

Significance
Many rare human genetic disorders are caused by mutations in genes that code for proteins of poorly characterized function. Determining the functions of these proteins is critical for understanding and devising potential treatments for human diseases. In this article, we discover using a combination of mouse genetic models, selective inhibitors, and lipid profiling that the DDHD2 enzyme, mutations of which cause a neurological disease termed complex hereditary spastic paraplegia (HSP), acts as a major brain triglyceride hydrolase. Mice lacking DDHD2 have elevated brain triglycerides and lipid droplet accumulation in neurons. We have thus discovered that the brain possesses a specialized pathway for triglyceride metabolism, disruption of which leads to biochemical and cellular changes that may contribute to complex HSP.
Abstract
Complex hereditary spastic paraplegia (HSP) is a genetic disorder that causes lower limb spasticity and weakness and intellectual disability. Deleterious mutations in the poorly characterized serine hydrolase DDHD2 are a causative basis for recessive complex HSP. DDHD2 exhibits phospholipase activity in vitro, but its endogenous substrates and biochemical functions remain unknown. Here, we report the development of DDHD2−/− mice and a selective, in vivo-active DDHD2 inhibitor and their use in combination with mass spectrometry-based lipidomics to discover that DDHD2 regulates brain triglycerides (triacylglycerols, or TAGs). DDHD2−/− mice show age-dependent TAG elevations in the central nervous system, but not in several peripheral tissues. Large lipid droplets accumulated in DDHD2−/− brains and were localized primarily to the intracellular compartments of neurons. These metabolic changes were accompanied by impairments in motor and cognitive function. Recombinant DDHD2 displays TAG hydrolase activity, and TAGs accumulated in the brains of wild-type mice treated subchronically with a selective DDHD2 inhibitor. These findings, taken together, indicate that the central nervous system possesses a specialized pathway for metabolizing TAGs, disruption of which leads to massive lipid accumulation in neurons and complex HSP syndrome.
Determining the genetic basis for rare hereditary human diseases has benefited from advances in DNA sequencing technologies (1). As a greater number of disease-causing mutations are mapped, however, it is also becoming apparent that many of the affected genes code for poorly characterized proteins. Assigning biochemical and cellular functions to these proteins is critical to achieve a deeper mechanistic understanding of human genetic disorders and for identifying potential treatment strategies.
Hereditary spastic paraplegia (HSP) is a genetically heterogeneous neurologic syndrome marked by spasticity and lower extremity weakness (2). Many genetic types of HSP have been identified and are numbered according to their order of discovery [spastic paraplegia (SPG) 1-72] (2, 3). Of these genetic variants, more than 40 have been mapped to causative mutations in protein-coding genes. HSP genes code for a wide range of proteins that do not conform to a single sequence- or function-related class. A subset of HSP genes, including PNPLA6 (or neuropathy-target esterase) (SPG39) (4), DDHD1 (SPG28) (5), and DDHD2 (SPG54) (3, 6–8), code for serine hydrolases. These enzymes have been designated as (lyso)phospholipases based on in vitro substrate assays (9–11), but their endogenous substrates and physiological functions remain poorly understood. The mutational landscape that affects these lipid hydrolases to cause recessive HSP is complex but collectively represents a mix of null and putatively null and/or functional mutations. Moreover, the type of HSP appears to differ in each case, with DDHD1 mutations causing uncomplicated HSP, whereas PNPLA6 and DDHD2 mutations lead to complex forms of the disease that exhibit additional phenotypes including, in the case of DDHD2, intellectual disability. Human subjects with DDHD2 mutations also displayed evidence of brain lipid accumulation as detected by cerebral magnetic resonance spectroscopy (6). Both rodent and human DDHD2 enzymes are highly expressed in the brain compared with most peripheral tissues (6, 9); however, the specific lipids regulated by DDHD2 in the central nervous system (CNS) have not yet been identified.
Determining the metabolic function of DDHD2 in the brain is an important step toward understanding how mutations in this enzyme promote complex HSP and for identifying possible therapeutic strategies for the disease. Toward this end, we report herein the generation and characterization of DDHD2−/− mice and a selective DDHD2 inhibitor. DDHD2−/− mice exhibit defects in movement and cognitive function. Mass spectrometry (MS)-based lipidomics (12, 13) revealed a striking and selective elevation in triglycerides (triacylglycerols, or TAGs) throughout the CNS, but not in peripheral tissues, of DDHD2−/− mice. This metabolic change correlated with pervasive lipid droplet (LD) accumulation in neuronal cell bodies of DDHD2−/− mice. Biochemical assays confirmed that DDHD2 possesses TAG hydrolase activity. Finally, wild-type mice treated subchronically with a DDHD2 inhibitor also exhibited significant elevations in CNS TAGs. These data, taken together, indicate that DDHD2 is a principal TAG hydrolase of the mammalian brain and point to deregulation of this pathway as a major contributory factor to complex HSP.
Results
Targeted Genetic Disruption of the Ddhd2 Gene.
