

Abstract
Hepatitis C Virus (HCV) affects 3% of the world’s population and causes serious liver ailments including chronic hepatitis, cirrhosis, and hepatocellular carcinoma. HCV is an enveloped RNA virus belonging to the family Flaviviridae. Current treatment is not fully effective and causes adverse side effects. There is no HCV vaccine available. Thus, continued effort is required for developing a vaccine and better therapy. An HCV cell culture system is critical for studying various stages of HCV growth including viral entry, genome replication, packaging, and egress. In the current procedure presented, we used a wild-type intragenotype 2a chimeric virus, FNX-HCV, and a recombinant FNX-Rluc virus carrying a Renilla luciferase reporter gene to study the virus replication. A human hepatoma cell line (Huh-7 based) was used for transfection of in vitro transcribed HCV genomic RNAs. Cell-free culture supernatants, protein lysates and total RNA were harvested at various time points post-transfection to assess HCV growth. HCV genome replication status was evaluated by quantitative RT-PCR and visualizing the presence of HCV double-stranded RNA. The HCV protein expression was verified by Western blot and immunofluorescence assays using antibodies specific for HCV NS3 and NS5A proteins. HCV RNA transfected cells released infectious particles into culture supernatant and the viral titer was measured. Luciferase assays were utilized to assess the replication level and infectivity of reporter HCV. In conclusion, we present various virological assays for characterizing different stages of the HCV replication cycle.
Keywords: Infectious Diseases, Issue 88, Hepatitis C Virus, HCV, Tumor-virus, Hepatitis C, Cirrhosis, Liver Cancer, Hepatocellular Carcinoma
Introduction
Hepatitis C virus (HCV) causes cirrhosis and liver cancer. It affects 170 million people worldwide with 350,000 people dying annually1-3. HCV is a positive strand RNA virus with a genome size of 9.6 kb. The HCV genome is translated as a single polyprotein of ~3,000 amino acid residues that is proteolytically cleaved by various cellular and viral proteases into 10 polypeptides. HCV is the prototype virus in the genus Hepacivirus and belongs to family Flaviviridae4. Upon exposure, HCV establishes chronic infection in 80% of the individuals. The infection is mostly asymptomatic and timely diagnosis can allow therapeutic intervention to prevent liver deterioration. Current treatment is suboptimal and no vaccine is available5,6.
The etiology of hepatitis C was first described in 1989 7. Studying HCV replication is important for hepatitis C vaccine and treatment research, but it had been long hampered by the lack of an efficient viral culture system. A molecular clone of HCV was shown to be infectious in chimpanzees upon intrahepatic inoculation8. Subsequently, HCV sub-genomic replicons were described, which allowed to dissect the viral genome replication stage in a cell culture system9,10. Discovery of a genotype 2a HCV isolate JFH-1 (Japanese Fulminant hepatitis-1), capable of infecting cell culture opened new avenues for HCV replication research11-13. Genotype 2a strain JFH-1 based inter- and intra-genotypic chimeric viruses and genotype 1 HCV based infectious culture systems are available as well14-18.
We have successfully used JFH-1 strain and HCV intragenotype 2a chimeric virus to obtain the high-resolution functional profiling map of protein domains and cis-acting RNA elements19,20. According to this, here we describe an effective culture system routinely used that allows studying various stages of the HCV replication cycle and host-pathogen interaction. We present virological assays to assess viral genome replication and de novo infectivity of intragenotype 2a HCV and a Renilla luciferase based reporter HCV.
Protocol
A general outline of the protocol is illustrated in Figure 1.
1. Cells
Prepare complete growth media that contains 10-15% fetal bovine serum (FBS), 10 mM nonessential amino acids, 10 mM Hepes, penicillin (100 units/ml), streptomycin (100 mg/ml), and 2 mM L-glutamine.
Maintain Huh-7.5.1 cells13 in complete growth media containing the above supplements for in vitro analysis of hepatitis C viral replication cycle.
Culture viral strains in Huh-7.5.1 cells with the specified supplemented growth media at 37 °C with 5% CO2.
2. Virus and Plasmid Constructs
Generate the complementary DNA (cDNA) form of HCV RNA and clone it into a plasmid vector for ease of genetic manipulation. Note: The discovery of the Japanese fulminant hepatitis 1, JFH-1 (genotype 2a HCV), isolate has been critical for HCV research and is primarily used to study the HCV replication cycle11-13. Inter- and intragenotypic chimeric viruses based on JFH-1 HCV are commonly used in research14-18. Construction of synthetic intragenotype 2a chimeric virus, FNX-HCV, and a monocistronic chimeric reporter strain was described previously19 and construction details are beyond the scope of this protocol.
Use the pFNX-HCV intragenotype 2a virus as it contains a 5’NTR, structural regions and p7, and part of the NS2 non-structural regions (nucleotides 1 to 2878) of the J6CF parental strain (NCBI accession no. AF177036), as well as the non-structural components of the JFH-1 viral strain (NCBI accession no. AB047639). Note: FNX-HCV was synthesized based on the sequence of Jc1 intragenotype 2a chimeric HCV reported previously15.
