

Significance
T-cell receptor recognition of antigen is an essential first step in the initiation of a T-cell response. This report demonstrates that CD4 T cells responding during an infection can discriminate between antigen affinity and antigen dose, resulting in distinct types of effector and memory cell generation. Moreover, memory T cells “remember” the strength of primary T-cell activation and maintain a biased recall response upon secondary infection. These data reveal that antigen affinity exerts an important influence on T-cell differentiation that is not compensated for by high antigen dose. Understanding the rules of CD4 T-cell differentiation is integral to effective vaccine design.
Keywords: follicular helper, infection, lymphocytes
Abstract
Cumulative T-cell receptor signal strength and ensuing T-cell responses are affected by both antigen affinity and antigen dose. Here we examined the distinct contributions of these parameters to CD4 T-cell differentiation during infection. We found that high antigen affinity positively correlates with T helper (Th)1 differentiation at both high and low doses of antigen. In contrast, follicular helper T cell (TFH) effectors are generated after priming with high, intermediate, and low affinity ligand. Unexpectedly, memory T cells generated after priming with very low affinity antigen remain impaired in their ability to generate secondary Th1 effectors, despite being recalled with high affinity antigen. These data challenge the view that only strongly stimulated CD4 T cells are capable of differentiating into the TFH and memory T-cell compartments and reveal that differential strength of stimulation during primary T-cell activation imprints unique and long lasting T-cell differentiation programs.
Following infection, T-cell receptor (TCR) interactions with foreign peptide/MHC (pMHC) drive the rapid clonal expansion and differentiation of T cells into distinct effector subsets specialized against different classes of microbes. An early bifurcation in CD4 T-cell responses results in the generation of T helper (Th)1 effectors, which regulate innate cell microbicidal function and follicular helper T (TFH) cells, which migrate to B-cell follicles to regulate germinal center (GC) responses and antimicrobial antibody production (1). After pathogen is cleared, T cells undergo a contraction phase during which the majority of effectors die by apoptosis, leaving behind a population of long-lived memory cells to provide protection upon subsequent reinfection. The decision to differentiate into Th1 and TFH lineages appears to occur very early in the immune response (2, 3). Initial T-cell priming by dendritic cells (DCs) is sufficient to induce fate-committed Th1 and TFH cells as early as 3 d after infection, whereas maintenance and further expansion of the TFH compartment depends on T-cell interactions with B cells (2). Similarly, memory T-cell differentiation occurs very early after infection and is critically dependent on B-cell interactions for optimal priming (4, 5). Importantly, CD4 T-cell differentiation is coupled to division, and unlike CD8 T-cell differentiation, requires constant antigen recognition (6, 7).
Although the strength of TCR–pMHC interactions has been shown to directly modulate T-cell expansion and clonal dominance within the Th cell compartment (8, 9), how this influences CD4 T-cell fate is not well understood. Cumulative TCR signaling can be influenced by both antigen affinity and antigen dose (10). In terms of proliferation, higher antigen dose can compensate for lower antigen affinity to some extent, but several reports have shown independent effects on T-cell responses both in vitro and in vivo (10–12). These data indicate that antigen affinity and antigen dose may promote qualitatively distinct TCR signals. Recently, modulation of the overall TCR signal by varying either TCR affinity or antigen dose was shown to influence the pattern of effector T-cell differentiation, with higher affinity ligands or higher antigen dose promoting TFH generation (13–15). However, another study examining high and low avidity CD4 T-cell responses during viral infection found significant differences in Th1 but not TFH generation (16). Sustained TCR–pMHC interactions have also been shown to promote memory T-cell differentiation, which is associated with increased TCR avidity (17, 18). These studies, however, have focused on the development of the Th1 memory compartment, which is phenotypically and functionally distinct from the TFH memory compartment (19, 20). Thus, although strong TCR signals resulting from high antigen affinity or high antigen dose can clearly affect the extent and quality of T-cell differentiation, whether or not T cells can discriminate these signals, and how this contributes to T-cell differentiation during infection, has not been determined.
To address this question, we infected mice with varying concentrations of Listeria expressing either high or low affinity antigens for the TCR. By normalizing the degree of proliferation induced by high and low affinity antigens we were able to discern distinct influences of antigen affinity and antigen dose on Th cell differentiation. We observed a strong positive correlation between antigen affinity and Th1 differentiation that occurs early and is dose independent. Importantly, high antigen dose does not compensate for the low efficiency of Th1 differentiation induced by low affinity antigen. In contrast, early TFH effector generation was observed after priming with high, intermediate, and low affinity antigen, but was not maintained at later time points under conditions of low antigen dose. In addition, we found that T cells activated by either high or low affinity antigen are equally capable of memory T-cell differentiation. Surprisingly, memory T cells generated by either low antigen affinity or low antigen dose maintained their biased effector lineages following recall activation with high affinity antigen. These data indicate that differential strength of stimulation during primary T-cell activation can imprint unique and long lasting T-cell differentiation programs.
Results
Establishing the TCR Ligand Affinity Hierarchy.