Mice with a targeted disruption of the Ddhd2 gene were generated by homologous recombination, where exon 8, which contains the catalytic serine nucleophile S351 of DDHD2 (9), was removed from the genome of C57BL/6 embryonic stem (ES) cells (SI Appendix, Fig. S1A). An ES clone bearing the targeted disruption was identified by Southern blotting (SI Appendix, Fig. S1B) and used to generate DDHD2−/− mice (Fig. 1 A–C). DDHD2−/− mice were viable and born at the expected Mendelian frequency. We confirmed loss of DDHD2 mRNA by RT-PCR of DDHD2−/− brain tissue (Fig. 1B) and loss of DDHD2 protein by quantitative proteomic experiments that combined activity-based protein profiling (ABPP) (14) with high-resolution MS. In brief, DDHD2+/+ and DDHD2−/− brain proteomes were treated with the serine hydrolase-directed activity-based probe fluorophosphonate (FP)-biotin, and probe-labeled serine hydrolases were enriched by streptavidin chromatography, digested on bead with trypsin, and the resulting tryptic peptides modified by reductive dimethylation of lysine residues using isotopically light and heavy formaldehyde, respectively (15) (also see SI Appendix, Fig. S2). The DDHD2+/+ and DDHD2−/− samples were then combined and analyzed by multidimensional liquid chromatography (LC)–MS (16) using an LTQ-Orbitrap Velos instrument. These experiments confirmed the complete loss of DDHD2 signals in brain tissue from DDHD2−/− mice (SI Appendix, Fig. S1C and Dataset S1), whereas the 40+ other serine hydrolases detected by ABPP were unchanged. Selective loss of DDHD2 in DDHD2−/− mice was further verified using DDHD2-directed (HT-01) and broad-spectrum [FP-rhodamine (FP-Rh)] fluorescent activity-based probes (17), where the former probe revealed the absence of a characteristic 75-kDa DDHD2 doublet (9) in DDHD2−/− brains, whereas other serine hydrolase signals were unaltered (Fig. 1C).
Fig. 1.

Confirmation of loss of DDHD2 expression in DDHD2−/− mice. (A) Confirmation by PCR genotyping of germ-line transfer for an ES cell clone with targeted replacement of exon 8 of the Ddhd2 gene with a neomycin (Neo) selection cassette (see SI Appendix, Fig. S1 for gene targeting information). (B) Absence of DDHD2 mRNA in DDHD2−/− mice was confirmed by RT-PCR analysis of brain tissue. (C) Gel-based ABPP with the HT-01 probe confirmed lack of DDHD2 activity in brain membrane proteomes from DDHD2−/− mice. ABPP with the broad-spectrum probe FP-Rh confirmed that other serine hydrolase activities were unchanged in DDHD2−/− brain proteomes. [Note that DDHD2 is too low in abundance for detection by gel-based ABPP with the FP-Rh probe, but other representative serine hydrolases are marked for reference (17).].
DDHD2−/− Mice Show Defects in Locomotion and Cognition.
DDHD2−/− mice were fertile and largely indistinguishable from their wild-type and heterozygous littermates in terms of normal cage behavior. DDHD2−/− mice, however, displayed significantly shorter stride lengths in gait measurement assays, and this locomotor defect was observed in both front and rear paws (SI Appendix, Fig. S3A). In assays of general locomotor activity, no genotype differences were observed in ambulation behavior, but DDHD2−/− mice showed significant reductions in rearing behavior, as measured by beam breaks that occur when a mouse moves vertically above the ground (SI Appendix, Fig. S3B). In the rotarod test, DDHD2−/− mice held their balance for a significantly shorter amount of time and fell off the rod at lower speeds compared with DDHD2+/+ mice (Fig. 2A), indicating that Ddhd2 gene disruption caused impairments in motor coordination.
Fig. 2.

Characterization of locomotor and cognitive functions in DDHD2−/− mice. (A) In a constant speed rotarod test, DDHD2−/− mice ran for a shorter time than DDHD2+/+ mice before falling (Left). In a rapidly accelerating speed rotarod test (Right), DDHD2−/− mice fell at lower speeds than DDHD2+/+ and DDHD2+/− mice. (B) In the Barnes maze spatial learning test, DDHD2−/− mice spent a shorter proportion of time in the quadrant with an escape chamber during training compared with DDHD2+/+ mice (Left). When the same mice were retested 2 wk later, the DDHD2−/− mice took longer to find the escape chamber compared with DDHD2+/+ and DDHD2+/− mice (Right). Data represent average values ± SEM. n = 13 mice per group. *P < 0.05 and **P < 0.01 for DDHD2+/+ versus DDHD2−/− mice; #P < 0.05 for DDHD2−/− versus DDHD2+/− mice.
We next assessed cognition and memory in DDHD2+/+ and DDHD2−/− mice. We first confirmed no significant genotype differences in vision (SI Appendix, Fig. S3C) and then measured cognition in the Y-maze test, which tests propensity of mice to explore new environments, and found no significant genotype difference in spontaneous alternations (SI Appendix, Fig. S3D). DDHD2−/− mice, on the other hand, showed impairments in the Barnes maze test (Fig. 2B), which assesses learning and memory of a target zone location based on visual cues in the surrounding environment (18). DDHD2−/− mice also exhibited defective long-term spatial memory as reflected in significantly longer times to find the target zone when re-evaluated 2 wk post-test phase (Fig. 2B).
These data demonstrate that DDHD2−/− mice display motor and cognitive impairments that resemble the clinical symptoms of SPG54 human subjects with DDHD2 mutations.
Triglyceride (TAG) Accumulation in DDHD2−/− Brains.
To identify endogenous lipids regulated by DDHD2, we performed lipidomic experiments of brain tissue from adult (2–4-mo-old) DDHD2+/+ and DDHD2−/− mice. In these studies, organic-soluble brain metabolites were analyzed by untargeted LC–MS and data processed using the XCMS program (19), resulting in identification of several metabolites that were significantly elevated in DDHD2−/− brains (Fig. 3A). The largest changes (∼3–5-fold) were observed for metabolites with m/z values of 948.8, 950.8, and 1,040.8 detected in positive-ion mode in the presence of ammonium formate (included as an ion pairing agent). The common retention times and differences in m/z values indicated that the changing metabolites represent structurally related members of a lipid class that differ in acyl chain length and degrees of unsaturation. Their large molecular weights and detection as [M+NH4]+ adducts further suggested that the metabolites are likely members of the glycero- (versus phospho)-lipid class.
