

Summary
Over the last decade, the known spectrum of CD4 T cell effect or subsets has become much broader and it has become clear that there are multiple dimensions by which subsets with a particular cytokine commitment can be further defined, including their stage of differentiation, their location and most importantly, their ability to carryout discrete functions. Here we focus on our studies that highlight the synergy among discrete subsets, especially those defined by helper and cytotoxic function, in mediating viral protection and on distinctions between CD4 T cell effectors located in spleen, draining lymph node, and in tissue sites of infection. What emerges is a surprising multiplicity of CD4 T cell functions that indicate a large arsenal of mechanisms by which CD4 T cells act to combat viruses.
Introduction
Since the 1980's, much has been learned about how CD4 T cells act during immune responses by studying polarized subsets defined by their cytokine production profiles (Th1, Th2, and much more recently Th17, and Th9). More recently, careful investigations have added to the spectrum of CD4 T cell activities by defining subsets of cells that are more appropriately characterized by their function rather than by their cytokine production. These include CD4 T cells that are specialized to help germinal center B cell responses (Tfh), cells that are specialized to kill target cells (ThCTL), cells with specialized regulatory function (Treg), as well as cells that induce inflammatory responses (innate inducer cells, or ThII). In addition, CD4 T cell subsets have been characterized based on whether they circulate through secondary lymphoid organs, or whether they are resident for extended periods in peripheral tissues. Finally, CD4 T cells have been grouped based on whether they are naïve – having not encountered cognate peptide antigen, are activated - at varying stages in a spectrum of differentiation, including effect or cells, or whether they are memory – resting cells that have responded specific antigen in the past. These different axes for classifying CD4 T cell subsets while helpful in fine definition of cells and their responses (Figure 1), can handicap the development of a simple integrated view of how CD4 T cells protect against pathogens.
Figure 1. Defining CD4 T cell subsets.
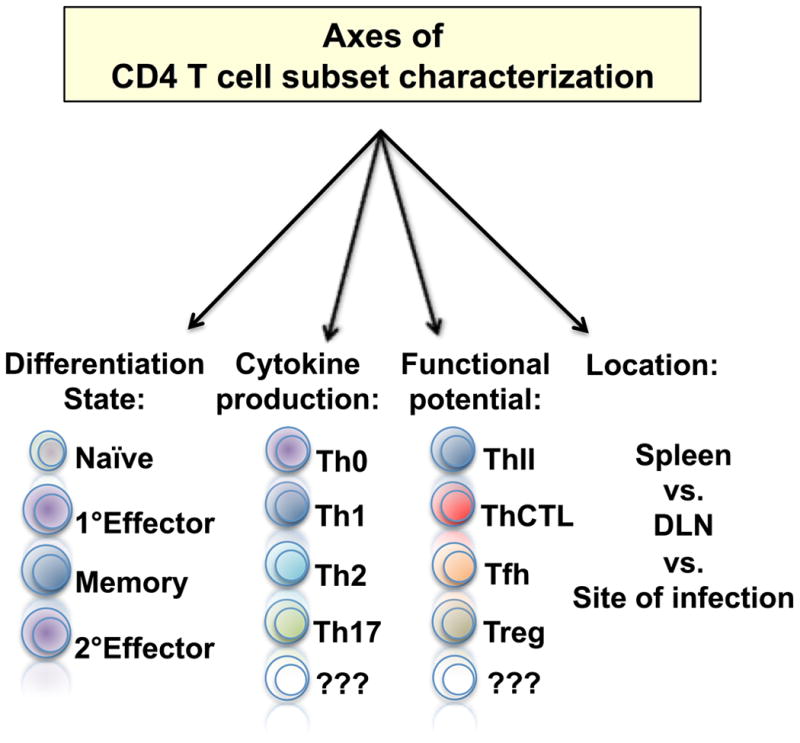
The state of activation, cytokine production profile, functional potential, and anatomical location can all be used to characterize and place CD4 T cells into distinct subsets.