Several models have been proposed to explain the sensitivity of TCR recognition of pMHC. The receptor occupancy model uses the affinity of the TCR for pMHC (KD) to predict ligand affinity, whereas the kinetic proofreading model suggests that ligand affinity is dependent on the half-life (t1/2) of TCR–pMHC interactions (10, 21–23). A recent variation of this model suggests that serial rebinding of pMHC results in an aggregate half-life of TCR binding (ta), that can allow antigens with fast on-rates to activate T cells more strongly than their t1/2 would predict (24). To study the effect of TCR ligand affinity on CD4 T-cell activation, we used B3K508 TCR transgenic T cells, which are specific for an H-2Ab bound peptide FEAQKAKANKAKAVD (3K). The KD, t1/2, and ta values for 3K and a panel of single amino acid substitutions 3K variants have been previously established (Fig. S1A) (24, 25). Although we use the term “affinity” to indicate the strength of TCR–pMHC interactions, the strength and hierarchy of the ligands used in our study is independently reflected by all three biophysical parameters. Importantly, these ligands have equivalent binding to H-2Ab but differ in their ability to stimulate B3K508 TCR transgenic T cells (24, 26). The thymic selection potential of 3K and its variants was recently assessed using fetal thymic organ culture (27). The ligands 3K, P5R, and P2A induced strong negative selection of developing B3K508 thymocytes, while the ligand P-1A elicited only partial thymocyte deletion (Fig. S1A). To further evaluate the affinity of these ligands, we compared CD69 expression on thymocytes and peripheral B3K508 T cells activated with 3K or variant ligands and confirmed that the affinity hierarchy remained consistent between the thymus and the periphery (Fig. S1 A and B). To examine the effect of TCR affinity on CD4 T cells responding to infection, we generated recombinant Listeria monocytogenes (Lm) strains engineered to express the 3K or a 3K variant peptide. All of the Lm strains were capable of inducing B3K508 T-cell expansion in vivo and a direct correlation between the number of B3K508 T cells recovered and the affinity of the priming variant was observed (Fig. 1A). However, unlike CD8+ T cells responding to threshold ligand in vivo (28, 29), B3K508 T cells activated by threshold ligand P-1A exhibited only minimal expansion (Fig. 1A), indicating that CD4 T cells may be more tightly regulated than CD8+ T cells in the periphery. Importantly, the extent of T-cell proliferation can be influenced by both antigen affinity and the amount of available antigen (pMHC dose), both of which can be further linked to the acquisition of effector function (30). To better discriminate the independent effects of ligand affinity on T-cell differentiation, we established the dose of high affinity 3K activation that elicits roughly similar expansion as low affinity ligand P2A, at both early and later time points after infection (Fig. 1B). To generate consistent levels of inflammation, mice infected with low dose Lm.3K were also infected with wild-type bacteria not expressing 3K.
Fig. 1.

Establishing the TCR affinity hierarchy. (A and B) Ten thousand naïve B3K508 T cells were transferred into CD45.1 congenic mice followed by infection with Lm.3K or Lm.variant strains. Low dose Lm.3K infection in B corresponds to 105 cfu. Mean number of B3K508 T cells recovered from spleen and lymph nodes over the first 8 d of infection. Data represent n ≥ 3 for each data point and are representative of two independent experiments.
Antigen Affinity Influences the Pattern of Effector T-cell Differentiation.
Infection results in the generation of two distinct effector populations. Th1 effector cells express high levels of the transcription factor T-bet, produce IFNγ, and are important for inducing macrophage microbicidal function (1). TFH cells express low levels of the surface marker Ly6c (20) and high levels of the chemokine receptor CXCR5, which directs T-cell migration to the B-cell areas of lymphoid structures where they provide signals to enhance B-cell antibody secretion (1). TFH cells expressing high levels of PD-1 and the transcription factor Bcl6 further migrate into B-cell germinal centers where they drive B-cell affinity maturation (31), whereas TFH cells that express low levels of PD-1 and intermediate levels of Bcl6 are suggested to be precursors to central memory cells (3, 31). To understand how ligand affinity affects CD4 effector T-cell differentiation, we examined the phenotype of B3K508 T cells responding to infection with high affinity Lm.3K or low affinity Lm.P2A. At day 6 after infection with high dose Lm.3K, B3K508 T cells exhibited heterogeneous effector differentiation with both Th1 (CXCR5−T-bethigh) and TFH (CXCR5+T-betlow) populations readily identifiable (Fig. 2A and Fig. S2A). T-bethigh cells expressed high levels of the surface marker Ly6c (Fig. 2B), while CXCR5+ TFH cells down-regulated Ly6c expression (Fig. 2C). Despite undergoing significantly less expansion, low dose infection with high affinity Lm.3K also resulted in the generation of Th1 and early TFH effectors. However, consistent with previous reports (13, 15), selectively decreasing antigen dose while keeping the level of inflammation constant was not sufficient to maintain high levels of PD-1 within the CXCR5+ compartment (Fig. 2D). In contrast, B3K508 T cells responding to low affinity P2A expressed lower levels of T-bet within the CXCR5− compartment, while differentiation into both PD-1+ and PD-1− CXCR5+ subpopulations was unimpaired (Fig. 2 A and D). Importantly, at day 8 after infection, TFH cells generated after P2A activation expressed comparable or higher levels of Bcl6 and TFH signature cytokines IL-21 and IL-4 as 3K generated TFH effectors (Fig. S2 B and C). In addition, B3K508 T cells responding to the P5R variant, which has an affinity intermediate to 3K and P2A (Fig. S1A), demonstrated an intermediate phenotype with decreased Th1 differentiation compared with high affinity 3K activation (Fig. S3A). In terms of cell numbers, 3K activation induced two- to threefold more Th1 effectors compared with P5R activation, while the number of TFH effectors was only marginally increased (Fig. S3 B and C). These data indicate a strong correlation between the accumulation of Th1 effectors and antigen affinity. In contrast, differentiation into the TFH compartment can occur in response to very weak, intermediate, and high affinity ligands.