Generate a monocistronic chimeric reporter viral construct, the pFNX-Rluc, based on the pFNX-HCV strain (above in step 2.2) by inserting a Renilla luciferase gene between the 5’ NTR and core gene (between nt 388 and 389). Subsequently, connect the luciferase gene and core gene of this construct using a foot and mouth disease virus 2A (F2A) peptide sequence, which would work as a cleavage signal.
Engineer the RNA polymerase-null (Pol-) viral construct using either the pFNX-HCV or the pFNX-Rluc viral background. Replace the NS5B polymerase catalytic residues, GDD (aa 2759-2761; nt 8615-8623), of either of these viral backbones with AAG amino acid residues.
3. In Vitro HCV RNA Transcription
Use an intragenotype 2a chimeric virus FNX-HCV and FNX-HCV Pol null (Pol-) HCV for evaluation of viral replication (Figure 2A).
Linearize the viral plasmids with XbaI restriction enzyme and then treat with mung bean nuclease to generate blunt ends. Purify the digested plasmids by anion-exchange chromatography. Verify the integrity of the linearized plasmid by subjecting DNA to agarose gel electrophoresis (Figure 2B).
Transcribe the linearized viral plasmids using the T7- RNA polymerase.
Purify the newly synthesized DNase-treated RNA using an RNA purification kit.
Verify the RNA production by agarose gel electrophoresis (Figure 2C).
Quantify the RNA by spectrophotometry.
Store the generated RNA in -80 °C in 10 μg working aliquots. Note: To minimize variation of RNA quality within each experimental design by transcribing all RNA for each viral sample and control at the same time.
4. HCV RNA Transfection and Sample Collection
Harvest the Huh-7.5.1 cells using trypsin.
To rinse cells, centrifuge and resuspend the suspension twice with cold low serum media. Resuspend the cells in low serum media at 1 x 107cells per ml.
Mix a total of 10 μg of transcribed viral RNA with 400 μl of resuspended cells (4 x 106 cells) in a 0.4-cm electroporation cuvette. Transfect the cells via electroporation at 270 V, 100 Ω, and 950 μF.
Resuspend the electroporated cells in 10 ml complete growth media with 15% FBS. Note: Increased survivability of electroporated cells is seen when the Huh-7.5.1 cells were cultured in 15% Fetal Bovine Serum.
Plate the cells in both T-25 flasks (~1.2 x 106 cells per flask) and 48-well plates (1 x 104 cells per well) for each of the following time points: 4, 48 and 96 hr.
Replace the media at 4-8 hr post-transfection with fresh supplemented growth media with 10% FBS to remove dead cell debris from the cultured flasks and plates.
Harvest cell culture supernatants at the 48, 96 hr time points and remove cellular debris from collected samples by centrifugation of cells at 1,500 rpm for 10 min at 4ºC.
Store the cell-free supernatants at -80 ºC. Lyse the cells for protein and RNA analysis by Western blot and reverse transcription-quantitative PCR at the indicated time points.
5. Reverse Transcription-quantitative PCR (RT-qPCR) for Assessing the HCV Genome Copies
Perform a two-step RT-qPCR to determine the HCV genomic RNA copy number.
Reverse transcribe 1 μg of total cellular RNA using reverse transcriptase and a primer specific for HCV sense strand that binds to 5’NTR (JFH RTQ R: 5’CCTATCAGGCAGTACCACA-3’) or a primer specific for housekeeping gene peptidylprolyl isomerase G (PPIG R: 5’-GTCTCTCCTCCTTCTCCTCCTATCTTT-3’). Also reverse transcribe FNX-HCV RNA (generated in Step 3) of known genome copies (101 to 109 standard) using HCV sense strand primer.
Carry out qPCR by using 50 ng of the resulting transcribed cDNA using specific HCV primers (JFH RTQ F: 5’CTGGGTCCTTTCTTGGATAA-3; JFH RTQ R: 5’CCTATCAGGCAGTACCACA-3’) and DNA binding green dye containing qPCR super mix. Perform qPCR for housekeeping gene PPIG as well (primers PPIG F: 5’-GAAGAGTGCGATCAAGAACCCATGAC-3’; PPIG R: 5’-GTCTCTCCTCCTTCTCCTCCTATCTTT-3’) Note: For calculating accurate intracellular HCV RNA level across samples, use cellular housekeeping gene, PPIG, expression level (Ct cycle) for normalization. Use the copy number of the FNX-HCV genome as the standard for the copy number determination.
Use the following conditions when running qPCR to determine HCV RNA copy number: 95 ºC for 15 sec and 60 ºC for 30 sec (40 cycles) using real-time PCR system.
See Figures 3A and 3B for genome replication results.
6. Western Blotting Analysis for Detecting HCV Protein Expression (Figure 3C)
Use cell lysates from viral RNA transfected at 96 hr post transfection for protein western blotting analysis.
Resolve the cell lysate using SDS-PAGE and transfer to polyvinylidene difluoride (PVDF) membrane.