While most often studied in isolation, it is increasingly clear that protective CD4 T cell responses against complex pathogens involve the actions of several different subsets of activated cells acting in concert and utilizing different mechanisms and often acting in different sites. These different specialized populations of CD4 T cells also act at different times following infection and they synergize to produce powerful responses made up of multiple layers of unique and redundant functions. Much of this complexity is generated during responses because of the plasticity of responding CD4 T cells, either naïve or memory, to develop multiple effector subsets. Our observations also highlight the power of multiple committed CD4 T subsets acting together for optimal protection. Inducing the whole spectrum of CD4 T cell responses by vaccination represents an attractive strategy to improve protection against pathogens, such as influenza A virus (IAV), where neutralizing antibody alone cannot provide reliable long-term immunity, but against which memory T cells with specificity for the highly conserved internal virus proteins, can mediate powerful protection (1).
Here, we discuss several observations that stress the importance of functional multiplicity in protective CD4 T cell responses, and review how these responses are generated and regulated. We suggest that functional multiplicity in the memory CD4 T cell response ensures redundant layers of protection, but complicates the definition of correlates of protection, given that different activities operating at different times and in different tissue sites might all contribute to an optimal multi-layered protective mechanism. Vaccines that induce such responses also need to target the sites of infection to generate local immunity, as tissue resident memory is most likely essential. While the signals that influence polarization of some subsets (i.e. Th1, Th2) occur early during the initial priming of T cells, our recent results studying ThCTL and the multi-step regulation of Tfh development (2) support a model in which later signals are required for the generation of key functionally specialized subsets. Thus, vaccines capable of inducing a broad spectrum of CD4 T cell functions likely need to be based on live pathogens that present antigens in the appropriate inflammatory milieu, in the right place, by the correct antigen presenting cells, and for the correct duration for the most effective protective immunity.
Protective CD4 T cell responses against influenza
T cell responses, especially those of memory CD4 T cells, play an integral role in protective immunity against viral pathogens. Key mechanisms include simultaneous help for B cell antibody (Ab) responses, help for CD8 T cells, late direct cytotoxic activity, and the early induction of innate inflammatory responses (3). Each of these different mechanisms has been identified as critical for protection against one or more different pathogens. Against some pathogens, only one CD4 T cell function (most often help for B cells) seems to be critical for optimal immunity, while against others, multiple mechanisms must be brought to bear for memory CD4 T cells to completely clear the infection (3).
IAV infection provides a powerful model in which to study how distinct mechanisms contribute to viral clearance, as memory CD4 T cells mediate strong protection when transferred to otherwise naïve mice (4-8), play an important role in heterosubtypic protection in mouse models (1, 9-12), and have been correlated with vaccine-induced protection (13) and improved responses against disease in humans (14). Multiple mechanisms have been found to contribute to, and correlate with, optimal protection against IAV mediated by memory CD4 T cells (15-17). We find that this wide functional diversity is best revealed when the CD4 T cells responding against IAV in different organs are examined (5, 18, 19). This partitioning by organ is a useful tool to partially separate different functional subsets, such as Tfh in the secondary lymphoid organs, and ThCTL and innate inducer cells in the infected lung (Figure 2). The organ specific location may also give us clues into how different subsets are obtained and what late signals drive their development. One theme of this review is that different aspects of protection take place in different anatomical sites.
Figure 2. Phases of memory CD4 T cell protective functions.
