

Significance
Molten globules—defined as compact protein conformations with significant secondary structure but only loosely packed tertiary structure—have been hypothesized to be general folding intermediates. In this work we investigate one folding intermediate long thought to be a molten globule and find significant evidence that it likely has a well-folded region, with closely packed tertiary structure. These results suggest that the evidence for moltenness in other protein folding intermediates should be revisited and that even for fairly simple, small proteins, exclusion of water can occur before the rate-limiting step to folding.
Abstract
The molten globule, a conformational ensemble with significant secondary structure but only loosely packed tertiary structure, has been suggested to be a ubiquitous intermediate in protein folding. However, it is difficult to assess the tertiary packing of transiently populated species to evaluate this hypothesis. Escherichia coli RNase H is known to populate an intermediate before the rate-limiting barrier to folding that has long been thought to be a molten globule. We investigated this hypothesis by making mimics of the intermediate that are the ground-state conformation at equilibrium, using two approaches: a truncation to generate a fragment mimic of the intermediate, and selective destabilization of the native state using point mutations. Spectroscopic characterization and the response of the mimics to further mutation are consistent with studies on the transient kinetic intermediate, indicating that they model the early intermediate. Both mimics fold cooperatively and exhibit NMR spectra indicative of a closely packed conformation, in contrast to the hypothesis of molten tertiary packing. This result is important for understanding the nature of the subsequent rate-limiting barrier to folding and has implications for the assumption that many other proteins populate molten globule folding intermediates.
Although many proteins populate intermediates early in the folding process, the role of such intermediates is still unclear. To what extent are they obligatory? What is the nature of the free-energy barrier that allows for the transient population of these states? Answering such questions requires detailed knowledge of the structure and dynamics of folding intermediates.
Many large single-domain proteins (>100 amino acids) populate a partially folded intermediate state within the burst-phase of stopped flow experiments (milliseconds, ms) that is thought to be a “molten globule” (1–4). Molten globules are defined as compact structures with a high degree of secondary structure that lack the tight tertiary interactions that are the hallmark of natively folded proteins (5). The molten globule has been proposed to be a general intermediate formed before the rate-limiting barrier (6), where later folding steps involve the exclusion of water and formation of close tertiary packing. [The state in which water has been excluded but close tertiary packing has not yet been achieved is a “dry” molten globule, hypothesized to be a general intermediate on the native side of the rate-limiting barrier (5, 7).] To evaluate the universality of molten globules in protein folding, it is important to clearly demonstrate the nature of the tertiary packing in early folding intermediates.
Molten globules were first observed in equilibrium studies in which they can be populated under extreme solution conditions (i.e., low pH, in the presence of chemical denaturant, and/or by removal of a cofactor such as heme) (8–10). In these studies, probes such as circular dichroism (CD), NMR, and ANS (1-anilino-8-naphthalene sulfonic acid) fluorescence revealed a conformational ensemble not well described by a native or unfolded state and therefore termed the molten globule conformation. Landmark hydrogen–deuterium (HD) exchange studies on the acid molten globule of apomyoglobin demonstrated secondary structure in three of eight helices from the native protein (A, G, and H) (11). Later, pulse-labeling hydrogen exchange showed that these same helices are also protected in the transient early (ms) folding intermediate under native conditions (3). The protein ribonuclease H (RNase H from Escherichia coli) also folds through a burst-phase intermediate and populates a molten globule under acidic conditions (9); again, the acid molten globule and transient kinetic intermediate have similar patterns of protection, indicating they form a similar subset of the native secondary structure (4). On the basis of these and other studies, the dominant hypothesis has been that the burst-phase folding intermediates are themselves molten globules.
It is particularly difficult, however, to assay the structural details of these transient folding intermediates to determine conclusively whether the protected region is molten or well folded. Structural probes such as CD or fluorescence typically yield only global information. Although HD exchange is a powerful way to gain site-specific information, it only directly monitors backbone hydrogen bonds. Orthogonal techniques, such as phi- and psi-value analyses (12, 13) and recently alkyl–proton exchange (14), have been used to probe the role of specific side-chain interactions in folding intermediates. However, these techniques cannot conclusively evaluate the side-chain packing in the structured region of a transient intermediate: resulting data consistent with moltenness do not rule out other possible hypotheses, such as a closely packed region that folds and refolds rapidly or has nonnative tertiary structure.
To address the question of specific tertiary packing in the structured region of transient folding intermediates, we have designed and interrogated different mimics of the kinetic folding intermediate of RNase H, an important protein folding model system with a well-characterized folding intermediate thought to be a molten globule. The folding process of E. coli RNase H has been characterized using CD, HD exchange, mutagenesis, single molecule force spectroscopy (optical tweezers), and a computational prediction algorithm (4, 15–18). These studies all lead to a picture of a folding intermediate (termed “Icore”) populated before the rate-limiting barrier to folding, with native-like secondary structure in a contiguous region of sequence from helix A to strand V (the “core” region of the protein), whereas the rest of the protein appears unfolded (Fig. 1). The acid molten globule state of RNase H, populated at very low pHs (<2), shows HD protection in the same helices (19).