Block the membrane using a blocking solution containing 5% skim milk, 0.2% Tween 20 in phosphate buffered saline (PBS).
Incubate the membrane with primary mouse monoclonal antibody NS3 (1 in 1,000 dilution) and beta-actin (1 in 5,000 dilution).
Add goat anti-mouse IgG conjugated to horseradish peroxidase (1 in 5,000 dilution) and detect by chemiluminescence.
7. Immunofluorescence Assay (IFA)
Fix the HCV-infected and transfected cells using methanol for 30 min at -20 ºC for immunofluorescence assay.
Wash the cell with PBS three times and block with IFA blocking buffer.
Use rabbit polyclonal anti-NS5A primarily antibody (kindly provided by Dr. Dasgupta, UCLA) or mouse monoclonal anti-dsRNA antibody J2 at a dilution of 1:200 (1 μg/ml) and incubate for 5 hr to overnight at 4ºC.
Wash the cells with PBS three times after the primary antibody.
Add goat anti-rabbit IgG-488 polyclonal secondary antibody or goat anti-mouse IgG-594 polyclonal secondary antibody at a 1:1,000 dilution (1 μg/ml) and incubate for 1 hr at room temperature.
Wash cells with PBS three times and stain nuclei using Hoechst dye and view using fluorescent microscope (Figure 3D).
8. Measuring Virus Titer
Plate naïve Huh-7.5.1 cells at approximately 3 x 103 cells/well using a 96-well plate. The next day, perform 10-fold serial dilutions of cell-free culture supernatant harvested from HCV RNA transfected cells using growth media and inoculate in triplicate onto Huh-7.5.1 cells.
Fix cells at 72 hr post-infection using methanol (for 30 min at -20ºC) and immunostain for HCV NS5A protein as stated in the section 7.
Use the highest dilution to count for the NS5A positive foci and calculate the average number of focus forming unit (FFU) per milliliter. See Figure 4 for HCV titer.
9. Renilla Luciferase Reporter Assay for Viral Genome Replication and Infectivity
For evaluating viral genome replication, plate HCV RNA-electroporated Huh-7.5.1 cells in triplicate in 48-well plates (1 x 104 cells/well).
Lyse the cells with 75 μl of passive lysis buffer at the 6 hr, 48 hr, and 96 hr post-transfection.
Rock the plates gently for 15 min at room temperature and store at -80 ºC.
To measure the Renilla luciferase enzymatic activity, thaw the protein lysate and Renilla luciferase assay reagents to room temperature.
Add 20 μl of protein lysate per well of a 96-well white luminescence plate and place the plate in a luminometer.
Dispense 100 μl of Renilla luciferase assay reagent (coelenterazine substrate + buffer) per well and after a 2 sec pre-read delay; integrate the emitted light for 10 sec.
Calculate the mean and standard deviation for each sample from the luciferase values of biological triplicates and subject the data to statistical analysis (Figure 5).
For assessing infectivity, inoculate 500 μl of cell-free supernatant obtained from HCV RNA-transfected cells for the indicated time points (48 hr and 96 hr) in triplicate onto naïve Huh-7.5.1 cells in 48-well plates.
Replace the viral inoculum with 500 μl of fresh medium per well after 6 hr post-infection.
Lyse the cells at 48 hr post-infection and subject to Renilla luciferase assay as described in steps 8.2 to 8.7 (Figure 5).
10. Statistical Analysis
The error bars in the graphs indicate standard deviations (SD). The P values were calculated by the unpaired t test.
Representative Results
Hepatitis C virus is a RNA virus. Thus for genetic manipulation purpose, the HCV genomic cDNA has been cloned into a bacterial plasmid vector. A T7 RNA polymerase promoter sequence was introduced immediately before the 5’ end of the HCV genome. A general outline of HCV analysis workflow is presented in Figure 1. To generate HCV genomic RNA with precise 3’ end, the HCV genome containing plasmid is cut with XbaI restriction enzyme and the generated single-stranded overhang was blunted with mung bean nuclease digestion. The quality of the linearized HCV plasmid was assessed by agarose gel electrophoresis (Figure 2B). The HCV DNA was subjected to T7 RNA polymerase mediated in vitro transcription and the resulting RNA yielded a single product at 9.6 kilobase (Figure 2C).
We tested the growth kinetics of an intragenotype 2a HCV (Figure 3). The Huh-7.5.1 cells were electroporated with in vitro transcribed RNA of wild-type (WT) and polymerase null viruses. We evaluated the viral sense genome replication by RT-qPCR. Results indicated that the WT virus replicated the genome efficiently (Figures 3A and 3B). The wild-type exhibited one to three log higher level of genome replication compared to that of Pol- virus. The WT virus produces the NS3 protein (Figure 3C) that is involved in viral protein cleavage (protease activity), and genome replication (helicase activity). Also the WT virus expressed NS5A protein as assessed by Immunofluorescence assay (Figure 3D). NS5A protein is part of the HCV RNA replicase complex. To visualize viral genome replication, we examined the presence of double- stranded (ds) HCV RNA using an antibody that specifically recognizes dsRNA. During HCV genome amplification, the sense strand RNA is copied to anti-sense genome, thus the double stranded RNA intermediates are present in the infected cell cytoplasm. The NS5A protein and dsRNA co-localizes in the cellular cytoplasm (Figure 3D) of WT transfected cells suggesting the active viral replication. Taken together, the WT virus established active replication in transfected Huh-7.5.1 cells.