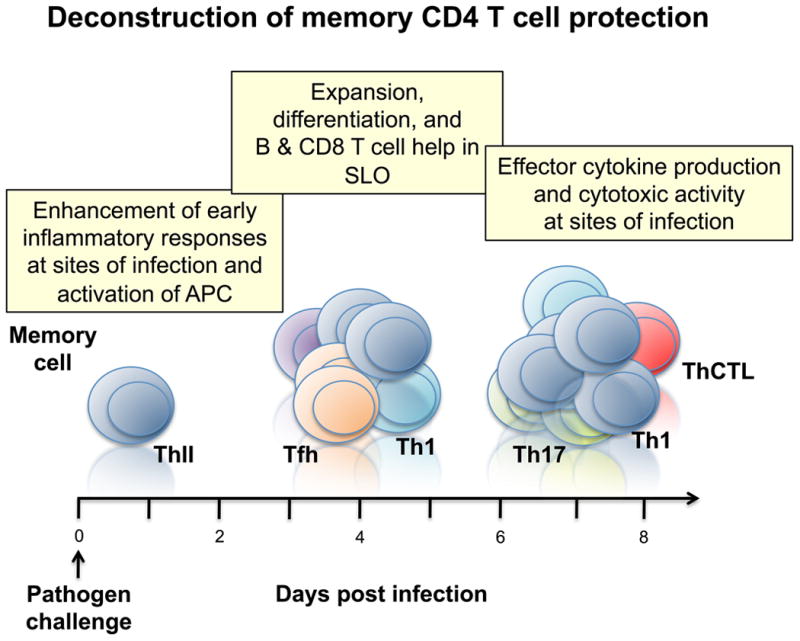
Following challenge with a pathogen, memory CD4 T cells mediate protection at different times and in different organs. Early at sites of infection, memory CD4 T cells resident in tissues, or inflammatory inducing memory T cells (ThII), enhance innate inflammatory responses and activate antigen presenting cells and other innate cells to control pathogen levels. This is followed by memory CD4 T cell provision of help for antibody producing B cells and help for CD8 T cells in the secondary lymphoid organs (SLO). At the peak of the response, memory CD4 T cells that migrate to sites of infection and produce cytokines and chemokines and kill infected cells to control and clear the pathogen.
Cytokine Responses of CD4 T cells during IAV challenge
When naïve or memory cells respond in vivo to IAV they generate large populations of effectors, many of which migrate to the lung (the site of infection), and carry out anti-viral activities. Effector CD4 T cell responses generated by IAV infection are prototypically ‘Th1’, characterized by strong IFN-γ production. Thus, it is not surprising that Th1-polarized CD4 T cell clones (20) or Th1 effector cells generated in vitro (21) provide protection against otherwise lethal challenges, as they largely mirror responses of effector populations primed by virus in vivo (22). Indeed, a common technique for quantifying human IAV-specific CD4 (and CD8) T cell responses is by the detection of IFN-γ-producing cells either by intracellular cytokine staining or by ELISPOT. Th2 responses are not protective (20, 21), and IL-4 production can exacerbate disease (23). However, this conventional Th1 designation belies the complexity of CD4 T cell responses against IAV and several other viral pathogens that have been identified in animal models in recent years. Below, we briefly touch on the major cytokines produced by CD4 T cells responding to IAV and their roles in protection, with an emphasis on the striking differences in cytokine production observed in different organs also outlined in Table I.
Table 1.
| CD4 effector | Spleen | dLN | Lung |
|---|---|---|---|
| Cytokine | |||
| IL-2 | ++ | +++ | +/- |
| IFN-γ | ++ | ++ | +++ |
| TNF | ++ | +++ | +/- |
| IL-10 | - | - | ++ |
| IL-17 | - | - | ++ |
| Phenotype | |||
| CD62L | ++ | ++ | - |
| CXCR5 | ++ | ++ | - |
| BTLA | ++ | ++ | - |
| CCR7 | ++ | ++ | - |
| IL-6Rα | ++ | ++ | + |
| GL-7 | ++ | ++ | - |
| CD49d | + | + | +++ |
| CD103 | + | + | ++ |
| VLA-4 | +/- | +/- | +++ |
| Granzyme B | + | + | +++ |
| CCR5 | ? | ? | ++ (BAL) |
IFN-γ and Th1 cytokines
IFN-γ is the most prominent cytokine produced by effector CD4 T cells responding to IAV in all organs. Surprisingly, the majority of studies have revealed that IFN-γ is dispensable for protection against IAV during both primary and heterosubtypic challenge (17). However, in certain experimental models where other mechanisms are removed, a key role for IFN-γ can be revealed. For example, while we found that there was no protective role for IFN-γ in protection mediated by memory CD4 T cells in WT mice, IFN-γ was critical for protection when memory cells were transferred to SCID hosts that lack endogenous T and B cells (4). Similarly, others found that an important anti-viral role for IFN-γ was revealed in mice deficient in nitric oxide synthase (24), which are unable to produce the potent inflammatory signaling molecule nitric oxide, suggesting that protective roles for IFN-γ become prominent only when other anti-viral, or regulatory, mechanisms are absent.