Fig. 2.

Antigen affinity and effector T-cell differentiation. Ten thousand B3K508 T cells were transferred into congenic mice followed by infection with Lm.3K or Lm.P2A. (A–D) Representative flow cytometry plots of B3K508 T cells at day 6. (E) Absolute number of Th1 (T-bethighCXCR5−) and TFH (T-betlowCXCR5+) effectors recovered from spleen and lymph nodes at day 6. Data represent n ≥ 3 and are representative of three independent experiments. (*P < 0.05, ***P < 0.0001).
T-cell Proliferation and IL-2 Activation.
Early after infection, a bifurcation of IL-2Rαhigh and IL-2Rαlow populations can be observed (2, 3). IL-2Rα signals are required for the differentiation of Th1 effector cells, whereas inhibition of IL-2Rα signals promotes TFH development (32). To address the possibility that decreased IL-2Rα expression on low affinity activated T cells precedes their failure to up-regulate T-bet, we examined T cells at early time points after infection. After 2 d, both high dose and low dose 3K-activated T cells expressed higher levels of IL-2Rα and produced more IL-2 (Fig. 3A and Fig. S4 A and B). In contrast, T cells activated by low affinity P2A antigen expressed lower levels of IL-2Rα and produced less IL-2 despite having undergone a similar degree of proliferation as low dose 3K-activated T cells. Three days after infection, T cells activated with 3K expressed high levels of T-bet, while the majority of P2A-activated T cells had low levels of T-bet (Fig. 3B and Fig. S4D). T cells activated with intermediate affinity antigen P5R had an intermediate phenotype with decreased early expression of IL-2Rα and T-bet (Fig. S3 C–E). Importantly, the generation of early (IL-2RαlowCXCR5+) TFH effectors was independent of antigen affinity and could be observed under all conditions (Figs. S3C and S4C). T cells activated in vitro demonstrated a similar correlation between antigen affinity and early IL-2 and T-bet expression at a wide range of antigen doses (Fig. S5). In contrast, T cells activated in vitro with low affinity antigen expressed more Bcl6, possibly due to decreased production of IL-2, which can negatively regulate Bcl6 expression (33). Indeed, blocking IL-2 in vitro led to further increased Bcl6 expression in low affinity activated T cells, particularly at antigen concentrations where IL-2 production was evident (Fig. S5). Taken together, these data reveal that antigen affinity is key to setting the early Th1 differentiation potential, whereas TFH differentiation is minimally impacted.
Fig. 3.

Antigen affinity and early IL-2 induction. Two hundred thousand carboxyfluorescein succinimidyl ester (CFSE)-labeled B3K508 T cells were transferred into congenic mice followed by infection with Lm.3K or Lm.P2A. CD25 proliferation (A) and T-bet expression (B) were assessed by flow cytometry after 2 (A) and 3 (B) d. T-bet MFI is shown for T cells that have undergone at least one division. Data represent n = 3 for each data point and are representative of two independent experiments. (**P < 0.001, ***P < 0.0001).
Location and Function of TFH and Th1 Effector Cells Generated by Low Affinity Ligand.
Following infection, Th1 effector cells migrate to splenic T-cell zones and red pulp, and TFH effector cells are sequestered in the B-cell follicles (1). To determine the location of CD4 T cells activated by high or low affinity ligands we analyzed frozen splenic sections by immunofluorescence microscopy. At day 6 after infection, T cells activated by high dose 3K ligand were identified in all three splenic areas (Fig. 4A). T cells activated by low dose 3K were predominantly located in splenic T-cell zones and were less abundant in B-cell areas (Fig. 4A), consistent with their decreased expression of PD-1. Consistent with high expression of TFH markers, P2A activated T cells were predominantly observed in splenic B-cell zones or at the B-cell–T-cell area border, although a reduced proportion could also be detected in the red pulp and T-cell zones (Fig. 4A). Although it is possible that Th1 effectors generated after low affinity activation emigrate prematurely from lymphoid organs, we did not observe a higher proportion of these cells in the splenic red pulp, a major site of T-cell trafficking to the blood. As the primary function of TFH effector cells is to help B cells, we also examined TFH helper function. TFH cells generated after Lm.3K or Lm.P2A infection were sorted based on high expression of CXCR5 and PD-1 and transferred into CD3ε−/− mice that had been s.c. immunized with 3K-OVA one day earlier. At day 6 after transfer, both 3K and P2A generated TFH effectors maintained high expression of CXCR5 and PD-1 and induced GC B cells in CD3ε−/− mice (Fig. 4B and Fig. S6 A and B). Although the proportion of GC B cells was slightly lower in mice transferred with P2A compared with 3K-generated TFH effectors, GC B-cell numbers correlated with the absolute number of TFH isolated at the time of harvest. These data indicate that TFH effectors generated by low affinity activation are capable of providing help to B cells. However, given the significantly lower number of effectors generated under these conditions, low affinity TFH cells are likely have a more limited impact on immune responses in an intact animal. To examine Th1 effector cell function in these differentially immunized populations, we analyzed IFNγ and IL-2 expression by B3K508 T cells isolated at day 6 after infection. The 3K-activated T cells had a high frequency of cytokine producers, while T cells activated with decreasing antigen affinity had fewer IFNγ and IL-2 producers, consistent with lower expression of T-bet (Fig. 4C and Fig. S3G). These data indicate that although low affinity activation results in a decreased proportion of Th1 effectors, the function of these cells is normal. T cells activated by low dose 3K also had a decreased proportion of IL-2+ responders compared with high dose 3K activation, but nevertheless remained competent to produce IFNγ.