Progress in HCV research had been limited due to lack of an infectious cell culture system. The discovery of JFH-1 strain of HCV and the subsequent characterization of chimeric laboratory strains allowed us to investigate the entire HCV replication cycle in cell culture including viral entry, RNA translation, RNA replication and the formation of infectious viral progeny. To assess the HCV titer and de novo infectivity, we inoculated the cell-culture supernatants harvested from HCV RNA transfected cells. The HCV infection was visualized by detecting the HCV NS5A protein and calculated the viral titer by counting the infectious foci (Figure 4). We considered isolated cluster of cells (over 2 cells) positive for NS5A as a single focus. Results indicated that the WT virus is infectious and produces over 104 foci forming units/ml of infectious particle.
We also established a HCV reporter virus harboring Renilla luciferase (Rluc) reporter gene (Figure 5A). A reporter HCV can be used for testing large number of mutant viruses as well as for high-content screening assays. Here we present the assessment of growth phenotypes of WT and Pol- reporter viruses. If the viral genome replicates, the luciferase activity would increase overtime post transfection. The increased genome replication levels can be deduced from increased luciferase activity. Hence, the luciferase activity indirectly provides the measurement of the level of genome replication. The lysates harvested from WT or Pol- viral RNA transfected cells at indicated time points were tested for luciferase activities. At 6 hr post-transfection, both WT and Pol- viruses had similar luciferase activities, indicating similar input level of transfected RNA that had been translated (Figure 5B). However at 48 hr and 96 hr post-transfection, the WT virus exhibited increased genome replication levels compared to that of Pol- virus. Also, the WT virus produced viral NS3 protein as verified by Western blotting analysis (Figure 5C). Subsequently, we tested the infectivity of WT and Pol- reporter viruses by inoculating naïve Huh-7.5.1 cells with the cell-free supernatants collected from 48 hr and 96 hr post-transfected cell cultures. The Pol- virus had base-level luciferase activity, whereas the WT virus had 2-3 log higher level of replication to that of Pol- reporter virus (Figure 5D). Results demonstrate that we have a robust HCV infectious cell culture system.
 Figure 1. General outline of HCV replication analysis workflow. Linearized HCV plasmid construct containing T7 RNA polymerase promoter (T7p) subjected to in vitro transcription. The purified HCV RNA genome is electroporated into Huh-7.5.1 cells and plated in flasks and 48-well plate. At 4 hr, 48 hr, and 96 hr post-transfection, the cellular RNA and culture supernatants are harvested from flasks. The cells from 48-well plate are utilized for harvesting protein lysate and are fixed for Immunofluorescence assay. Genome copy number analysis by RT-qPCR, Western blotting and measuring viral titer are done to assess the HCV replication.
Figure 1. General outline of HCV replication analysis workflow. Linearized HCV plasmid construct containing T7 RNA polymerase promoter (T7p) subjected to in vitro transcription. The purified HCV RNA genome is electroporated into Huh-7.5.1 cells and plated in flasks and 48-well plate. At 4 hr, 48 hr, and 96 hr post-transfection, the cellular RNA and culture supernatants are harvested from flasks. The cells from 48-well plate are utilized for harvesting protein lysate and are fixed for Immunofluorescence assay. Genome copy number analysis by RT-qPCR, Western blotting and measuring viral titer are done to assess the HCV replication.
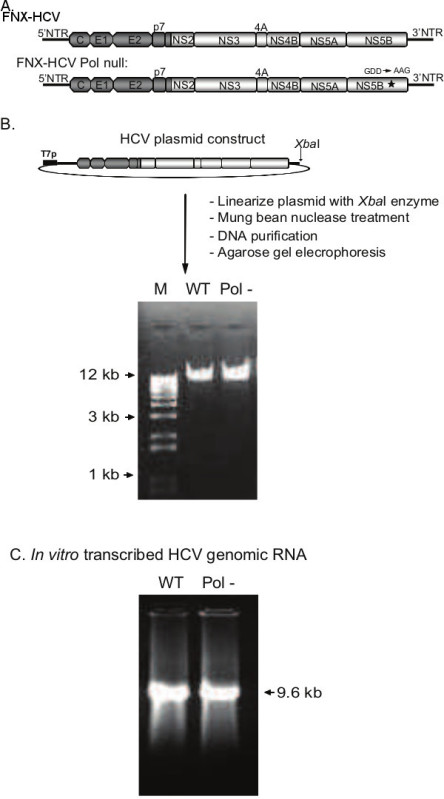 Figure 2. Production of HCV genomic RNAs by in vitro transcription of constructed plasmid DNAs. A) Genomic organization of the J6CF/JFH-1 intragenotype 2a chimeric viruses, FNX-HCV and FNX-HCV Pol null. The J6CF strain region (5’NTR to part of NS2) is depicted in dark gray and the JFH-1 strain region (part of NS2 to 3’NTR) is displayed in light gray. NS5B polymerase catalytic domain mutation (GDD to AAG) is indicated with an asterisk. B) Steps involved in generating linearized HCV plasmid. Gel picture shows the linearized, blunt ended plasmid DNAs produced by XbaI and mung bean nuclease digestion, ready for in vitro transcription. 0.8% agarose gel was used for resolving the DNA. C) Gel picture depicts the HCV genomic RNAs produced by in vitro transcription using the T7 RNA polymerase system. WT: wild-type; Pol-: Polymerase null; M: marker.