Fig. 1.
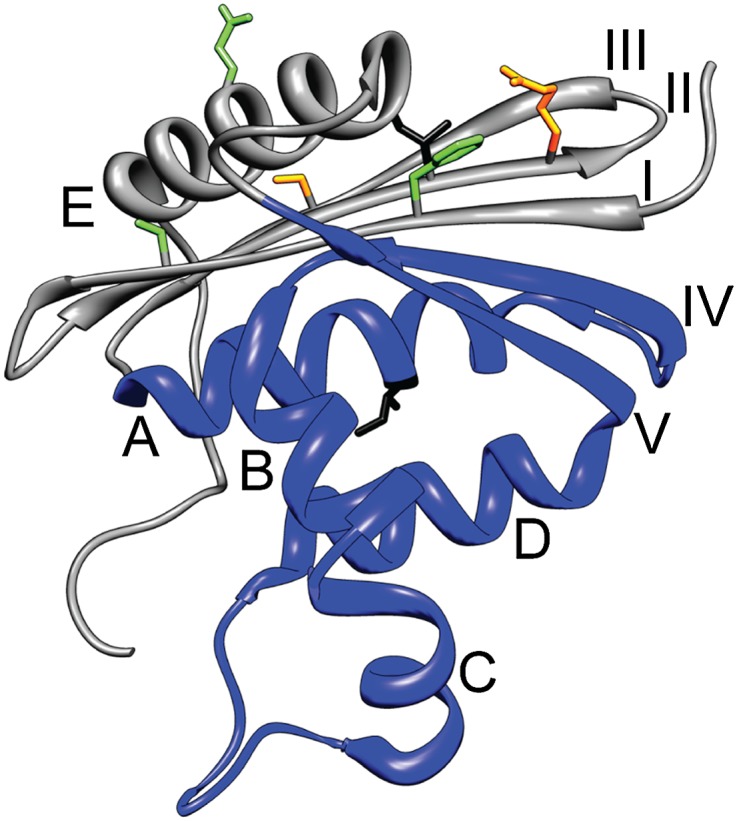
Structure of E. coli RNase H (Protein Data Bank 2RN2). Helices are labeled with letters and β-strands with Roman numerals. The region comprising the Icore fragment is colored blue. Residues I25 (on strand II) and I53 (on helix A) are shown in black stick. Other residues shown in stick are involved in the full-length Icore mimics (FL1 contains mutations at I25 and the orange residues, FL2 contains mutations at the orange and green residues).
In addition to similarities to the acid state molten globule, other observations have been interpreted to support the hypothesis that the structured region of Icore is molten. The I53L mutation in the core region destabilizes the native state but does not notably affect the stability of Icore (20). This was interpreted to indicate loose association of hydrophobic side chains in the intermediate but is also consistent with nonnative tertiary structure in Icore accommodating the mutation. In a separate study, a single destabilizing mutation (I25A) was shown to cause extensive population of the Icore intermediate under equilibrium conditions (21). In the presence of ∼2 M urea, Icore is the dominant species at equilibrium for I25A, but no peaks corresponding to the structured region of the intermediate could be observed in NMR HSQC (1H-15N heteronuclear single quantum coherence) experiments under these conditions, suggesting that the structured region of Icore is very dynamic. The dynamics could be due to a poorly packed, heterogeneous structured region, or due to fast folding and unfolding of a closely packed structure.
Here we probe the structured region of the RNase H Icore intermediate directly by creating equilibrium mimics of the folding intermediate that are populated under native conditions. First, we generate a fragment of E. coli RNase H comprising the sequence from helix A to strand V (Fig. 1). The fragment folds in isolation and mimics the known behavior of the kinetic intermediate. We find, however, that the fragment is not molten and forms a closely packed structure. In a second approach, we create several destabilizing mutations in the periphery (the presumably unfolded region of Icore), to populate the intermediate under native conditions in the context of the full-length protein and find that the structured region is again well folded. Finally, we show that our results are compatible with the previous data that were originally interpreted to support the hypothesis of a molten structure.
In sum, our data imply that the structured region of the E. coli RNase H kinetic intermediate (Icore) is well folded, with closely packed side chains. It remains possible that the region lacking protection in the kinetic HD exchange experiments, previously assumed to be unstructured in the folding intermediate, may itself have a molten globule character, and/or that the protein forms a molten intermediate before this well-folded intermediate. This work suggests that the rate-limiting step in the folding of E. coli RNase H is not the packing down of existing tertiary interactions in the Icore intermediate, but is more likely related to assembly of the rest of the protein or a reorganization of the folded region. These results also suggest that the evidence for moltenness in the folding intermediates of other proteins is worth revisiting.