Fig. 4.

Function and location of low affinity activated T cells. (A) Splenic sections were stained for B220 (red) to identify B-cell zones, F4/80 (blue) to identify red pulp area, and CD45.2 (green) to identify B3K508 donor T cells. Percentage of B3K508 T cells located in the indicated areas at day 6. (B) Th1 and TFH cells were sorted from infected mice and transferred into CD3ε−/− mice immunized 1 d earlier with 3K-OVA and alum. GC B-cell numbers correlate with TFH cell numbers. (C) Intracellular cytokine production was assessed by flow cytometry. Data represent n ≥ 3 mice per group and at least two independent experiments.
Memory T Cells Generated by Low-Affinity pMHC–TCR Interactions Exhibit Distinct Recall Properties.
CD4 memory T-cell differentiation is suggested to be driven by the strength of TCR signaling during the primary response (17, 18). However, CXCR5+ T cells present at early time points after infection have also been suggested to be precursors to the long-lived central memory T-cell compartment (3). As low affinity P2A activation resulted in efficient differentiation of CXCR5+ CD4 T-cell effectors, we wondered if low affinity interactions were sufficient to generate long-lived memory T cells. Similar to 3K-activated T cells, P2A-activated T-cell numbers peaked at ∼5–6 d after infection followed by a rapid contraction phase between days 6 and 25 (Fig. S7A). P2A-activated T cells underwent less contraction compared with high dose 3K-activated T cells, and both populations could be identified in the long-lived memory T-cell compartment. Unlike polyclonal memory T cells, which have been shown to differentiate into distinct T-bethighCXCR5− and T-betlowCXCR5+ populations (3), the majority of B3K508 memory T cells had high expression of CXCR5 regardless of the nature of their primary stimulation (Fig. S7B), possibly due to the intrinsic nature of the B3K508 TCR (15). However, 3K memory T cells generated by both high and low antigen doses had higher overall expression of T-bet and a larger proportion of Ly6chighT-bethigh cells, while P2A memory T cells had an approximately threefold lower proportion of Ly6clowT-betlow cells that were mostly confined to the CXCR5+ compartment (Fig. S7B). Both CXCR5+T-betlow and Ly6clow memory T-cell subsets have been suggested to belong to the central memory compartment and have an increased ability to further differentiate following a recall infection (3, 20, 34). To determine whether recall capacity differed between 3K memory T cells and P2A memory T cells, mice were given a secondary infection with Listeria expressing high affinity 3K ligand. Four days after secondary infection, almost all high dose 3K memory T cells were T-bethighLy6chigh and approximately half of the T cells had down-regulated CXCR5 expression (Fig. 5A and Fig. S7C). Low dose 3K memory T cells also demonstrated a strong T-bethighLy6chigh bias, although a significant proportion of T-betlowLy6clow cells were also present (Fig. 5 and Fig. S7C). Surprisingly, P2A memory T cells further down-regulated T-bet and Ly6c expression, and had a larger proportion of CXCR5+T-betlo cells, despite receiving a high affinity secondary stimulation (Fig. 5 and Fig. S7C). These data indicate that memory T cells generated by low affinity activation remain impaired in their ability to up-regulate T-bet, despite receiving a high affinity recall stimulus. Recall responses following infection with recombinant vaccinia virus expressing 3K ligand showed a similar trend, with a higher proportion of P2A memory T cells expressing low levels of T-bet and Ly6c compared with 3K memory T cells (Fig. S8). Consistent with its intermediate priming affinity, P5R memory T cells generated an intermediate proportion of Th1 and TFH effectors (Fig. S9). Of note, memory T cells generated after weak priming by low dose Lm.3K and Lm.P2A expanded much more than memory T cells generated by high dose Lm.3K (Fig. S7D), indicating an inverse relationship between the magnitude of the primary and secondary responses. Taken together, these data show that robust and strikingly biased recall responses can be generated following primary infection with low antigen affinity versus low antigen dose, and suggest that a differentiated recall capacity is imprinted during the primary response.
Fig. 5.