Fig. 3.

TAG accumulation in DDHD2−/− brain tissue. (A) Lipidomic profile of brain tissue from DDHD2+/+ and DDHD2−/− mice. Metabolites were extracted from tissue of adult mice using a chloroform/methanol mixture and analyzed by LC–MS in both positive and negative ion mode as described in Materials and Methods. The XCMS algorithm identified features that were significantly different between DDHD2+/+ and DDHD2−/− brains, including a set of lipids that accumulated in DDHD2−/− brains and corresponded to the [M+NH4]+ adduct of triglycerides (TAGs). Fragmentation analysis and LC migration times were used to confirm structural assignment of TAGs (SI Appendix, Figs. S4 and S5). Other major lipid classes, including DAGs, monoglycerides (MAGs), free fatty acids (FFAs), and phospholipids [phosphatidic acids (PAs), phosphatidylcholines (PCs), and phosphatidylethanolamines (PEs)] were unchanged in DDHD2−/− brain tissue. (B) Targeted LC–MS analysis verified widespread elevations in TAGs in brain tissue from DDHD2−/− mice. (C) Comparison of TAGs in different tissues from DDHD2+/+ and DDHD2−/− mice (Sp. cord, spinal cord). Data represent average values ± SEM. n = 5–9 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 for DDHD2−/− versus DDHD2+/+ mice.
Tandem MS analysis revealed that the DDHD2-regulated metabolites all produced characteristic fragmentation patterns of triglycerides (TAGs), including a diagnostic diglyceride (DAG) fragment ion formed from loss of one acyl chain (20, 21) (SI Appendix, Fig. S4). Reanalysis of the lipidomics data identified additional candidate TAGs that accumulated in DDHD2−/− brains, including a species with an m/z value of 824.8, which corresponds to tripalmitoylated (C16:0/C16:0/C16:0) TAG (SI Appendix, Table S1). Consistent with this structural assignment, the natural m/z 824.8 and 1,040.8 metabolites and synthetic tripalmitate and docosahexaenoate TAG standards, respectively, showed matching LC elution times and fragmentation spectra (SI Appendix, Fig. S5). In contrast to the elevations found for TAGs in DDHD2−/− brains, other major lipid classes (e.g., phospholipids, lysophospholipids, free fatty acids, monoglycerides, and DAGs) were unchanged in lipidomic comparisons of brain tissue from DDHD2+/+ and DDHD2−/− mice (SI Appendix, Table S1).
We next used targeted, multiple reaction monitoring (MRM) LC–MS to confirm the changes in TAGs in DDHD2−/− mice, which revealed accumulation of numerous TAG species containing combinations of both saturated (C16:0 and C18:0) and unsaturated (C18:1, C20:4, C22:5, and C22:6) fatty acyl chains in brains of adult (10-mo-old) DDHD2−/− mice (Fig. 3B and SI Appendix, Table S2). We also analyzed DDHD2+/− mice and found that brain TAGs did not accumulate in these animals (SI Appendix, Fig. S6). The age dependency of TAG changes was assessed by analyzing brains from 3-wk- and 2-mo-old mice, which showed accumulation of many TAGs in the latter but not former DDHD2−/− mice (SI Appendix, Fig. S7A). The basis for the age-dependent accumulation of TAGs in DDHD2−/− mice is not clear, but we note that DDHD2 activity as detected by ABPP with the HT-01 probe was much lower in embryonic and early postnatal brain tissue compared with adult brain (SI Appendix, Fig. S7B).
Humans with deleterious DDHD2 mutations present principally with nervous system-related deficits (3, 6–8). We therefore wondered whether the TAG changes observed in DDHD2−/− mice were restricted to the brain or more broadly distributed throughout central and peripheral tissues. Targeted LC–MS studies revealed that TAGs were generally unaltered in peripheral tissues of adult DDHD2−/− mice (6–10 mo), with the exception of white adipose tissue, which exhibited a modest (∼40%) increase in TAGs (Fig. 3C and SI Appendix, Fig. S8). We also did not observe changes in serum TAGs in DDHD2−/− mice (SI Appendix, Fig. S8). In contrast, significant TAG increases were observed in spinal cord tissue from DDHD2−/− mice, indicating that this metabolic change occurs broadly throughout the CNS (Fig. 3C and SI Appendix, Fig. S8).
Taken together, our metabolomic findings indicate that DDHD2 plays a specialized role in regulating the metabolism of TAGs in the CNS.
DDHD2 Disruption Causes LD Accumulation in Neurons.
Elevations in TAGs in peripheral tissues can lead to an accumulation of LDs and an increase in the size of these LDs (22). We therefore next compared DDHD2+/+ and DDHD2−/− sagittal brain slices treated with osmium tetroxide, which reacts with unsaturated lipid acyl chains to enable visualization of LDs by electron microscopy (EM) (23). Lower magnification EM images revealed substantial accumulation of LDs in brain tissue from DDHD2−/− mice (Fig. 4A). Examples of regions with massively increased LDs were located in the pons (region 1), mostly within the pontine central gray, and ventral to the corpus callosum (region 2) in the septal nuclei (Fig. 4A). Light microscopy studies of brain region sections (2-µm thick, osmicated and counterstained with toluidine blue) confirmed extensive LD accumulation in DDHD2−/− mice (Fig. 4B) and enabled quantification of these LDs, which showed a significant increase in number, area, and diameter in DDHD2−/− brains (Fig. 4C). LD accumulation was detected in young (3 mo; Fig. 4A) and older (6–9 mo; Fig. 4B) DDHD2−/− mice.