Results
A Fragment Mimic of the Transient Folding Intermediate, Icore.
Previous pulse-labeling hydrogen exchange, computational, and protein engineering studies have identified residues 43–122 as the structured region of Icore (Fig. 1) (4, 15, 16, 18). The sequence of E. coli RNase H constituting this region was cloned, expressed, and purified from E. coli. [In this study, E .coli RNase H refers to a well-studied cysteine-free variant (9).] The resulting fragment is soluble and is shown to adopt a stable fold; the CD spectrum appears to show double minima close to 208 nm and 222 nm, consistent with helix formation (Fig. 2A). Equilibrium urea-induced denaturation of the fragment, monitored by the CD signal at 222 nm, shows a cooperative folding transition (Fig. 2B) with a ΔGunf of 3.0 ± 0.3 kcal/mol, and an m-value of 1.16 ± 0.05 kcal·mol−1·M−1 using a two-state assumption and linear-extrapolation model (Table 1) (22). The stability of the transient folding intermediate Icore is ∼3.5 kcal/mol (with an m-value of 1.2 kcal·mol−1·M−1) as determined by stopped-flow CD studies fit to an on-pathway three-state model (4). Thus, both the CD spectrum and the energetics of the fragment are consistent with those measured for the transient kinetic folding intermediate (4).
Fig. 2.

The RNase H Icore fragment is well folded. (A) CD spectrum of the fragment measured at 4-μM protein concentration. (B) Representative equilibrium denaturation curves of the fragment monitored by CD and normalized to fraction folded, at 3.6 μM (black) and 36 μM (red). (C) Overlay of 1H-15N HSQC spectra of the fragment measured at 25 μM (black) and 200 μM (red). The red peaks were shifted slightly to the right for easier comparison. A subset of the tryptophan side chain peaks have been enlarged for illustration.
Table 1.
Stabilities of Icore fragments compared with kinetic intermediates
| Variant | Fragment equilibrium denaturation | Kinetic burst-phase intermediate (20) | ||
| ΔGunf (kcal/mol) | munf (kcal·mol−1·M−1) | ΔGunf (kcal/mol) | munf (kcal·mol−1·M−1) | |
| WT | 3.0 ± 0.3 | 1.16 ± 0.05 | 3.5 | 1.2 |
| I53A | 1.1* | 1.16* | 1.7 | 1.2 |
| I53L | 2.7 ± 0.3 | 1.15 ± 0.06 | 3.6 | 1.2 |
| I53V | 3.4 ± 0.9 | 1.2 ± 0.3 | 4.0 | 1.2 |
Using an m-value fixed to that of WT.
The Fragment Folds as a Monomer at CD Concentrations.
To confirm that the folded fragment is a monomer under our conditions, we used equilibrium analytical ultracentrifugation (AUC). The AUC data fit well to a monomer–dimer model, with a dissociation constant of ∼75 μM (Fig. S1). On the basis of this Kd, the above CD studies (3–4 μM) contain ∼95% monomer, indicating that we are effectively measuring properties of the monomer, and therefore the monomer is folded. Additionally, because dimerization will be attenuated by urea, the stability reported from the urea denaturation should be a reasonable approximation of the monomeric stability. To underscore this, the melt was repeated with a 10-fold higher protein concentration (36 μM), whereby ∼75% monomer is expected in the 0 M urea sample (Fig. 2B), and yielded a ΔGunf and m-value of 3.1 kcal/mol and 1.2 kcal·mol−1·M−1, within error of the result at 3.6-μM protein concentration.
NMR Suggests the Interior of the Fragment is Closely Packed.
We used HSQC NMR studies to evaluate the structural heterogeneity of the folded fragment. These experiments, however, require a minimum concentration near the Kd of dimerization. To distinguish the monomer signal from dimer signal, spectra were measured at two different protein concentrations: 200 μM and 25 μM, predicted to contain ∼50% monomer and ∼80% monomer, respectively (Fig. 2C).
Both HSQC spectra exhibit sharp, well-dispersed peaks. In each spectrum, many more peaks are observed than expected if only one molecular species were present, but the peaks can be categorized into two groups that can be attributed to monomer and dimer. In the 200-μM sample, both sets of peaks have similar intensities; however, in the 25-μM sample, the monomer peaks dominate and the dimer peaks persist only at low intensities. The fact that all peaks are sharp and well dispersed indicates that both the monomer and dimer are unique, well-folded structures. Fortuitously, dimerization kinetics are slow enough to allow clear resolution of peaks from both species.
Mutational Analysis of the Fragment.