Low affinity TCR–pMHC ligands support memory T-cell differentiation. Phenotype (A) and absolute cell number (B) of B3K508 T cells recovered at day 4 after recall infection with virulent Lm.3K. Data represent n ≥ 3 and are representative of three independent experiments. (*P < 0.05, ***P < 0.0001).
T Cells Activated by Low Affinity Antigen Depend on B Cells for Differentiation.
The observation that low affinity activation is sufficient to induce memory T-cell differentiation was unexpected, because decreased functional avidity has been previously correlated with a failure to differentiate into the memory compartment (17). In the previous study, however, mice were infected with a virulent strain of Listeria that results in lower antigen presentation by B cells, weak TFH generation, and decreased serum antibody titers. Given that both TFH and CXCR5+ memory T-cell differentiation depends on B-cell antigen presentation (3, 35), we wondered if P2A activated T cells could differentiate and survive long term in the absence of B cells. To examine this, the differentiation of B3K508 T cells in B-cell deficient (mb1-cre) mice was assessed. At day 6 after infection, B3K508 T cells activated with either Lm.3K or Lm.P2A in B-cell–deficient hosts underwent less expansion compared with B3K508 T cells activated in wild-type controls (Fig. S10A). T cells activated with high dose Lm.3K had a higher proportion of Th1 effectors (Fig. 6) and a higher mean fluorescence intensity (MFI) of T-bet (Fig. S10B), suggesting that B cells may play a role in limiting Th1 differentiation. In contrast, T cells weakly activated with either low dose Lm.3K or Lm.P2A in B-cell–deficient hosts had a lower T-bet MFI compared with priming in wild-type hosts (Fig. S10B), indicating that B cells are required for both optimal expansion and T-bet expression in response to weak TCR stimulation. At late time points after infection, memory B3K508 T cells were nearly undetectable in B-cell–deficient hosts infected with either Lm.3K or Lm.P2A (Fig. S10C), indicating that B cells are critically required for the optimal generation of B3K508 T-cell memory. However, upon secondary infection with Lm.3K, T-bethigh memory T cells could be recovered from all B-cell–deficient hosts, with the greatest number of secondary effector T cells generated by high dose 3K memory T cells (Fig. S10 D and E). These data demonstrate that T cells activated by low affinity antigen are critically dependent on B cells for differentiation into both TFH effector and memory T-cell compartments.
Fig. 6.
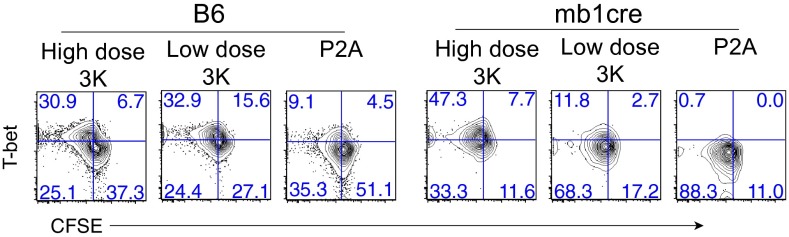
Effector and memory T-cell differentiation in B-cell–deficient mice. Phenotype of B3K508 T cells recovered from spleen and lymph nodes of B6 or mb-1cre mice at day 6 after infection. Data representative of two independent experiments.
Discussion
The experiments presented here underscore the importance of TCR affinity in directing Th cell fate. We found that T cells activated by increasing affinity ligands undergo more T-cell expansion and increased Th1 differentiation. In contrast, TFH differentiation and memory T-cell generation is not limited by antigen affinity and can occur in response to very weak, intermediate, and high affinity activation. Our results were unexpected in light of previous reports that increased TCR signal strength results in enhanced TFH cell and memory T-cell generation (15, 17). However, the complex relationship between T-cell expansion and differentiation makes it difficult to discern the potentially distinct contributions of pMHC affinity and pMHC dose in these studies. By normalizing the degree of proliferation induced by high and low affinity ligands, we observed distinct influences on T-cell differentiation during an infection. Regardless of antigen dose, high affinity antigen induced efficient Th1 differentiation, whereas low affinity antigen did not. Despite impaired Th1 differentiation, however, low affinity activated T cells were able to up-regulate TFH-associated surface markers, home to splenic B-cell areas, and were capable of providing help to germinal center B cells in an adoptive transfer model. Importantly, however, strong stimulation conditions, which induce the most T-cell expansion (i.e., high dose of high affinity antigen), result in the highest number of TFH effectors that are likely to provide more help to B cells. Although our model allows us to track very low affinity antigen-specific T-cell responses, studying a monoclonal population has inherent limitations and cannot address the relative impact of low affinity T-cell responses in a polyclonal setting, where low affinity T cells might make a greater contribution to antigen-specific responses (discussed further below) (36).