Fig. 4.

Ultrastructural analysis of LD accumulation in DDHD2−/− brains. (A) Brains from adult DDHD2+/+ and DDHD2−/− mice were fixed, sectioned sagittally, and treated for 3 h with OsO4 before imaging with electron and light microscopy. Lower magnification images of thin (70 nm) sections of the pontine central gray (region 1) and septal nuclei (region 2) of 3-mo-old mice were captured by transmission EM, which revealed dark staining LDs in both regions in DDHD2−/− brains, but not in DDHD2+/+ brains. (Scale bar, 20 μm in all images.) (B) Thick, resin-embedded sections (2 μm) of the same regions were taken from 6- to 9-mo-old mice and counterstained with toluidine blue. Extensive LD accumulation was detected in both regions of DDHD2−/−, but not in DDHD2+/+ brains. (Scale bar, 50 μm in the Upper images and 10 μm in the Lower insets taken from the boxed regions of Upper images.) (C) The LD margins were identified using Image-Pro Plus 7.0 as described in Materials and Methods and used to quantify an increased number (Left), area (Center), and diameter (Right) of LDs in DDHD2−/− brains. (D) Ultrastructural EM analysis showing LD accumulation in neurons from DDHD2−/− mice. LD accumulation was observed either as multiple smaller droplets (Left) or one large droplet (Right) in the cytoplasm of neurons. Arrowhead denotes LDs. M, mitochondria; N, nucleus. See SI Appendix, Fig. S9 for additional images. Data represent average values ± SEM. n = 4 mice per group. **P < 0.01 and ****P < 0.0001 for DDHD2−/− versus DDHD2+/+ mice.
Brain regions of adult mice (3–9 mo) were analyzed at higher magnification by EM, which revealed that LDs were primarily found within neuronal compartments of DDHD2−/− mice (Fig. 4D). Neurons were positively identified by the presence of synapses, and the accumulating LDs were found in different segments of the neurons, including both axons and dendrites (SI Appendix, Fig. S9A). In contrast, LD accumulation was not observed in glial cells of DDHD2−/− mice. In regions 1 and 2, multiple LDs could be found within the same neuronal cell (Fig. 4D). The size of these LDs varied from a few hundred nm to >5 µm (Fig. 4D and SI Appendix, Fig. S9). In region 2, we observed noticeable swelling of the processes that occurred to accommodate large LDs. The detected LDs were often surrounded by organelles, including mitochondria (SI Appendix, Fig. S9C).
Our combined light and EM studies demonstrated that DDHD2−/− mice exhibit substantial LD accumulation in multiple brain regions and these LDs appear to be primarily localized to intracellular compartments of neurons.
Development of a Selective and In Vivo-Active DDHD2 Inhibitor.
DDHD2 is a multidomain protein that may have both enzymatic and nonenzymatic functions in cells (9, 24). To ascertain whether brain TAG accumulation in DDHD2−/− mice was due to loss of catalytic function of DDHD2, we sought to develop a selective inhibitor of this enzyme for in vivo pharmacological studies. We previously discovered that acyclic phenethyl-1,2,3-triazole ureas, including the HT-01 probe, react with DDHD2 in a covalent, irreversible manner (17). A survey of analogs of HT-01 revealed that KLH45 bearing a cyclohexyl group in place of the extended BODIPY fluorophore of HT-01 (Fig. 5A) potently inhibited DDHD2 as measured by competitive ABPP (SI Appendix, Fig. S10 A and B). KLH45, a 2,4-substituted regioisomer, showed enhanced potency and selectivity for DDHD2 over the corresponding 1,4-isomer (KLH45b; SI Appendix, Fig. S10C). KLH45 exhibited an IC50 value of 1.3 nM (SI Appendix, Fig. S10B) for DDHD2 and excellent selectivity against other serine hydrolases in the mouse neuroblastoma Neuro2A proteome as assessed by gel-based competitive ABPP, showing cross-reactivity with only a single detected enzyme ABHD6 (SI Appendix, Fig. S10 A and B). Further exploration of HT-01 analogs identified an inactive-control inhibitor KLH40 (Fig. 5A), which showed negligible activity against DDHD2 (IC50 > 10 µM), but comparable cross-reactivity with ABHD6 (IC50 ∼0.4–0.6 µM; SI Appendix, Fig. S10 A and B).
Fig. 5.

Mice treated subchronically with a selective DDHD2 inhibitor show TAG accumulation in the CNS. (A) Structural modifications to HT-01 yielded a DDHD2-selective inhibitor KLH45 and inactive-control inhibitor KLH40. (B) Targeted LC–MS analysis revealed accumulation of TAGs in brain and spinal cord (spinal) tissues from mice treated subchronically for 4 d with KLH45 versus vehicle or KLH40 (inhibitors were administered at 20 mg⋅kg−1 compound, i.p., every 12 h). (C) Competitive ABPP experiments confirmed the inactivation of DDHD2 in KLH45-treated but not KLH40-treated mice. Both KLH45 and KLH40 partially inhibited ABHD6 and FAAH but showed negligible cross-reactivity with other brain serine hydrolases. Data represent average values ± SEM. n = 4 mice per group. *P < 0.05 and ***P < 0.001 for KLH45-treated versus vehicle-treated mice.