The mechanisms of IFN-γ action involved in protection are unclear. IFN-γ may act by directly activating cell types such as macrophages (25), NK cells (26), and neutrophils (27, 28), all of which have been shown to contribute to protection against severe disease caused by IAV. Another possibility is that IFN-γ is required for the establishment of cytokine and chemokine gradients that are responsible for maximizing the efficiency of the influx of anti-viral populations (7, 29). IFN-γ may also regulate the degree of immunopathology caused by IAV infection (29, 30). Finally, IFN-γ is required for the efficient generation of IgG2a responses, and this Ab isotype may play unique roles in protection (31).
Many CD4 T cells responding to IAV produce TNF, often in conjunction with IFN-γ (5). However, effectors responding in different organs produce very different levels of TNF, with the cells in the lung producing the least at the peak of both primary and secondary responses (5). TNF can have both pro- and anti-inflammatory roles. On the one hand, TNF may constrain the production of cytokines and chemokines such as monocyte chemoattractant protein-1 that can exacerbate inflammation following IAV challenge (32). On the other, studies have identified a direct pro-inflammatory role for TNF, with improved responses against IAV in its absence (33, 34). TNF does not seem to contribute significantly to viral clearance. We find that TNF production by CD4 T cells is greatly enhanced in the spleen and draining lymph node compared to the lung (5). This could reflect a preponderance of terminal effector cells in the lung that have lost the capacity to produce TNF.
Th17 cytokines
Both CD4 and CD8 T cells responding to IAV produce low levels of IL-17 and this is almost exclusively restricted to the lung (35, 36). The transfer of either well-polarized Th17 or Tc17 polarized CD4 or CD8 T cell effectors can protect otherwise naïve mice against lethal doses of IAV (35, 36), but as with Th1 effector-derived IFN-γ, the mechanisms responsible remains unclear. It is unlikely that IL-17 itself serves a direct protective or anti-viral role as no decrease in protection is observed when Th17 polarized effectors are transferred to IL-17 receptor-deficient hosts (Figure 3A). Protection, if anything, is improved as IL-17 receptor-deficient mice lost significantly less weight than WT recipients (Figure 3B). This finding correlates well with a study from Kolls' group demonstrating a critical role for IL-17 in contributing to immunopathology associated with IAV infection (37). Our studies, however, do suggest that both Th17 (CD4) and Tc17 (CD8), protect through mechanisms distinct from Th1 or Tc1 cells, since the removal of prototypical Th1/Tc1 effector molecules IFN-γ and perforin did not abrogate their protective ability (35, 38). Th17 cells often also co-produce IL-22. Recent studies suggest that IL-22 may be critical in facilitating epithelial regeneration in the lung following infection, indicating that Th17 responses may be more important to mediate optimal repair rather than to carryout antiviral functions (39-41). Finally, Th17 cells can also produce IL-21. IL-21 has been found to play an important role in facilitating optimal Ab production against IAV (42, 43), and it is thus possible that Th17 cells could help aspects of the protective Ab response.
Figure 3. Th17 effector protection does not require IL-17 production.
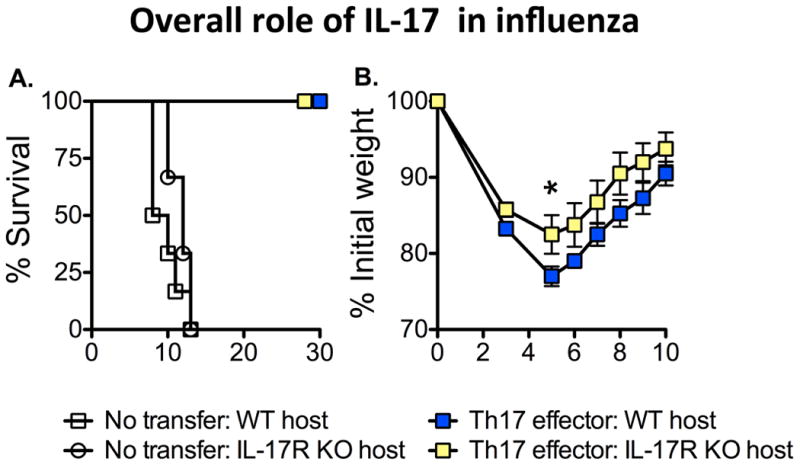
5×106 Th17-polarized effector CD4 T cells (OT-II transgenic) recognizing IAV were transferred to either WT or IL-17 receptor-deficient (IL-17R KO) hosts. All mice were challenged with a lethal (2 LD50) dose of IAV expressing OVAII peptide and survival and weight loss monitored. Th17 effector transfer rescued both WT and IL-17R KO host survival (left), but IL-17R KO mice lost less weight (right) (n=4-6 mice/group).