We used mutational analysis to make a direct comparison between the properties of the fragment mimic and the kinetic intermediate. In the native state of RNase H, helix A forms buried hydrophobic contacts with helix D—interactions that are also predicted to be present in the kinetic intermediate (Fig. 1). Because the effect of mutations at residue 53 on the folding and stability of RNase H have been previously characterized (20), we chose to construct and analyze a subset of these mutations (I53A, I53L, and I53V) in the fragment and determine their effect on its stability. All three single-site variants fold and show similar CD spectra to the wild-type fragment (Fig. 3A). All were evaluated by equilibrium urea-induced denaturation and show cooperative unfolding transitions (Fig. 3B). The denaturation curves for fragments I53L and I53V were fit using the standard two-state assumption. For the fragment I53A, the folded state was too destabilized to obtain a folded baseline. To determine the approximate stability of this variant, the data were fit to a two-state model with the m-value fixed to that obtained for the WT fragment (1.16 kcal·mol−1·M−1). This fit yielded reasonable values for the slope of the folded baseline and the native-state signal, comparable to those for the other fragments. These results are summarized in Table 1 and demonstrate that the fragment mirrors the transient kinetic intermediate in terms of the effects of these mutations. (Please note that there is substantial error in the stabilities calculated from burst phase intermediates, so the trends are more meaningful than the exact numbers.)
Fig. 3.

The effect of mutations at residue 53 in the RNase H Icore fragment. (A) CD spectra of Icore fragment single-site variants I53A (black circles), I53L (orange circles), and I53V (gray circles) compared with wild type (line), measured at ∼3.5 μM protein concentration. (B) Representative equilibrium denaturation curves of the Icore fragment variants I53A (black circles), I53L (orange circles), and I53V (gray circles) normalized to fraction folded, compared with wild type (line alone). Two-state fits are shown. (The I53A fit was performed with a fixed m-value.) All experiments were measured at ∼3.5 μM. (C) Overlay of HSQC spectra of the I53L fragment variant measured at 50 μM (black) and 420 μM (orange). The orange peaks were shifted slightly to the right for easier comparison. A subset of peaks has been enlarged for illustration.
NMR Analysis of a Single-Site Variant Suggests That the Variant Is Also Well Folded.
As seen in the transient kinetic intermediate, I53L has no significant effect on fragment stability, even though I53L destabilizes the native state by 1.4 kcal/mol (20). (The fragment is also not destabilized at all by I53V, but neither is the native state.) One interpretation of the I53L result could be a loosely packed core without steric constraints in the fragment. To gain further structural insight into the effect of the I53L mutation on side-chain packing in the core of the fragment, we determined the HSQC spectrum of this variant.
The spectrum of the I53L variant shows many well-dispersed peaks at similar (but not identical) chemical shifts to the wild-type fragment (Fig. 3C). Measuring spectra at two protein concentrations again demonstrated two sets of peaks with relative intensities dependent on protein concentration. These data suggest that the I53L variant has a similar monomer–dimer equilibrium as the wild-type fragment and that both the monomer and dimer are well folded.
The Urea Dependence of the HSQC Spectrum.
Previous NMR studies on the single-site RNase H variant I25A, which preferentially populates the Icore intermediate in the presence of ∼2 M urea, show no folded peaks corresponding to the structured region of Icore (21). To investigate whether these results could be consistent with our fragment mimic, we measured HSQC spectra of the wild-type fragment as a function of urea (Fig. 4).
Fig. 4.

Urea dependence of the RNase H Icore fragment 1H-15N HSQC spectrum. Overlay of HSQC spectra measured in the presence of 1 M urea (red) and 2 M urea (black). Spectra were measured at ∼100-μM protein concentration. A subset of peaks has been enlarged for illustration.
The HSQC spectrum of the wild-type fragment in 1 M urea looks essentially identical to the spectrum measured in 0 M urea. However, in the 2 M urea spectrum, only unfolded state peaks are visible, and only at very low signal intensity. On the basis of our CD studies, we expect greater than 80% of the molecules are folded in these conditions (Fig. 2B). These data resemble the I25A variant and therefore suggest that at 2 M urea the rate of fragment unfolding increases sufficiently to cause significant exchange broadening of the folded peaks. Thus, the lack of folded Icore peaks in the I25A variant NMR spectrum is not necessarily indicative of a molten heterogeneous structure.
Full-Length Icore Mimics via Selective Destabilization of the Native State.
Although the fragment mimic allows us to evaluate properties of the structured region of the transient intermediate, it lacks the region predicted to be unstructured. To evaluate the role of this region, we took a second approach to making an equilibrium mimic of Icore, starting from the full-length protein.