Figure 2. Production of HCV genomic RNAs by in vitro transcription of constructed plasmid DNAs. A) Genomic organization of the J6CF/JFH-1 intragenotype 2a chimeric viruses, FNX-HCV and FNX-HCV Pol null. The J6CF strain region (5’NTR to part of NS2) is depicted in dark gray and the JFH-1 strain region (part of NS2 to 3’NTR) is displayed in light gray. NS5B polymerase catalytic domain mutation (GDD to AAG) is indicated with an asterisk. B) Steps involved in generating linearized HCV plasmid. Gel picture shows the linearized, blunt ended plasmid DNAs produced by XbaI and mung bean nuclease digestion, ready for in vitro transcription. 0.8% agarose gel was used for resolving the DNA. C) Gel picture depicts the HCV genomic RNAs produced by in vitro transcription using the T7 RNA polymerase system. WT: wild-type; Pol-: Polymerase null; M: marker.
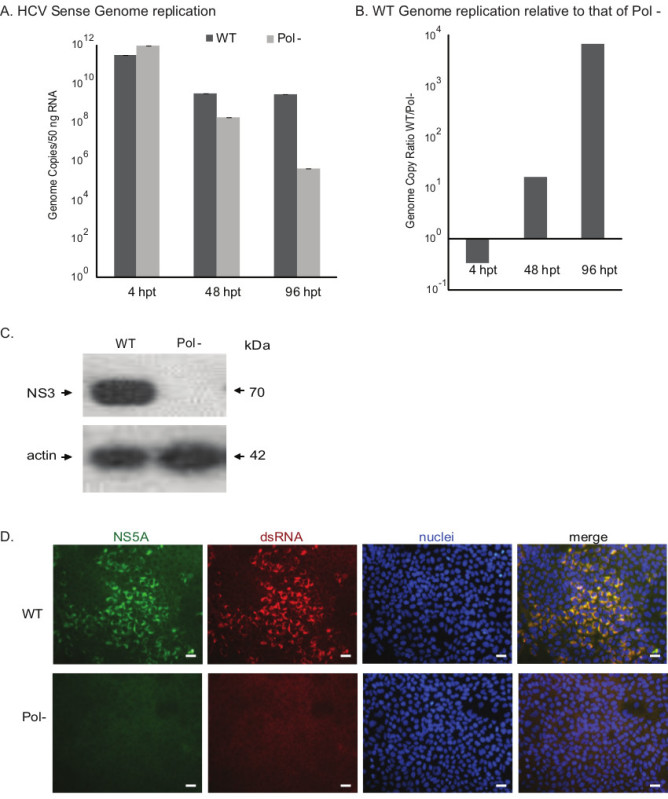 Figure 3. Assessing the growth phenotypes of HCV constructs. A) Evaluating the genome replication kinetics of wild-type (WT) and polymerase null (Pol-) viruses. The genome copy numbers of sense RNA strand assessed by RT-qPCR are presented in the bar graph. The viral genome copies of Pol- virus declined over the period of time indicating replication deficient phenotype. B) Relative genome replication level of wild-type HCV is compared to that of Pol- virus. C) Western blot analysis of HCV protein expression. HCV protein NS3 is detected and beta-actin is used as a cellular control. D) Immunofluorescence assay for investigating viral replication. At 96 hr post-electroporation the cells were fixed and subjected to immunostaining for HCV NS5A protein and double-stranded RNA (ds RNA), a marker for HCV RNA replication intermediates. The nuclei were visualized with Hoechst stain (Scale bar 50 μm). hpt: hours post-transfection. Please click here to view a larger version of this figure.
Figure 3. Assessing the growth phenotypes of HCV constructs. A) Evaluating the genome replication kinetics of wild-type (WT) and polymerase null (Pol-) viruses. The genome copy numbers of sense RNA strand assessed by RT-qPCR are presented in the bar graph. The viral genome copies of Pol- virus declined over the period of time indicating replication deficient phenotype. B) Relative genome replication level of wild-type HCV is compared to that of Pol- virus. C) Western blot analysis of HCV protein expression. HCV protein NS3 is detected and beta-actin is used as a cellular control. D) Immunofluorescence assay for investigating viral replication. At 96 hr post-electroporation the cells were fixed and subjected to immunostaining for HCV NS5A protein and double-stranded RNA (ds RNA), a marker for HCV RNA replication intermediates. The nuclei were visualized with Hoechst stain (Scale bar 50 μm). hpt: hours post-transfection. Please click here to view a larger version of this figure.
