle TR, Shah MH, Dusich CL, Lectka T. J. Am. Chem. Soc. 2004;126:4245. doi: 10.1021/ja039046t. [DOI] [PubMed] [Google Scholar]; (c) Lee EC, McCauley KM, Fu GC. Angew. Chem. Int. Ed. 2007;46:977. doi: 10.1002/anie.200604312. [DOI] [PubMed] [Google Scholar]
- 49.(a) Bruice TC, Kundu NT. J. Am. Chem. Soc. 1966;88:4097. doi: 10.1021/ja00956a026. [DOI] [PubMed] [Google Scholar]; (b) Lienhard G. J. Am. Chem. Soc. 1966;88:5642. doi: 10.1021/ja00969a017. [DOI] [PubMed] [Google Scholar]; (c) Owen TC, Harris JN. J. Am. Chem. Soc. 1990;112:6136. [Google Scholar]; (d) Owen TC, Richards A. J. Am. Chem. Soc. 1987;109:2520. [Google Scholar]
- 50.See references 42 and 44.
- 51.Han C, Lee JP, Lobkovsky E, Porco JA. J. Am. Chem. Soc. 2005;127:10039. doi: 10.1021/ja0527976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carpino LA. J. Am. Chem. Soc. 1993;115:4397. [Google Scholar]
- 53.Vora HU, Rovis T. J. Am. Chem. Soc. 2007;129:13796. doi: 10.1021/ja0764052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bode JW, Sohn SS. J. Am. Chem. Soc. 2007;129:13798. doi: 10.1021/ja0768136. [DOI] [PubMed] [Google Scholar]
- 55.Chiang P-C, Kim Y, Bode JW. Chem. Commun. 2009:4566. doi: 10.1039/b909360e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vora HU, Moncecchi JR, Epstein O, Rovis T. J. Org. Chem. 2008;73:9727. doi: 10.1021/jo8020055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Czekelius C, Tzschucke CC. Synthesis. 2010;4:543. and references cited therein. [Google Scholar]
- 58.Takikawa H, Suzuki K. Org. Lett. 2007;9:2713. doi: 10.1021/ol070929p. [DOI] [PubMed] [Google Scholar]
- 59.Vora HU, Rovis T. J. Am. Chem. Soc. 2010;132:2860. doi: 10.1021/ja910281s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grondal C, Jeanty M, Enders D. Nature Chem. 2010;2:167. doi: 10.1038/nchem.539. [DOI] [PubMed] [Google Scholar]
- 61.Lathrop SP, Rovis T. J. Am. Chem. Soc. 2009;131:13628. doi: 10.1021/ja905342e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Filloux CM, Lathrop SP, Rovis T. Proc. Nat. Acad. Sci. 2010;107:20666. doi: 10.1073/pnas.1002830107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao G-L, Córdova A. Tetrahedron Lett. 2007;48:5976. [Google Scholar]
- 64.Jiang H, Gschwend B, Albrecht L, Jörgensen KA. Org. Lett. 2010;12:5052. doi: 10.1021/ol102164y. [DOI] [PubMed] [Google Scholar]