Single-site amino acid changes (to glycine or alanine) were generated in the unstructured region of the Icore model. For each single point mutant, the equilibrium urea-induced denaturation was monitored by its CD signal at 222 nm, and fit as previously described to calculate its effect on the stability of the protein (Table S1). To create full-length intermediate mimics, these point mutations were combined such that the native state would be less stable than the intermediate state assuming additive destabilizations (Fig. 5A, Inset). Two different full-length mimics were created using different combinations of mutations. The full-length mimic 1 (FL1) contains the mutations I25A/R27A/S36G, and full-length mimic 2 (FL2) contains F8A/S12G/R27A/S36G/E135G (Fig. 1).
Fig. 5.

Full-length Icore mimics made by selective destabilization of the native state are well folded. (A) CD spectra of the FL1 mimic (black circles) and FL2 mimic (gray circles) measured at ∼1.5-μM protein concentration, compared with full-length wild-type RNase H (line). (Inset) The native state is selectively destabilized so that Icore becomes the ground state. (B) Equilibrium denaturation curves of the FL1 mimic (black circles) and FL2 mimic (gray circles) measured at ∼2-μM protein concentration, normalized to fraction folded, compared with full-length wild-type RNase H (line alone). Two-state fits are shown. (C) HSQC spectrum of the FL1 mimic measured at ∼100 μM.
Both full-length mimics display CD spectra notably different from the native protein (Fig. 5A). Like the fragments, the spectra appear to have double minima close to 222 nm and 208 nm, whereas the full-length native protein has a single minimum around 215 nm. Equilibrium urea-induced denaturation was monitored by CD signal at 222 nm and showed cooperative transitions with ΔGunf of 3.2 ± 0.4 kcal/mol and m-value of 1.2 ± 0.1 kcal·mol−1·M−1 for FL1 and ΔGunf of 3.3 ± 0.5 kcal/mol and m-value of 1.2 ± 0.2 kcal·mol−1·M−1 for FL2 (Fig. 5B). The m-value of 1.2 kcal·mol−1·M−1 is much lower than the m-value of the native protein, 2.0 kcal·mol−1·M−1, indicating that the full-length mimics bury much less solvent-exposed surface area than the native states. These values mimic those measured in the equilibrium denaturation of the fragment discussed above.
The Full-Length Mimics Are Structurally Similar to the Fragment.
The HSQC spectra of the full-length mimics show well-dispersed peaks and very similar chemical shifts as the fragment HSQC spectrum, as illustrated by the spectrum of FL1 (Fig. 5C). Specifically, the full-length mimic HSQC peaks match the chemical shifts of the peaks identified previously as belonging to the fragment monomer, indicating that the structured regions of the full-length mimics are similar to the fragment mimic. The most notable difference is the presence of a large number of collapsed peaks along the hydrogen axis in the full-length mimic spectra compared with the fragment spectrum. We interpret these collapsed peaks as corresponding to the unfolded region of the full-length mimics. Interestingly, peaks at chemical shifts matching those of the fragment dimer are visible at low intensity in the full-length mimic spectra.
The Intermediate Mimics Do Not Bind and Increase the Fluorescence of ANS.
Our last experiment to evaluate the tertiary packing of the Icore mimics was to monitor binding to ANS by fluorescence, a traditional hallmark of molten globules. Under acid-state conditions (i.e., the previously identified molten globule), the wild-type protein shows a large increase in ANS fluorescence consistent with it being a molten globule. In contrast, under native conditions, we observe that the fragment and full-length mimics all exhibit low ANS fluorescence similar to natively folded wild-type, supporting that they adopt a well-folded structure (Fig. 6). These experiments were performed using 2 μM protein and 50 μM ANS. Molten globule-like fluorescence with the fragment and full-length mimics [as well as I25A (21)] could, however, be induced by increasing the ANS concentration to 500 μM. However, at high ANS concentration the observed fluorescence intensity correlates with precipitation of a protein–ANS aggregate and is not a true measure of monomer tertiary packing.
Fig. 6.

The Icore mimics do not bind the hydrophobic dye ANS. Fluorescence emission spectra of ANS in the presence of (A) the Icore fragment (red) and (B) the Icore full-length mimics FL1 (solid red) and FL2 (dashed red). For comparison, spectra in the presence of full-length wild-type RNase H at pH 1.2 (top black curve), pH 5.5 (middle black curve), and 6 M urea (bottom black curve) are shown in both plots. All experiments were performed with 2 μM protein and 50 μM ANS. The fluorescence emission of ANS in buffer alone has been subtracted from the data.
Discussion
In this work, we set out to investigate the claim that the early folding (ms) intermediate of RNase H (Icore) is a molten globule. Tertiary packing is very difficult to probe in transient intermediates, so we used protein engineering to create mimics of Icore that can be studied with equilibrium tools. We created fragment and full-length mimics of Icore by either fully removing or making mutations in the periphery of RNase H. In all cases the equilibrium properties of the mimics were consistent with known properties of the kinetic intermediate. Additionally, the mimics exhibited cooperative equilibrium denaturation and good dispersion in NMR HSQC experiments—hallmarks of well-folded proteins. Finally, previous results used to support a molten globule hypothesis—fast structural dynamics and a high tolerance for core mutations—are exhibited by the well-folded fragment, indicating these results are compatible with well-folded structure. We conclude that the RNase H Icore folding intermediate is likely not a molten globule.