IL-10
Like Th17 cytokines, IL-10 production during IAV challenge is largely limited to the lung during the peak of infection. The majority of IL-10 is made by T cells that co-produce IFN-γ (35, 44). IL-10 production can profoundly impact the outcome of IAV infection, though in dramatically different ways. We found that IL-10-deficient mice, or WT mice treated with IL-10 receptor blocking Ab, responded equivalently to low doses of IAV as did WT mice but demonstrated dramatically enhanced survival compared to WT mice when challenged with higher doses of virus (35). Improved protection in the absence of IL-10 was not due to differences in viral titer, nor the magnitude of T cell responses, but correlated with selective up-regulation of several Th17-associated cytokines including IL-17A, IL-17F, IL-21, and IL-22. Interestingly, we observed that IL-10-deficient CD4 T cells responding against IAV in WT adoptive hosts developed strikingly enhanced IL-17 responses, suggesting an autocrine inhibition of IL-17 responses by IL-10 (35). Similar enhanced IL-17 production has been observed in the absence of IL-10 in other settings (45-47), suggesting that this mechanism acts generally to constrain physiological Th17 responses (48). As discussed above, it is unclear exactly how increased levels of Th17-associated cytokines may facilitate improved protection, but accelerated repair of damaged lung epithelium may be key. IL-10 was also found to impair protection against IAV in a study from Metzger's lab (44). In this case, a more rapid production of local Ab at the site of infection was found to contribute to the enhanced protection in the absence of IL-10, suggesting IL-10 may participate in down-regulating T cell help and/or B cell differentiation, especially in the lung.
Another study addressing the role of IL-10 during IAV infection concluded that survival of mice was impaired in the absence of IL-10 and correlated with enhanced pulmonary inflammation and lung damage (49). It is possible that the different outcomes in studies addressing the role of IL-10 during IAV infection are at least in part influenced by the relative ability of mice to generate strong IL-17 responses. IL-17 production is strongly influenced by constituents of the normal flora (50), suggesting one variable that might account for differences in the strength of Th17 responses during IAV challenge seen by different groups. Interestingly, we also observed enhanced immunopathology in the lungs of IL-10-deficient mice challenged with higher doses of IAV compared to controls (35). We thus propose that the increased inflammation observed in the absence of IL-10 may drive enhanced immunopathology that could lead to death if not countered by strong up-regulation of Th17 cytokines, that could lead to improved repair making enhanced inflammatory responses bearable.
Effectors are highly diverse
In vitro, under polarizing conditions, relatively uniform populations of highly differentiated CD4 T cell effectors can be generated that appear similar in terms of phenotype, division history, and cytokine production potential. When CD4 T cell effectors are generated in vivo by IAV infection, the effectors produced are a surprisingly heterogeneous population of activated cells marked by wide differences in phenotype and cytokine production (5, 18). We find that the distinctions among IAV-specific effectors isolated from different organs are pronounced. For example, while lung-resident effectors are almost uniformly CD62Llo and produce IFN-γ, many effectors responding in the spleen and draining lymph node retain high levels of CD62L and produce IL-2 and TNF in addition to IFN-γ (18) and Table I.
One possible interpretation of these results is that the effectors in the lung constitute the most differentiated cells, and that the effectors present in secondary lymphoid organs are mostly at earlier stages of differentiation. This reasoning predicts that an optimal population of highly activated effectors (those in the lung), are best suited to combat IAV while those cells present in the spleen and draining lymph nodes are at earlier stages of differentiation. Indeed, highly-polarized Th1 effectors, that most resemble the effectors isolated from the lung, can mediate potent viral clearance and protect against supra lethal doses of IAV when transferred to unprimed mice while less-differentiated Th0 effectors cannot (21) and our unpublished observations.