 Figure 4. Examining the infectivity of wild-type HCV and measuring virus titer. A) Naïve Huh-7.5.1 cells were used for infection studies. The cell-free supernatant collected at 48 and 96 hr post-transfection (hpt) of HCV RNA were subjected to 10-fold serial dilution and added to the cells in a 96-well plate in triplicate. 72 hr post-infection, the cells were fixed and immunostained for HCV NS5A protein. The cells with NS5A positive staining (red) are infected with virus. For assessing Foci Forming Unit (FFU), the positive foci at the highest dilution were counted. Representative panels of images are shown (Scale bar 100 μm). B) Mean values and standard deviations of viral titer in FFU per milliliter are shown in the graph. The polymerase null mutant did not produce infectious particles. WT: wild-type; Pol-: Polymerase null. Please click here to view a larger version of this figure.
Figure 4. Examining the infectivity of wild-type HCV and measuring virus titer. A) Naïve Huh-7.5.1 cells were used for infection studies. The cell-free supernatant collected at 48 and 96 hr post-transfection (hpt) of HCV RNA were subjected to 10-fold serial dilution and added to the cells in a 96-well plate in triplicate. 72 hr post-infection, the cells were fixed and immunostained for HCV NS5A protein. The cells with NS5A positive staining (red) are infected with virus. For assessing Foci Forming Unit (FFU), the positive foci at the highest dilution were counted. Representative panels of images are shown (Scale bar 100 μm). B) Mean values and standard deviations of viral titer in FFU per milliliter are shown in the graph. The polymerase null mutant did not produce infectious particles. WT: wild-type; Pol-: Polymerase null. Please click here to view a larger version of this figure.
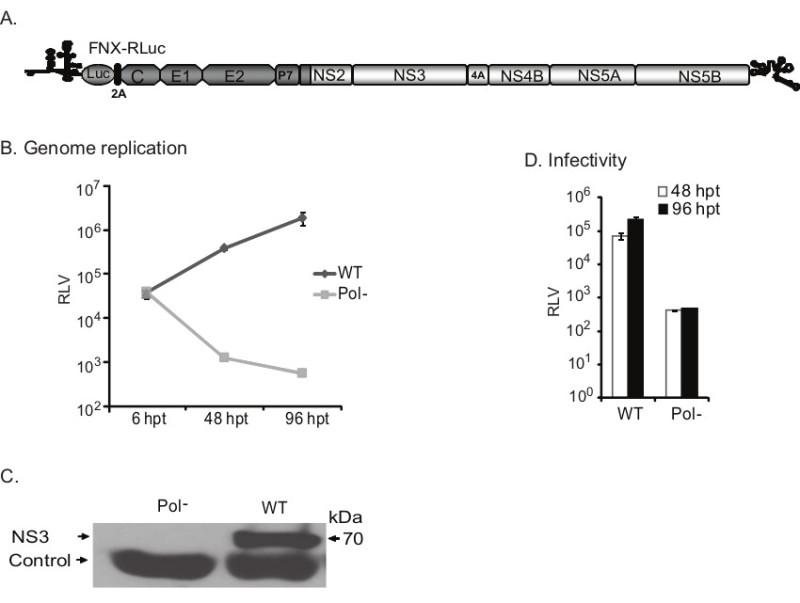 Figure 5. Evaluating the genome replication and infectivity of reporter HCV. A) A cartoon of intragenotype 2a chimeric reporter virus is presented. The Renilla luciferase gene is inserted inframe between 5’NTR and core. B) The genome replication kinetics of wild-type and Pol- mutant reporter viruses at indicated time points post-transfection is shown in the graph. Protein lysates were harvested at 6 hr, 48 hr and 96 hr time points for measuring Renilla luciferase enzymatic activity. The mean and standard deviation calculated from triplicate Renilla luciferase values (RLV) for each virus are presented in the graph. C) Western blotting panel shows the HCV NS3 protein expression. A non-specific antigen detected by NS3 primary antibody acts as a loading control. Wild-type (WT) reporter virus produces high level of NS3 protein. D) Analysis of virus infectivity. Naïve Huh-7.5.1 cells were inoculated with the cell-free supernatant obtained from transfected culture at 48 hr and 96 hr time points. Renilla luciferase activities of infected cells were measured at 48 hr post-infection. Mean values with standard deviation are shown in the graph. The Pol- reporter virus infected cells had only background level of luciferase activity, whereas WT reporter virus exhibited high level of infectivity.