KLH45 inactivated DDHD2 and ABHD6 in Neuro2A cells with low nanomolar in situ potency (<10 nM; SI Appendix, Fig. S11A), whereas treatment of these cells with KLH40 inhibited ABHD6 but not DDHD2 (SI Appendix, Fig. S11A). Higher resolution, quantitative MS analyses using the ABPP–stable isotope labeling by amino acids in cell culture method (17) confirmed that KLH45 (25 nM, 4 h) completely inactivated DDHD2 (>95% inhibition) in Neuro2A cells and showed no cross-reactivity with any of the other 40+ detected serine hydrolases with the exception of ABHD6 (SI Appendix, Fig. S11B and Dataset S2). KLH40 showed similar inhibitory activity against ABHD6 (∼90% blockade) and partial inhibition of FAAH and PLA2G15 (50–70%), but negligible cross-reactivity with DDHD2 (SI Appendix, Fig. S11B). Among the serine hydrolases that were not affected by KLH45 were DDHD1 and Sec23ip (SI Appendix, Fig. S11B), indicating that this inhibitor shows good selectivity for DDHD2 over other sequence-related DDHD enzymes.
We next tested whether KLH45 could inhibit DDHD2 in vivo. Mice were treated with a dose range of KLH45 or KLH40 (5–40 mg⋅kg−1, i.p.) for 4 h, killed, and brain tissue collected, processed, and analyzed by gel-based competitive ABPP using the HT-01 probe. A clear dose-dependent blockade of brain DDHD2 activity was observed in KLH45-treated animals, with near-complete loss of activity occurring at the 40 mg⋅kg−1 dose (SI Appendix, Fig. S12). KLH45 also inhibited ABHD6 in vivo, but this off-target was similarly inhibited by KLH40, which did not block DDHD2 (SI Appendix, Fig. S12).
These data indicate that KLH45 and KLH40 constitute a suitable pair of active and inactive control probes to investigate the function of DDHD2 in cell and animal models. More details on the medicinal chemistry that led to the discovery of these probes will be provided in a separate manuscript in due course.
Pharmacological Inactivation of DDHD2 Elevates Brain TAGs.
We next examined whether acute or subchronic treatments with KLH45 affected TAG metabolism in the mouse brain. Mice treated acutely with KLH45 (40 mg⋅kg−1, 4 h) did not show altered brain TAGs (SI Appendix, Fig. S13A). In contrast, mice treated twice daily with KLH45 (20 mg⋅kg−1; administered every 12 h) for a total of 4 d exhibited significant elevations in several of the TAGs that accumulated in the brains of DDHD2−/− mice (Fig. 5B and SI Appendix, Fig. S13B). Mice treated with KLH40 under the same dosing regimen showed no changes in brain TAGs relative to a vehicle-treated control group (Fig. 5B and SI Appendix, Fig. S13B). Similar changes in TAGs were observed in the spinal cord of mice subchronically treated with KLH45 (Fig. 5B and SI Appendix, Fig. S13B). Competitive ABPP studies confirmed complete inactivation of brain DDHD2 in mice treated subchronically with KLH45, whereas no changes in brain DDHD2 activity were observed in mice treated subchronically with the control probe KLH40 (Fig. 5C). Both probes showed minimal cross-reactivity with other brain serine hydrolases in the subchronic dosing paradigm, with each inhibitor producing partial blockade of ABHD6 and FAAH (Fig. 5C).
These results, taken together, demonstrate that prolonged pharmacological blockade of DDHD2 promotes elevations in brain TAGs, indicating that this metabolic change is likely due to disruption of DDHD2 activity.
DDHD2 Exhibits TAG Hydrolase Activity.
To test whether DDHD2 functions as a TAG hydrolase, we transfected HEK293T cells with a cDNA encoding FLAG epitope-tagged mouse DDHD2 (wild type, WT) and confirmed expression of the enzyme by HT-01 labeling and Western blotting (SI Appendix, Fig. S14A). A catalytically inactive serine to alanine mutant (S351A) of DDHD2 exhibited similar expression in transfected cells and showed no HT-01 labeling (SI Appendix, Fig. S14A). Recombinant DDHD2 was inhibited by KLH45 with a similar potency to that measured for endogenous DDHD2 in the Neuro2A proteome (IC50 values of 3.6 and 1.3 nM, respectively) and was not affected by the inactive control probe KLH40 (SI Appendix, Fig. S14 B and C). Transfected HEK293T cell lysates were next incubated with a 14C-labeled C18:1/C18:1/C18:1 TAG substrate (22 µM, 90 min), and conversion of this lipid to C18:1/C18:1 DAG, C18:1 MAG and C18:1 fatty acid (oleic acid) products was monitored by a radiolabeled TLC assay following established protocols (25). We observed a significant increase in all three products in WT–DDHD2-transfected but not S351A–DDHD2-transfected cell lysates compared with mock-transfected or heat-denatured cell lysates (Fig. 6A and SI Appendix, Fig. S15A). Pretreatment with KLH45, but not KLH40 (2 µM, 30 min), blocked the increased TAG hydrolytic activity in WT–DDHD2-transfected cells (Fig. 6A). Increased TAG hydrolysis activity was also observed for WT–DDHD2-transfected cells compared with control groups using an LC–MS assay that monitored the conversion of unlabeled C18:1/C18:1/C18:1 TAG to oleic acid (Fig. 6B). We next assayed brain lysates from DDHD2−/− mice and found that they exhibited significantly lower TAG hydrolysis activity compared with brain lysates from DDHD2+/+ mice (Fig. 6C). Finally, we also found that DDHD2 hydrolyzed a C18:1/C18:1 DAG substrate (SI Appendix, Fig. S15B), indicating that the enzyme can act as both a TAG and DAG hydrolase (similar to some other TAG hydrolases, such as hormone-sensitive lipase) (25).