Our data support a model in which CD4 T-cell fate diversification begins at the earliest stages of naïve T-cell priming with DCs. T cells activated by high affinity antigen at both high and low antigen doses differentiate into distinct IL-2Rαhigh and IL-2Rαlow populations that are likely to be the precursors to Th1 and TFH effectors, respectively (37). These data indicate that the early generation of Th1 and TFH effectors is dose independent. In contrast, the early generation of Th1 effectors is strongly linked to antigen affinity. T cells activated by low affinity ligand demonstrate impaired Th1 differentiation as early as 3 days after infection, while the induction of early (CXCR5+IL-2Rαlow) TFH effectors occurs normally. TFH generation is thought to be a two-step process with early differentiation initiated by T-cell–DC interactions followed by a maintenance phase that is dependent on T-cell–B-cell interactions. Although we observed early TFH induction under all conditions, T cells activated by low antigen dose had a decreased proportion of TFH effectors at later time points after infection. The PD-1high TFH compartment, believed to contain germinal center TFH cells, was particularly affected, indicating the importance of antigen dose for sustained T-cell help to B cells (13, 15). One possible explanation for this is that low antigen dose results in decreased antigen availability during the maintenance phase of TFH differentiation. It is plausible that increased T-cell competition for limited amounts of antigen might inhibit late phase TFH expansion, resulting in a higher proportion of Th1 effectors. Of note, ongoing interactions between T and B cells at the peak of infection with lymophocytic choriomeningitis virus were recently shown to result in multiple rounds of TFH cell proliferation, while Th1 cells were largely refractory to stimulation at this later time point (37). These data suggest that temporal requirements for antigen may allow antigen dose and antigen affinity to set different thresholds for TFH development. B cells have also been suggested to play an active role in suppression of the Th1 response, which may be an alternate way in which they promote TFH generation (16). Our observation that T cells activated with high dose 3K in the absence of B cells express higher levels of T-bet support these findings.
In a study published last year, the aggregate dwell time (ta) of an antigen was suggested to be a better predictor of Th differentiation than antigen affinity (15). The authors reported that antigens with increased ta values (2.8 and 3.1 s) supported greater accumulation of TFH effectors, whereas an antigen with an intermediate ta value (2.3 s) supported maximal Th1 differentiation. Similar to our study, the authors also observed TFH generation in response to antigens with lower ta values. Although our results are largely in agreement with this study, it is possible that we do not see maximal Th1 differentiation with intermediate strength ligand because the ta value of the intermediate ligand in our study (0.9 s) is much lower than the intermediate ligand used in ref. 15 (2.3 s). Alternatively, small differences in the experimental systems, such as the number of transferred precursors or the time point of analysis might play a role. Tubo et al. (15) analyzed differentiation at day 7 after infection, which might not account for differences in the expansion and/or survival of Th1 and TFH effectors. For example, high antigen doses have been shown to induce more activation-induced cell death, particularly in Th1 effectors, which might in turn elevate the proportion of Th1 effectors at later time points (38).
An important but unresolved issue is how TFH differentiation can be supported by both strong and weak TCR signals. One possibility is that high and low affinity antigens are permissive for TFH differentiation at the level of IL-2 signaling. Strong TCR signals have been shown to transiently block IL-2R signaling, whereas weak TCR signals result in less IL-2/IL-2R expression (39). Both of these conditions would be permissive for Bcl6 up-regulation and TFH generation, which are negatively regulated by IL-2 (3, 32, 33). Of note, high affinity TCR signals have been shown to promote more stable T-cell contacts with antigen-presenting cells (APCs) compared with low affinity TCR signals (10). These data suggest that longer T-cell–APC contacts are required for the accumulation of signaling intermediates that facilitate IL-2 expression and Th1 differentiation. Long T-cell–APC contacts are also required for the induction of asymmetric cell division (28), which might lead to the generation of IL-2Rαhigh and IL-2Rαlow daughter cells that subsequently differentiate into Th1 and TFH effectors. In contrast, low affinity antigens that induce shorter T-cell–APC contacts might still support the accumulation of signaling intermediates required for TFH differentiation.
Importantly, our data also reveal that T cells activated by low affinity ligand can give rise to long-lived memory T cells capable of undergoing robust recall responses. Several studies demonstrated a relationship between CXCR5+ T cells present at early time points after infection and memory T-cell generation (3, 19, 20, 37). Consistent with this, low affinity activation supports both predominant differentiation of CXCR5+ T-cell effectors as well as memory T-cell development. Surprisingly, memory T cells generated by low affinity activation demonstrate a bias toward TFH phenotype upon recall, whereas memory T cells generated by high affinity activation are biased toward a Th1 recall response. By comparing memory T cells generated after priming with either high dose/low affinity or low dose/high affinity antigen, our data additionally show that T cells with a similar history of primary expansion have a similar capacity to expand during secondary infection, independent of effector lineage. These data are consistent with a recent report showing an inverse correlation between primary T-cell expansion and memory T-cell development (40).
To date, efforts to understand the contribution of low affinity antigens to polyclonal CD4 T-cell responses have been limited by the use of tetramers for the identification of antigen-reactive T cells. In addition to the observation that various peptides can bind MHC-II molecules in different registers, a recent study using a 2D binding assay showed that low affinity T cells, below detection with MHC-II tetramer, are present at a greater frequency and comprise approximately half of the effector cytokine response compared with tetramer positive cells (36). Thus, the contribution of low affinity CD4 T cells to immune responses is likely to be underestimated. Although our model reveals efficient TFH generation in response to high, intermediate, and low affinity ligand, there is a clear impact of antigen affinity on the number of effectors that can be generated. These data raise the question: To what end is the propagation of a low avidity T-cell response useful? Our data suggest the possibility that low affinity T cells might be particularly relevant to host protection against chronic pathogens or pathogens that are able to vary their antigenic motifs. In the case of chronic infection, resistance to exhaustion (41) coupled with a bias toward TFH differentiation might position low affinity T cells to provide help to B cells producing protective neutralizing antibody responses. Alternatively, the preservation of low affinity CD4 T cells in the memory T-cell compartment might confer broader host protection to pathogens such as influenza, which undergo frequent antigenic drift. Thus, understanding the cellular and molecular mechanisms by which low affinity antigens contribute to and shape immune responses has important implications for enhancing T-cell clonal diversity, boosting B-cell responses, and improving long-term protection against reinfection.