To further investigate organ-specific distinctions between effectors, we transferred naïve, CFSE-labeled CD4 T cells recognizing IAV to unprimed hosts and challenged with virus. Seven days later, we sort-purified effector cells by collecting only donor cells that fit the criteria of ‘effector’ (having undergone at least 5 divisions) from the spleen, draining lymph node, and lung, and analyzed their transcriptome by whole genome microarray. There was a surprising degree of heterogeneity between effectors responding in different organs consistent with previous observations of phenotypic distinctions (5). We also transferred Th1-like memory cells isolated from IAV infected animals (5) or well-polarized in vitro-generated Th1 memory cells (51). Most surprisingly, the secondary effectors that developed from these already polarized memory cells generated in vitro were characterized by very similar, and nearly as extensive organ-specific differences in gene expression (Figure 4) as that which characterized effectors generated from naïve cells, or in vivo IAV-primed memory cells (5). This demonstrates that even memory cells generated from effector cells well-polarized for cytokine production (i.e. Th1) can further differentiate in vivo in response to IAV to give rise to distinct subsets of cells that each have unique functional attributes (5).
Figure 4. Secondary effectors derived from in vitro generated Th1 memory CD4 T cells show organ-specific differences in gene expression following IAV challenge.
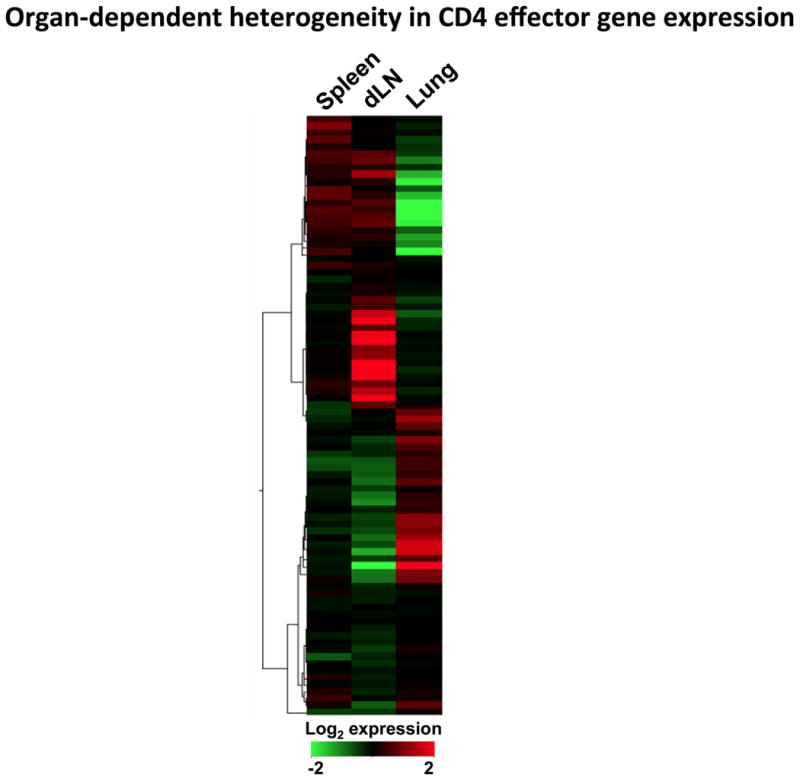
Heat map, generated using GeneSifter (GeoSpiza), showing the signal strength of genes differentially expressed by secondary CD4 T cell effectors isolated from the spleen, draining lymph node (dLN), and lung 7 days post IAV infection.
Compartmentalized functions of CD4 T cells
Analysis of the microarray data revealed several patterns that suggest that the effectors in the spleen and draining lymph node may not simply be at a less-differentiated functional state compared to effectors responding in the lung. One viewpoint is that effectors that migrate to tissues/sites of infection are effectors that have inherently greater protective capacity compared to effectors in the secondary lymphoid organs that then become effector memory, while those present in the secondary lymphoid organs instead become central memory that have more potential for future development. We support an alternate view, that many of the effectors in each site are highly specialized to perform functions that differ between the sites so that cells responding to IAV in both the secondary lymphoid organs and the lung perform distinct functions as illustrated in Figure 5. For example, we observed an enrichment for genes associated with Th1 effect or potential from the cells isolated from the lung including ifng, ccl3, ccl4, ccl5, ccl9, cxcl9, cxcl10, as well as grB, grC, encoding granzymes B and C and pfn1 encoding perforin (5). This pattern is consistent with observations that effectors with the most potent anti-viral function are almost exclusively found in the lung and BAL following IAV challenge. The spectrum of genes expressed higher in the lung than the spleen and draining lymph nodes also correlates with the lung-restricted location of CD4 T cells with cytolytic activity (ThCTL) (22, 52). The ThCTL depend on perforin for cytotoxicity and express high levels of granzyme B (21), as described in more detail below.