Fig. 6.
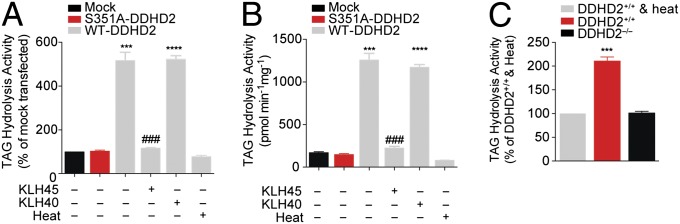
DDHD2 exhibits TAG hydrolase activity. (A and B) Soluble lysates from HEK293T cells transiently transfected with a WT–DDHD2 cDNA showed greater C18:1/C18:1/C18:1 TAG hydrolytic activity measured by either a radiolabeled TLC (A) or LC–MS (B) assay compared with lysates from mock-transfected cells, heat-denatured WT–DDHD2-transfected lysates, or cells transfected with an S351A–DDHD2 mutant cDNA. Both assays report formation of C18:1 fatty acid. For measurement of 14C-C18:1 MAG and 14C-C18:1/C18:1 DAG formation in the radiolabeled 14C-TAG substrate assay, see SI Appendix, Fig. S15. In both substrate assays, KLH45 but not KLH40 blocked the TAG hydrolase activity of DDHD2. (C) Soluble brain lysates from DDHD2−/− mice show reduced TAG hydrolysis activity compared with soluble brain lysates from DDHD2+/+ mice measured by a radiolabeled substrate assay following conversion of C18:1/C18:1/C18:1 TAG to C18:1 fatty acid. Heat-denatured DDHD2+/+ brain lysates were assayed as a control and displayed a similar signal to those observed in DDHD2−/− lysates. Data represent average values ± SEM for three experimental replicates per group. ***P < 0.001 and ****P < 0.0001 for WT–DDHD2 versus S351A–DDHD2 transfected groups or DDHD2+/+ versus DDHD2−/− groups; ###P < 0.001 for KLH45-treated versus DMSO-treated WT–DDHD2 groups.
These data indicate that DDHD2 is a principal brain TAG hydrolase, providing a plausible biochemical mechanism to explain the accumulation of TAGs in the CNS of DDHD2-disrupted animals.
Discussion
The enzymes that regulate TAG hydrolysis have been mapped in peripheral tissues, where adipose triglyceride lipase (ATGL or PNPLA2) plays a major role in this process (22, 25, 26). Mutations in the PNPLA2 gene cause neutral lipid storage disease with myopathy associated with substantial peripheral elevations in TAGs (27, 28). In contrast, TAG metabolism in the CNS has remained poorly characterized. PNPLA2−/− mice have elevated TAGs in brain tissue, but these changes are restricted to barrier regions of the CNS (e.g., cerebrovascular cells, choroid plexus) (29). LDs were not found to accumulate in neurons of PNPLA2−/− mice, indicating that these cells may express distinct TAG hydrolytic enzymes. Our findings indicate that DDHD2 serves as a principal TAG hydrolase in the mammalian brain, where deletion of this enzyme leads to massive LD accumulation in neurons.
DDHD2 is a member of a small clan of three sequence-related serine hydrolases in humans [the other two enzymes are termed DDHD1 (10) and Sec23ip (30)]. DDHD hydrolases have been annotated as phospholipases based on in vitro substrate assays (9, 10). We did not, however, observe significant changes in brain phospholipids in DDHD2−/− mice. Although bulk brain levels of phospholipids were unchanged, it is possible that DDHD2 regulates phospholipids in specific brain cell types in vivo. DDHD2 also possesses a noncatalytic phosphatidylinositol 4-phosphate–binding domain (24) and regulates organellar morphology when overexpressed in mammalian cells (9), functions that could, in principle, indirectly impact TAG trafficking and metabolism. However, mice treated with a selective DDHD2 inhibitor also showed elevations in brain TAGs, indicating that this metabolic change is likely due to blockade of the catalytic activity of DDHD2, rather than the indirect outcome of eliminating DDHD2 protein. That DDHD2 exhibited TAG hydrolase activity in vitro supports this conclusion. Interestingly, previous studies have identified the main TAG hydrolase from the fat body of the insect Manduca sexta as a protein that shares sequence homology with DDHD2 and other mammalian DDHD proteins (31). This finding raises the possibility that DDHD1 and Sec23ip may also function as TAG hydrolases. On the other hand, HSP patients with mutations in the DDHD1 gene do not show gross lipid accumulation in the brain as measured by magnetic resonance imaging (32), suggesting that disruption of DDHD1 and DDHD2 produce distinct metabolic effects in the human nervous system.