Our work demonstrates that the core region of RNase H is capable of autonomously forming a well-folded structure, representing a partially folded intermediate on the energy landscape. However, is this intermediate necessarily the Icore folding intermediate? What if the close-packed equilibrium conformation of the mimics represents a conformation formed on the native side of the rate-limiting barrier to folding, with a molten conformation populated before the rate-limiting barrier? This can be addressed by kinetic studies monitoring tertiary structure formation in the mimics. Ongoing experiments using an ultrarapid mixing technique to directly observe the first 10 ms of folding of the fragment mimic look very similar to early folding of the wild-type protein, which strongly suggests that the fragment achieves its equilibrium conformation on the same timescale as the formation of Icore, and that the fragment is a good mimic of Icore.
What is the nature of the slow, rate-limiting barrier if not the packing down of tertiary interactions? Our mutagenesis results suggest that the rate-limiting barrier may be related to a reorganization of the folded core. The I53L mutation destabilizes the native state, but not the transient Icore intermediate nor the fragment mimic, even though the fragment is well folded. This may indicate that the folded region reorganizes upon formation of the native state (i.e., the folded core is not quite native-like in the Icore intermediate, and therefore mutations have a different effect on stability than in the native state). [Nonnative interactions in on-pathway kinetic intermediates have been observed for other model systems (23–25).] Alternatively, it is also possible that the folded region of Icore is natively packed in the wild-type protein but can repack to accommodate the I53L mutation with no penalty in stability (the impact on stability occurs only when the rest of the protein assembles onto the repacked core to form the native state). In this situation, the folding barrier may simply be the unfavorable conformational entropy for assembly of the β-sheet. More work will be needed to distinguish between these scenarios.
Our work is consistent with other studies that have demonstrated that acid molten globules are not accurate models for transient folding intermediates under native conditions. Extensive experiments on apomyoglobin have shown that the hydrogen-exchange protection in the acid state molten globule follows a slightly different pattern and has a different stability distribution than observed in the kinetic intermediate (26, 27). In a related protein, apoleghemoglobin, there are substantial structural differences between the protection in the acid state molten globule and the kinetic intermediate (28). Though conformational heterogeneity has been observed in the kinetic intermediate of apomyoglobin, such observations are not incompatible with a region of structure that remains well folded (25, 27, 29). Rapid formation of native-like tertiary contacts has been observed in other model systems, which also calls into question the generality of early folding to a molten globule state (30, 31).
Last, a previous NMR study on Thermus thermophilus RNase H suggests that it may have a folding intermediate with closely packed side chains. In a study from Yawen Bai’s laboratory, the authors made a fragment of T. thermophilus RNase H (composed of the region we have identified as the structured region of the folding intermediate plus β-strand I connected with a nonnative junction) and observed that their fragment is well folded (32). This fragment did not have the complication of dimerization, and so they were able to solve the NMR structure (32). The overall topology of the fragment looks completely native-like, although a close analysis reveals deviations in side-chain packing compared with the crystal structure of wild-type T. thermophilus RNase H (33). However, it is not clear whether these deviations are significant, or indeed whether this fragment truly mimics the intermediate before the rate-limiting barrier to folding.
Overall, the RNase H data suggest that many observed protein folding intermediates may have regions of closely packed structure, contrary to a widespread assumption of molten globule kinetic intermediates. Experimental data interpreted to support moltenness should be examined carefully for other possible interpretations.
Materials and Methods
Construction and Purification of RNase H Variants.
The fragment was constructed by subcloning from the pSM101 vector, with insertion into a pET27 vector. All other variants were generated by QuikChange Mutagenesis starting with pSM101 or the fragment vector. For the FL mimics, a hexahistidine tag was added to the C terminus. Expressions were performed in Rosetta2(DE3)pLysS. Expression of 15N-labeled protein was done by initial growth in LB with a switch to M9 with 15NH4Cl as the sole nitrogen source before a 3-h induction. Labeling efficiency was ∼90% as evaluated by mass spectrometry. All constructs expressed insolubly and were purified from inclusion bodies, as detailed in SI Materials and Methods. After purification, all proteins were dialyzed into experiment buffer conditions: 20 mM NaOAc (pH 5.5) and 50 mM KCl. Protein concentrations were determined on the basis of the extinction coefficient, calculated according to the number of Trp and Tyr residues (34).
CD Experiments.
All CD data were measured on an Aviv 410 CD spectropolarimeter. All experiments were performed with a 1-cm path length cuvette, except melts with 10× protein concentration used a 1-mm path length. Signal was averaged 20 s at each wavelength for spectra and 60 s for each melt sample. Further details are provided in SI Materials and Methods.