Figure 5. Evaluating the genome replication and infectivity of reporter HCV. A) A cartoon of intragenotype 2a chimeric reporter virus is presented. The Renilla luciferase gene is inserted inframe between 5’NTR and core. B) The genome replication kinetics of wild-type and Pol- mutant reporter viruses at indicated time points post-transfection is shown in the graph. Protein lysates were harvested at 6 hr, 48 hr and 96 hr time points for measuring Renilla luciferase enzymatic activity. The mean and standard deviation calculated from triplicate Renilla luciferase values (RLV) for each virus are presented in the graph. C) Western blotting panel shows the HCV NS3 protein expression. A non-specific antigen detected by NS3 primary antibody acts as a loading control. Wild-type (WT) reporter virus produces high level of NS3 protein. D) Analysis of virus infectivity. Naïve Huh-7.5.1 cells were inoculated with the cell-free supernatant obtained from transfected culture at 48 hr and 96 hr time points. Renilla luciferase activities of infected cells were measured at 48 hr post-infection. Mean values with standard deviation are shown in the graph. The Pol- reporter virus infected cells had only background level of luciferase activity, whereas WT reporter virus exhibited high level of infectivity.
Discussion
This illustration describes a method for analyzing the hepatitis C Virus replication cycle. HCV is a human pathogen and the prescribed biosafety protocol will have to be strictly followed. Infectious HCV cell culture systems have been described previously11-13,16,17. There are few crucial points we implement when following the illustrated protocol. First, it is of high importance to have good quality of intact full length viral genomic RNA for downstream studies. The input plasmid carrying the viral cDNA has to be linearized and blunted carefully. The mung-bean nuclease employed to blunt the XbaI overhang is a non-specific nuclease and prolonged incubation will result in damage or degradation of 3’ end of viral genome. The nuclease has to be removed immediately after the specified 30 min incubation by phenol-chloroform extraction or anion exchange column purification. Gel extraction of nuclease treated DNA may not be recommended as the enzyme is active during gel electrophoresis process.
For subsequent in vitro RNA transcription steps, generally, one will also take broad-spectrum precautions for minimizing ribonuclease (RNase) contamination. All the reagents and materials used should be RNase free. To generate high-quality genomic RNA with minimum RNA strand breaks, the RNA need to be subjected to less physical stress by avoiding harsh vortexing and repeated pipetting during purification and downstream handling steps. After completion of RNA transcription, the input plasmid DNA templates have to be removed from transcribed RNA mix by DNase (RNase free) treatment. Incomplete removal can result in carryover of plasmid DNA into transfected cells and affect the quantification of absolute genome copy number of replicated viral RNA. Hence use DNase enzyme at a concentration of 1 unit per microgram of DNA and also having extra 15 min of DNase incubation at 37ºC can help. The transcribed RNA can be purified by either phenol-chloroform extraction or anion exchange column. Care should be exercised to avoid carryover of phenol or other reagents to the purified RNA sample, which can be cytotoxic and interfere with molecular assays.
For electroporation purposes, the purified RNA has to be dissolved in RNase-free sterile water. If the RNA pellet is difficult to dissolve, heating the sample to 65 ºC for 5 min will help. Once transcription is completed, it is important to store the RNA immediately in 10 μg of working aliquots made in RNase free water at -80 ºC. The purpose of the aliquots is to prevent repeated freeze/thaw mediated RNA degradation. For consistent experimental results, it is recommended to transcribe all samples and controls at the same time. High quality RNA is recommended for a higher viral titer yield. For transfecting HCV genomic RNA, polymer and lipid based transfection agents can also be used21,22.
Viral and host factors greatly influence the growth kinetics of HCV strains. Human hepatoma cell line (Huh-7) derivatives Huh-7.5, Huh-7.5.1 and Huh7-Lunet cell lines are commonly used for evaluating viral growth11-13,15. The peak viral production of JFH-1 strain in Huh7-Lunet cells is at 3-4 days post-transfection, whereas in Huh-7.5.1 cells it is at 21 days post-transfection13,15. The intragenotype 2a chimeric virus FNX-HCV having non-structural genes from JFH-1 strain has peak titer at 4 days post-transfection in Huh-7.5.1 cells19,20. For viral production from in vitro transcribed RNA, early passage human hepatoma cell line is preferred. Viral production is significantly reduced while using Huh-7.5.1 cells over passage number 20. To ensure increased cell survivability after electroporation, thawed Huh-7.5.1 cells are allowed a two week recovery period before conducting an experiment and maintained in 12%-15% FBS. Along with this criterion, resuspension of the cells in growth media containing 15% FBS immediately after electroporation increases the cell survival allowing for better viral production. After the first 8 hr time point, the cells are then switched to media containing 10% FBS. To minimize the carry over cellular debris, the viral culture supernatant is subjected to centrifugation and filtration through 4 micron filter. The unconcentrated or concentrated viral supernatants are aliquoted and stored in -80ºC for further in vitro and in vivo experiments. These precautions and suggestions can help researchers for successfully performing the analysis of HCV replication cycle.
Another variation of reporter HCV that harbors a marker gene such as fluorescent protein and a secreted form of Gaussia luciferase (Gluc) are described for HCV research18,23,24. For researchers focusing on investigating HCV genome replication, HCV sub-genomic replicon (lacking the genes core to NS2) is a valuable tool9,10. HCV Replicons are non-infectious, thus require less bio-safety regulations and easier to work with compared to the infectious cell culture HCV and clinical isolates. HCV deficient in NS5B polymerase activity is a critical control for studying HCV replication cycle11,12. HCV lacking envelope glycoproteins (delta E1E2), or p7-NS2 activity can be useful controls for studies involving HCV entry, virion morphogenesis and egress12,19,25-27.