Based on our results, the most parsimonious explanation for the abnormal lipid peak observed by cerebral magnetic resonance spectroscopy in human subjects with DDHD2 mutations (6) is that it reflects elevated TAGs, although it remains possible that DDHD2 could regulate distinct classes of lipids in the mouse and human brain. We also cannot explain, at present, why subchronic treatment with a DDHD2 inhibitor is needed to produce TAG elevations in the mouse brain, but we should note that acute treatment with an ATGL inhibitor also causes more limited changes in TAGs in peripheral tissues compared with chronic (genetic) disruption of ATGL (22, 33). Projecting forward, it will be important to evaluate how deregulated TAG metabolism and LD accumulation affect neuronal cell biology and, ultimately, HSP neuropathology. We believe that DDHD2−/− mice and DDHD2-selective inhibitors should serve as valuable tools for probing this mechanistic relationship. Could, for instance, DDHD2-regulated LDs sequester and/or disrupt the trafficking of key proteins and signaling lipids to impair neuronal function and the assembly of synaptic machinery? Are there differences in the neuronal or behavioral phenotypes caused by lifelong, genetic deletion of DDHD2 versus pharmacological inactivation of the enzyme in adult animals? Although DDHD2 disruption had a less severe impact on peripheral TAGs, it would be worthwhile to investigate whether this enzyme contributes to peripheral metabolism under conditions of metabolic stress (e.g., exposure to a high-fat diet). Finally, might inhibitors of enzymes that produce TAGs, such as diacylglycerol transferases (DGATs) (34, 35), counteract DDHD2 disruption to normalize brain TAG content and behavior? DGAT inhibitors are under investigation for the treatment of peripheral metabolic disorders (36); our findings suggest that examination of these inhibitors in models of DDHD2-related neurological syndromes is warranted.
Materials and Methods
An extended section is provided in SI Appendix, SI Materials and Methods.
Generation of DDHD2−/− Mice.
Disruption of the Ddhd2 gene was achieved on the C57BL/6 genetic background using standard gene targeting techniques as described in SI Appendix, SI Materials and Methods.
Biochemical Studies.
ABPP and substrate assays of mouse brain and transfected cell lysates were performed as described previously (37). Metabolomic and proteomic analyses of brain homogenates and cell lines were performed as described previously (37) and in SI Appendix, SI Materials and Methods.
Behavioral Studies.
Tests for locomotor activity and cognitive function were performed as described in SI Appendix, SI Materials and Methods.
Electron and Light Microscopy Analysis.
Mice were exsanguinated by perfusion with saline followed by a mixture of 4% (vol/vol) paraformaldehyde and 1.5% glutaraldehyde in 0.1 M Na cacodylate buffer (pH 7.3). Following dissection of whole brains, fixation continued overnight and brains were then sectioned and analyzed as described in SI Appendix, SI Materials and Methods.
Chemical Synthesis and Characterization.
The DDHD2 inhibitor KLH45 and inactive control compound KLH40 were synthesized and characterized as described in SI Appendix, SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank S. Tully and J. Blankman for helpful discussions and technical assistance and S. Kupriyanov and G. Martin [The Scripps Research Institute (TSRI) Mouse Genetics Core], A. Roberts (TSRI Mouse Behavioral Assessment Core), A. San-Soucie (TSRI Histology Core), and H. Pugh for technical assistance. This work was supported by the National Institutes of Health (DA033760 and DK099810, to B.F.C.; DA035864, to K.-L.H.; and GM109315, to A.V.).
Footnotes
Conflict of interest statement: The authors declare competing financial interests. B.F.C. is cofounder and advisor for a biotechnology company interested in developing inhibitors for serine hydrolases as therapeutic targets.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1413706111/-/DCSupplemental.
References
- 1.Rabbani B, Mahdieh N, Hosomichi K, Nakaoka H, Inoue I. Next-generation sequencing: Impact of exome sequencing in characterizing Mendelian disorders. J Hum Genet. 2012;57(10):621–632. doi: 10.1038/jhg.2012.91. [DOI] [PubMed] [Google Scholar]
- 2.Fink JK. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013;126(3):307–328. doi: 10.1007/s00401-013-1115-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Novarino G, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343(6170):506–511. doi: 10.1126/science.1247363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rainier S, et al. Neuropathy target esterase gene mutations cause motor neuron disease. Am J Hum Genet. 2008;82(3):780–785. doi: 10.1016/j.ajhg.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tesson C, et al. Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am J Hum Genet. 2012;91(6):1051–1064. doi: 10.1016/j.ajhg.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuurs-Hoeijmakers JH, et al. FORGE Canada Consortium Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am J Hum Genet. 2012;91(6):1073–1081. doi: 10.1016/j.ajhg.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez M, et al. Mutations in phospholipase DDHD2 cause autosomal recessive hereditary spastic paraplegia (SPG54) Eur J Hum Genet. 2013;21(11):1214–1218. doi: 10.1038/ejhg.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Citterio A, et al. Mutations in CYP2U1, DDHD2 and GBA2 genes are rare causes of complicated forms of hereditary spastic paraparesis. J Neurol. 2014;261(2):373–381. doi: 10.1007/s00415-013-7206-6. [DOI] [PubMed] [Google Scholar]
- 9.Nakajima K, et al. A novel phospholipase A1 with sequence homology to a mammalian Sec23p-interacting protein, p125. J Biol Chem. 2002;277(13):11329–11335. doi: 10.1074/jbc.M111092200. [DOI] [PubMed] [Google Scholar]