Materials and Methods
Mice.
All animal work was approved by the Cantonal Veterinary Office of the City of Basel. See SI Materials and Methods.
Infections.
Recombinant ActA-deficient L. monocytogenes [Lm] stably expressing 3K or variant ligands were generated following established protocols. For detailed L. monocytogenes preparation, see SI Materials and Methods.
Immunofluorescent Microscopy.
Purification of T Cells, Adoptive Cell Transfer, and Cell Labeling.
Flow Cytometry.
All samples were analyzed on BD FACSCalibur or BD Fortessa cytometers. See SI Materials and Methods.
Analysis of TFH Function.
Data Analyses.
Flow cytometry data were analyzed with FlowJo software (Tree Star). Graphs were prepared with Prism 6 (GraphPad Software) using the unpaired two-tailed Student t test.
Supplementary Material
Acknowledgments
We thank Ed Palmer and Michelle Linterman for helpful discussions and critical review of the manuscript, Barbara Hausmann for excellent technical support, and Elias Hobeika and Michael Reth for providing mb1-Cre mice. The work was supported by an Ambizione Grant from the Swiss National Science Foundation (to C.G.K.) and research grants from the Swiss Vaccine Research Institute and the Swiss National Science Foundation (PP00P3_144883 and PDFMP3_137128) (to D.Z.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. M.K.J. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1403271111/-/DCSupplemental.
References
- 1.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 2.Choi YS, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34(6):932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pepper M, Pagán AJ, Igyártó BZ, Taylor JJ, Jenkins MK. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity. 2011;35(4):583–595. doi: 10.1016/j.immuni.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitmire JK, et al. Requirement of B cells for generating CD4+ T cell memory. J Immunol. 2009;182(4):1868–1876. doi: 10.4049/jimmunol.0802501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corbin GA, Harty JT. Duration of infection and antigen display have minimal influence on the kinetics of the CD4+ T cell response to Listeria monocytogenes infection. J Immunol. 2004;173(9):5679–5687. doi: 10.4049/jimmunol.173.9.5679. [DOI] [PubMed] [Google Scholar]
- 6.Bird JJ, et al. Helper T cell differentiation is controlled by the cell cycle. Immunity. 1998;9(2):229–237. doi: 10.1016/s1074-7613(00)80605-6. [DOI] [PubMed] [Google Scholar]
- 7.Obst R, van Santen HM, Mathis D, Benoist C. Antigen persistence is required throughout the expansion phase of a CD4(+) T cell response. J Exp Med. 2005;201(10):1555–1565. doi: 10.1084/jem.20042521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gett AV, Sallusto F, Lanzavecchia A, Geginat J. T cell fitness determined by signal strength. Nat Immunol. 2003;4(4):355–360. doi: 10.1038/ni908. [DOI] [PubMed] [Google Scholar]
- 9.Malherbe L, Hausl C, Teyton L, McHeyzer-Williams MG. Clonal selection of helper T cells is determined by an affinity threshold with no further skewing of TCR binding properties. Immunity. 2004;21(5):669–679. doi: 10.1016/j.immuni.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Gottschalk RA, et al. Distinct influences of peptide-MHC quality and quantity on in vivo T-cell responses. Proc Natl Acad Sci USA. 2012;109(3):881–886. doi: 10.1073/pnas.1119763109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hosken NA, Shibuya K, Heath AW, Murphy KM, O’Garra A. The effect of antigen dose on CD4+ T helper cell phenotype development in a T cell receptor-alpha beta-transgenic model. J Exp Med. 1995;182(5):1579–1584. doi: 10.1084/jem.182.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tao X, Grant C, Constant S, Bottomly K. Induction of IL-4-producing CD4+ T cells by antigenic peptides altered for TCR binding. J Immunol. 1997;158(9):4237–4244. [PubMed] [Google Scholar]
- 13.Baumjohann D, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity. 2013;38(3):596–605. doi: 10.1016/j.immuni.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 14.Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol. 2009;10(4):375–384. doi: 10.1038/ni.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tubo NJ, et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell. 2013;153(4):785–796. doi: 10.1016/j.cell.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ploquin MJ, Eksmond U, Kassiotis G. B cells and TCR avidity determine distinct functions of CD4+ T cells in retroviral infection. J Immunol. 2011;187(6):3321–3330. doi: 10.4049/jimmunol.1101006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams MA, Ravkov EV, Bevan MJ. Rapid culling of the CD4+ T cell repertoire in the transition from effector to memory. Immunity. 2008;28(4):533–545. doi: 10.1016/j.immuni.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim C, Wilson T, Fischer KF, Williams MA. Sustained interactions between T cell receptors and antigens promote the differentiation of CD4+ memory T cells. Immunity. 2013;39(3):508–520. doi: 10.1016/j.immuni.2013.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weber JP, Fuhrmann F, Hutloff A. T-follicular helper cells survive as long-term memory cells. Eur J Immunol. 2012;42(8):1981–1988. doi: 10.1002/eji.201242540. [DOI] [PubMed] [Google Scholar]