Regarding limitations, currently the infectious cell culture system is available only for HCV genotypes 1 and 2. Authentic viral strains belong to genotypes 3 to 6 that are capable of growing efficiently in cell culture yet to be identified. Intergenotypic recombinant viruses are useful alternatives for studying replication cycle of various genotypes. Replication competent chimeric virus harboring core to NS2 region from genotypes 1 to 7 and rest of the sequence from JFH-1 genotype 2a genome were described15,18. Another limitation is that the viral yield of JFH-1 and its derivative chimeric strains is in the range of 103 to 106 per ml, which is relatively low compared to that of other RNA viruses such as polio and influenza. HCV strain with robust growth needs to be developed. Future investigation includes the use of the reporter HCV for high-throughput library screening to identify potent antivirals and focus on developing vaccine candidates for preventing HCV infection.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank F. Chisari for providing Huh-7.5.1 cell line. We would like to thank Justine Ho for editing the manuscript. This work was supported by Cedars-Sinai Medical Center Institutional Programmatic Research Award and National Center for Advancing Translational Sciences, Grant UL1TR000124 to V.A.
References
- Alter M. J. Epidemiology of hepatitis C. Hepatology. 1997;26(3 Suppl 1) doi: 10.1002/hep.510260711. [DOI] [PubMed] [Google Scholar]
- Alter M. J. Epidemiology of hepatitis C virus infection. World J Gastroenterol. 2007;13(17):2436–2441. doi: 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavanchy D. The global burden of hepatitis C. Liver Int. 2009;29 (Suppl 1):74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- Lindenbach BD, Thiel HJ, Rice CM. Flaviviridae: viruses and their replication in Fields Virology. Philadelphia, PA: 2007. pp. 1101–1152. [Google Scholar]
- Ciesek S, Manns MP. Hepatitis in 2010: the dawn of a new era in HCV therapy. Nat Rev Gastroenterol Hepatol. 2011;8(2):69–71. doi: 10.1038/nrgastro.2010.219. [DOI] [PubMed] [Google Scholar]
- Ghany MG, et al. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54(4):1433–1444. doi: 10.1002/hep.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo QL, et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244(4902):359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- Kolykhalov AA, et al. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science. 1997;277(5325):570–574. doi: 10.1126/science.277.5325.570. [DOI] [PubMed] [Google Scholar]
- Lohmann V, et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285(5424):110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290(5498):1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- Lindenbach BD, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309(5734):623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- Wakita T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11(7):791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102(26):9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, et al. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J Virol. 2007;81(2):629–638. doi: 10.1128/JVI.01890-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietschmann T, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A. 2006;103(19):7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. Highly efficient full-length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc Natl Acad Sci U S A. 2012;109(48):19757–19762. doi: 10.1073/pnas.1218260109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Lemon S. Genotype 1a HCV (H77S) infection system. Methods Mol Biol. 2009;510:337–346. doi: 10.1007/978-1-59745-394-3_25. [DOI] [PubMed] [Google Scholar]
- Gottwein JM, et al. Development and application of hepatitis C reporter viruses with genotype 1 to 7 core-nonstructural protein 2 (NS2) expressing fluorescent proteins or luciferase in modified JFH1 NS5A. J Virol. 2011;85(17):8913–8928. doi: 10.1128/JVI.00049-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugaswami V, et al. High-resolution functional profiling of hepatitis C virus genome. PLoS Pathog. 2008;4(10) doi: 10.1371/journal.ppat.1000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D, et al. Systematic analysis of enhancer and critical cis-acting RNA elements in the protein-encoding region of the hepatitis C virus genome. J Virol. 2013;87(10):5678–5696. doi: 10.1128/JVI.00840-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloum S, et al. Rab18 binds to hepatitis C virus NS5A and promotes interaction between sites of viral replication and lipid droplets. PLoS Pathog. 2013;9(8) doi: 10.1371/journal.ppat.1003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez G, et al. Selection of an optimal RNA transfection reagent and comparison to electroporation for the delivery of viral RNA. J Virol Methods. 2007;145(1):14–21. doi: 10.1016/j.jviromet.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan T, et al. Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J Virol. 2009;83(17):8379–8395. doi: 10.1128/JVI.00891-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller T, et al. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J Virol. 2007;81(9):4591–4603. doi: 10.1128/JVI.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, et al. Production of hepatitis C virus lacking the envelope-encoding genes for single-cycle infection by providing homologous envelope proteins or vesicular stomatitis virus glycoproteins in trans. J Virol. 2011;85(5):2138–2147. doi: 10.1128/JVI.02313-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CT, et al. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol. 2007;81(16):8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmann E, et al. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007. [DOI] [PMC free article] [PubMed]