- 10.Higgs HN, Han MH, Johnson GE, Glomset JA. Cloning of a phosphatidic acid-preferring phospholipase A1 from bovine testis. J Biol Chem. 1998;273(10):5468–5477. doi: 10.1074/jbc.273.10.5468. [DOI] [PubMed] [Google Scholar]
- 11.van Tienhoven M, Atkins J, Li Y, Glynn P. Human neuropathy target esterase catalyzes hydrolysis of membrane lipids. J Biol Chem. 2002;277(23):20942–20948. doi: 10.1074/jbc.M200330200. [DOI] [PubMed] [Google Scholar]
- 12.Gross RW, Han X. Lipidomics at the interface of structure and function in systems biology. Chem Biol. 2011;18(3):284–291. doi: 10.1016/j.chembiol.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saghatelian A, et al. Assignment of endogenous substrates to enzymes by global metabolite profiling. Biochemistry. 2004;43(45):14332–14339. doi: 10.1021/bi0480335. [DOI] [PubMed] [Google Scholar]
- 14.Simon GM, Cravatt BF. Activity-based proteomics of enzyme superfamilies: Serine hydrolases as a case study. J Biol Chem. 2010;285(15):11051–11055. doi: 10.1074/jbc.R109.097600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson-Grady JT, Haas W, Gygi SP. Quantitative comparison of the fasted and re-fed mouse liver phosphoproteomes using lower pH reductive dimethylation. Methods. 2013;61(3):277–286. doi: 10.1016/j.ymeth.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 16.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19(3):242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 17.Hsu KL, et al. Development and optimization of piperidyl-1,2,3-triazole ureas as selective chemical probes of endocannabinoid biosynthesis. J Med Chem. 2013;56(21):8257–8269. doi: 10.1021/jm400898x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnes CA. Memory deficits associated with senescence: A neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93(1):74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- 19.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78(3):779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 20.Hsu FF, Turk J. Structural characterization of triacylglycerols as lithiated adducts by electrospray ionization mass spectrometry using low-energy collisionally activated dissociation on a triple stage quadrupole instrument. J Am Soc Mass Spectrom. 1999;10(7):587–599. doi: 10.1016/S1044-0305(99)00035-5. [DOI] [PubMed] [Google Scholar]
- 21.Murphy RC, et al. Detection of the abundance of diacylglycerol and triacylglycerol molecular species in cells using neutral loss mass spectrometry. Anal Biochem. 2007;366(1):59–70. doi: 10.1016/j.ab.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haemmerle G, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312(5774):734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 23.Fujimoto T, Ohsaki Y, Suzuki M, Cheng J. Imaging lipid droplets by electron microscopy. Methods Cell Biol. 2013;116:227–251. doi: 10.1016/B978-0-12-408051-5.00012-7. [DOI] [PubMed] [Google Scholar]
- 24.Inoue H, et al. Roles of SAM and DDHD domains in mammalian intracellular phospholipase A1 KIAA0725p. Biochim Biophys Acta. 2012;1823(4):930–939. doi: 10.1016/j.bbamcr.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Zimmermann R, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306(5700):1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 26.Zimmermann R, Lass A, Haemmerle G, Zechner R. Fate of fat: The role of adipose triglyceride lipase in lipolysis. Biochim Biophys Acta. 2009;1791(6):494–500. doi: 10.1016/j.bbalip.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Fischer J, et al. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet. 2007;39(1):28–30. doi: 10.1038/ng1951. [DOI] [PubMed] [Google Scholar]
- 28.Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R. Neutral lipid storage disease: Genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab. 2009;297(2):E289–E296. doi: 10.1152/ajpendo.00099.2009. [DOI] [PubMed] [Google Scholar]
- 29.Etschmaier K, et al. Adipose triglyceride lipase affects triacylglycerol metabolism at brain barriers. J Neurochem. 2011;119(5):1016–1028. doi: 10.1111/j.1471-4159.2011.07498.x. [DOI] [PubMed] [Google Scholar]
- 30.Tani K, Mizoguchi T, Iwamatsu A, Hatsuzawa K, Tagaya M. p125 is a novel mammalian Sec23p-interacting protein with structural similarity to phospholipid-modifying proteins. J Biol Chem. 1999;274(29):20505–20512. doi: 10.1074/jbc.274.29.20505. [DOI] [PubMed] [Google Scholar]
- 31.Arrese EL, Patel RT, Soulages JL. The main triglyceride-lipase from the insect fat body is an active phospholipase A(1): Identification and characterization. J Lipid Res. 2006;47(12):2656–2667. doi: 10.1194/jlr.M600161-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Liguori R, et al. Impairment of brain and muscle energy metabolism detected by magnetic resonance spectroscopy in hereditary spastic paraparesis type 28 patients with DDHD1 mutations. J Neurol. 2014;261(9):1789–1793. doi: 10.1007/s00415-014-7418-4. [DOI] [PubMed] [Google Scholar]
- 33.Mayer N, et al. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat Chem Biol. 2013;9(12):785–787. doi: 10.1038/nchembio.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yen CL, Stone SJ, Koliwad S, Harris C, Farese RV., Jr Thematic review series: Glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res. 2008;49(11):2283–2301. doi: 10.1194/jlr.R800018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harris CA, et al. DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes. J Lipid Res. 2011;52(4):657–667. doi: 10.1194/jlr.M013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naik R, et al. Therapeutic strategies for metabolic diseases: Small-molecule diacylglycerol acyltransferase (DGAT) inhibitors. ChemMedChem. 2014 doi: 10.1002/cmdc.201402069. [DOI] [PubMed] [Google Scholar]
- 37.Blankman JL, Long JZ, Trauger SA, Siuzdak G, Cravatt BF. ABHD12 controls brain lysophosphatidylserine pathways that are deregulated in a murine model of the neurodegenerative disease PHARC. Proc Natl Acad Sci USA. 2013;110(4):1500–1505. doi: 10.1073/pnas.1217121110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.