Equilibrium Analytical Ultracentrifugation.
Sedimentation equilibrium experiments were performed with a Beckman XL-I analytical ultracentrifuge, at 24,500, 30,000 and 37,000 rpm. Details are given in the SI Materials and Methods.
HSQC Spectra.
Two-dimensional 1H-15N HSQCs were recorded on a Bruker Avance II 900-MHz spectrometer equipped with a TCI cryoprobe at 25 °C. For samples with ≥100 μM protein concentration, 16 or 32 scans were collected. Otherwise, 64 or 128 scans were collected, in all cases with 1,024 points in the direct dimension and 256 points in the indirect dimension. The data were processed and viewed using either mNOVA or NMRpipe and CARA.
ANS Binding.
Samples containing 50 μM ANS in buffer with and without 2 μM protein were prepared and equilibrated for ≥3 h. Fluorescence emission spectra were collected from 430 to 650 nm with excitation at 370 nm. The spectrum of ANS in buffer alone was subtracted from the data with protein.
Supplementary Material
Acknowledgments
We thank J. Pelton for guidance in NMR data collection and analysis; D. Wemmer for additional help with interpretation of NMR data; the QB3 MacroLab (and C. Jeans) for help with equilibrium AUC data collection and analysis; K. Fleming and C. Kimberlin for additional advice regarding the AUC experiments; and the entire Marqusee Lab for helpful comments and discussion. This work was supported by National Institutes of Health Grant GM50945 (to S.M.) and a Molecular Biophysics Training Grant, and a National Science Foundation Graduate Research Fellowship (to L.E.R.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1410630111/-/DCSupplemental.
References
- 1.Arai M, Kuwajima K. Rapid formation of a molten globule intermediate in refolding of alpha-lactalbumin. Fold Des. 1996;1(4):275–287. doi: 10.1016/s1359-0278(96)00041-7. [DOI] [PubMed] [Google Scholar]
- 2.Fujiwara K, et al. Folding-unfolding equilibrium and kinetics of equine β-lactoglobulin: Equivalence between the equilibrium molten globule state and a burst-phase folding intermediate. Biochemistry. 1999;38(14):4455–4463. doi: 10.1021/bi982683p. [DOI] [PubMed] [Google Scholar]
- 3.Jennings PA, Wright PE. Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science. 1993;262(5135):892–896. doi: 10.1126/science.8235610. [DOI] [PubMed] [Google Scholar]
- 4.Raschke TM, Marqusee S. The kinetic folding intermediate of ribonuclease H resembles the acid molten globule and partially unfolded molecules detected under native conditions. Nat Struct Biol. 1997;4(4):298–304. doi: 10.1038/nsb0497-298. [DOI] [PubMed] [Google Scholar]
- 5.Baldwin RL, Rose GD. Molten globules, entropy-driven conformational change and protein folding. Curr Opin Struct Biol. 2013;23(1):4–10. doi: 10.1016/j.sbi.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Barrick D, Baldwin RL. Stein and Moore Award address. The molten globule intermediate of apomyoglobin and the process of protein folding. Protein Sci. 1993;2(6):869–876. doi: 10.1002/pro.5560020601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baldwin RL, Frieden C, Rose GD. Dry molten globule intermediates and the mechanism of protein unfolding. Proteins. 2010;78(13):2725–2737. doi: 10.1002/prot.22803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuwajima K. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins. 1989;6(2):87–103. doi: 10.1002/prot.340060202. [DOI] [PubMed] [Google Scholar]
- 9.Dabora JM, Marqusee S. Equilibrium unfolding of Escherichia coli ribonuclease H: Characterization of a partially folded state. Protein Sci. 1994;3(9):1401–1408. doi: 10.1002/pro.5560030906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griko YV, Privalov PL, Venyaminov SY, Kutyshenko VP. Thermodynamic study of the apomyoglobin structure. J Mol Biol. 1988;202(1):127–138. doi: 10.1016/0022-2836(88)90525-6. [DOI] [PubMed] [Google Scholar]
- 11.Hughson FM, Wright PE, Baldwin RL. Structural characterization of a partly folded apomyoglobin intermediate. Science. 1990;249(4976):1544–1548. doi: 10.1126/science.2218495. [DOI] [PubMed] [Google Scholar]
- 12.Matouschek A, Kellis JT, Jr, Serrano L, Fersht AR. Mapping the transition state and pathway of protein folding by protein engineering. Nature. 1989;340(6229):122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 13.Krantz BA, Sosnick TR. Engineered metal binding sites map the heterogeneous folding landscape of a coiled coil. Nat Struct Biol. 2001;8(12):1042–1047. doi: 10.1038/nsb723. [DOI] [PubMed] [Google Scholar]