- 20.Hale JS, et al. Distinct memory CD4+ T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity. 2013;38(4):805–817. doi: 10.1016/j.immuni.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aleksic M, et al. Dependence of T cell antigen recognition on T cell receptor-peptide MHC confinement time. Immunity. 2010;32(2):163–174. doi: 10.1016/j.immuni.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Merwe PA, Dushek O. Mechanisms for T cell receptor triggering. Nat Rev Immunol. 2011;11(1):47–55. doi: 10.1038/nri2887. [DOI] [PubMed] [Google Scholar]
- 23.Zehn D, King C, Bevan MJ, Palmer E. TCR signaling requirements for activating T cells and for generating memory. Cell Mol Life Sci. 2012;69(10):1565–1575. doi: 10.1007/s00018-012-0965-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Govern CC, Paczosa MK, Chakraborty AK, Huseby ES. Fast on-rates allow short dwell time ligands to activate T cells. Proc Natl Acad Sci USA. 2010;107(19):8724–8729. doi: 10.1073/pnas.1000966107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huseby ES, Crawford F, White J, Marrack P, Kappler JW. Interface-disrupting amino acids establish specificity between T cell receptors and complexes of major histocompatibility complex and peptide. Nat Immunol. 2006;7(11):1191–1199. doi: 10.1038/ni1401. [DOI] [PubMed] [Google Scholar]
- 26.Vanguri V, Govern CC, Smith R, Huseby ES. Viral antigen density and confinement time regulate the reactivity pattern of CD4 T-cell responses to vaccinia virus infection. Proc Natl Acad Sci USA. 2013;110(1):288–293. doi: 10.1073/pnas.1208328110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stepanek O, et al. Co-receptor scanning by the T-cell receptor provides a kinetic mechanism for T-cell tolerance. 2014 doi: 10.1016/j.cell.2014.08.042. Cell, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King CG, et al. T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity. 2012;37(4):709–720. doi: 10.1016/j.immuni.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Enouz S, Carrié L, Merkler D, Bevan MJ, Zehn D. Autoreactive T cells bypass negative selection and respond to self-antigen stimulation during infection. J Exp Med. 2012;209(10):1769–1779. doi: 10.1084/jem.20120905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hemmer B, Stefanova I, Vergelli M, Germain RN, Martin R. Relationships among TCR ligand potency, thresholds for effector function elicitation, and the quality of early signaling events in human T cells. J Immunol. 1998;160(12):5807–5814. [PubMed] [Google Scholar]
- 31.Ansel KM, McHeyzer-Williams LJ, Ngo VN, McHeyzer-Williams MG, Cyster JG. In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J Exp Med. 1999;190(8):1123–1134. doi: 10.1084/jem.190.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. 2012;209(2):243–250. doi: 10.1084/jem.20111174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oestreich KJ, Mohn SE, Weinmann AS. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat Immunol. 2012;13(4):405–411. doi: 10.1038/ni.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marshall HD, et al. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity. 2011;35(4):633–646. doi: 10.1016/j.immuni.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goenka R, et al. Cutting edge: dendritic cell-restricted antigen presentation initiates the follicular helper T cell program but cannot complete ultimate effector differentiation. J Immunol. 2011;187(3):1091–1095. doi: 10.4049/jimmunol.1100853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabatino JJ, Jr, Huang J, Zhu C, Evavold BD. High prevalence of low affinity peptide-MHC II tetramer-negative effectors during polyclonal CD4+ T cell responses. J Exp Med. 2011;208(1):81–90. doi: 10.1084/jem.20101574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi YS, et al. Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol. 2013;190(8):4014–4026. doi: 10.4049/jimmunol.1202963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, et al. Unequal death in T helper cell (Th)1 and Th2 effectors: Th1, but not Th2, effectors undergo rapid Fas/FasL-mediated apoptosis. J Exp Med. 1997;185(10):1837–1849. doi: 10.1084/jem.185.10.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamane H, Zhu J, Paul WE. Independent roles for IL-2 and GATA-3 in stimulating naive CD4+ T cells to generate a Th2-inducing cytokine environment. J Exp Med. 2005;202(6):793–804. doi: 10.1084/jem.20051304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weber KS, et al. Distinct CD4+ helper T cells involved in primary and secondary responses to infection. Proc Natl Acad Sci USA. 2012;109(24):9511–9516. doi: 10.1073/pnas.1202408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caserta S, Kleczkowska J, Mondino A, Zamoyska R. Reduced functional avidity promotes central and effector memory CD4 T cell responses to tumor-associated antigens. J Immunol. 2010;185(11):6545–6554. doi: 10.4049/jimmunol.1001867. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.