- 14.Bernstein R, Schmidt KL, Harbury PB, Marqusee S. Structural and kinetic mapping of side-chain exposure onto the protein energy landscape. Proc Natl Acad Sci USA. 2011;108(26):10532–10537. doi: 10.1073/pnas.1103629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu W, et al. Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2013;110(19):7684–7689. doi: 10.1073/pnas.1305887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raschke TM, Kho J, Marqusee S. Confirmation of the hierarchical folding of RNase H: A protein engineering study. Nat Struct Biol. 1999;6(9):825–831. doi: 10.1038/12277. [DOI] [PubMed] [Google Scholar]
- 17.Cecconi C, Shank EA, Bustamante C, Marqusee S. Direct observation of the three-state folding of a single protein molecule. Science. 2005;309(5743):2057–2060. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 18.Fischer KF, Marqusee S. A rapid test for identification of autonomous folding units in proteins. J Mol Biol. 2000;302(3):701–712. doi: 10.1006/jmbi.2000.4049. [DOI] [PubMed] [Google Scholar]
- 19.Dabora JM, Pelton JG, Marqusee S. Structure of the acid state of Escherichia coli ribonuclease HI. Biochemistry. 1996;35(37):11951–11958. doi: 10.1021/bi9611671. [DOI] [PubMed] [Google Scholar]
- 20.Spudich GM, Miller EJ, Marqusee S. Destabilization of the Escherichia coli RNase H kinetic intermediate: Switching between a two-state and three-state folding mechanism. J Mol Biol. 2004;335(2):609–618. doi: 10.1016/j.jmb.2003.10.052. [DOI] [PubMed] [Google Scholar]
- 21.Connell KB, Horner GA, Marqusee S. A single mutation at residue 25 populates the folding intermediate of E. coli RNase H and reveals a highly dynamic partially folded ensemble. J Mol Biol. 2009;391(2):461–470. doi: 10.1016/j.jmb.2009.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santoro MM, Bolen DW. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry. 1988;27(21):8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 23.Capaldi AP, Kleanthous C, Radford SE. Im7 folding mechanism: Misfolding on a path to the native state. Nat Struct Biol. 2002;9(3):209–216. doi: 10.1038/nsb757. [DOI] [PubMed] [Google Scholar]
- 24.Feng H, Zhou Z, Bai Y. A protein folding pathway with multiple folding intermediates at atomic resolution. Proc Natl Acad Sci USA. 2005;102(14):5026–5031. doi: 10.1073/pnas.0501372102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishimura C, Dyson HJ, Wright PE. Identification of native and non-native structure in kinetic folding intermediates of apomyoglobin. J Mol Biol. 2006;355(1):139–156. doi: 10.1016/j.jmb.2005.10.047. [DOI] [PubMed] [Google Scholar]
- 26.Nishimura C, Dyson HJ, Wright PE. Enhanced picture of protein-folding intermediates using organic solvents in H/D exchange and quench-flow experiments. Proc Natl Acad Sci USA. 2005;102(13):4765–4770. doi: 10.1073/pnas.0409538102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uzawa T, et al. Hierarchical folding mechanism of apomyoglobin revealed by ultra-fast H/D exchange coupled with 2D NMR. Proc Natl Acad Sci USA. 2008;105(37):13859–13864. doi: 10.1073/pnas.0804033105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishimura C, Dyson HJ, Wright PE. The kinetic and equilibrium molten globule intermediates of apoleghemoglobin differ in structure. J Mol Biol. 2008;378(3):715–725. doi: 10.1016/j.jmb.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meinhold DW, Wright PE. Measurement of protein unfolding/refolding kinetics and structural characterization of hidden intermediates by NMR relaxation dispersion. Proc Natl Acad Sci USA. 2011;108(22):9078–9083. doi: 10.1073/pnas.1105682108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizukami T, Xu M, Cheng H, Roder H, Maki K. Nonuniform chain collapse during early stages of staphylococcal nuclease folding detected by fluorescence resonance energy transfer and ultrarapid mixing methods. Protein Sci. 2013;22(10):1336–1348. doi: 10.1002/pro.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orevi T, et al. Fast closure of N-terminal long loops but slow formation of β strands precedes the folding transition state of Escherichia coli adenylate kinase. Biochemistry. 2014;53(19):3169–3178. doi: 10.1021/bi500069w. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Z, Feng H, Ghirlando R, Bai Y. The high-resolution NMR structure of the early folding intermediate of the Thermus thermophilus ribonuclease H. J Mol Biol. 2008;384(2):531–539. doi: 10.1016/j.jmb.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishikawa K, et al. Crystal structure of ribonuclease H from Thermus thermophilus HB8 refined at 2.8 A resolution. J Mol Biol. 1993;230(2):529–542. doi: 10.1006/jmbi.1993.1169. [DOI] [PubMed] [Google Scholar]
- 34.Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6(7):1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.