Figure 5. Compartmentalization of CD4 T cell effector functions during IAV challenge.
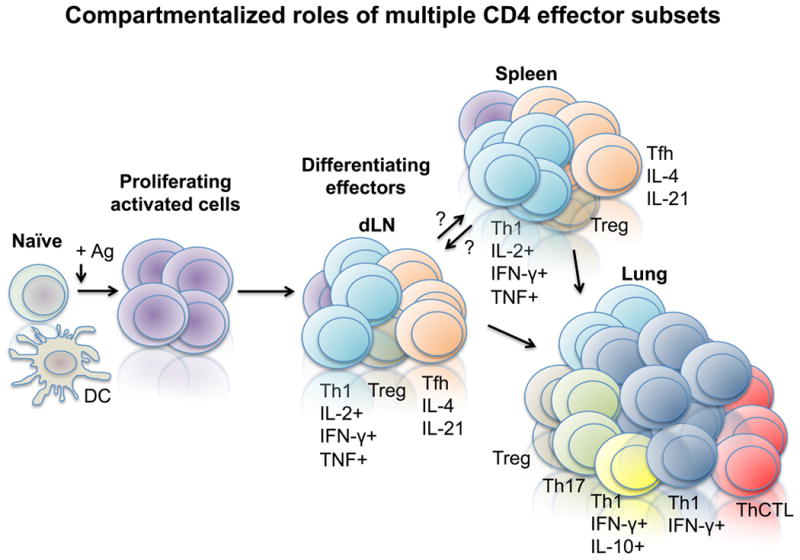
Following activation of naive cells in the draining lymph node (dLN) by antigen presenting cells (APC) displaying viral antigen (Ag), activated effector cells develop that traffic to the spleen and lung. Functional analysis reveals that effectors present in secondary lymphoid organs (spleen and dLN) are strikingly different from the effector cells responding in the lung.
There was a broad up-regulation of Tfh-associated genes in the effectors that were isolated from the spleen and draining lymph node vs. the lung including btla, cxcr5, bcl6, IL6ra, slamf6, sh2d1a encoding SAP, and CD200 (5). An increased presence of Tfh phenotype cells in the secondary lymphoid organs in comparison to the lung following primary IAV challenge was confirmed by surface marker expression of CXCR5, PD-1, and intracellular expression of Bcl-6 (5), and is in agreement with other recent studies of Tfh responses during primary IAV challenge (53, 54). These data are most compatible with the concept that, once activated, CD4 T cells give rise to multiple subsets of highly specialized effector cells that each contribute important and unique protective functions in their resident tissue. Interestingly, we found that secondary effectors, derived from memory cells, contained Tfh in the spleen, draining lymph nodes, and the lung (5), suggesting that secondary responses may be less functionally compartmentalized by organ than primary responses against pathogens.
In addition to specialized subsets of effectors that contribute to viral clearance such as Tfh and ThCTL, induced Tregs have also been characterized upon IAV challenge. These cells can potently suppress T cell responses and inflammatory responses, and seem to play a critical role during recall responses (55). Interestingly, Tregs can be found in the lung, draining lymph node, and spleen (55) and our unpublished observations, and at least in in vitro assays, all populations display similar functions (55), suggesting that their development and function are relatively uniform across organs. It is unclear how Tregs function during IAV challenge to reduce immunopathology, but it is unlikely to involve IL-10 production as little is detected from Tregs generated by IAV (35, 55).
Regulation of compartmentalization by transcription factors
The compartmentalization of effector functions is further supported by differential expression by effector CD4 T cells of genes involved in transcriptional regulation in distinct organs (Figure 6). Some of the most relevant of such differences in expression, and how such differences might contribute to functional distinctions between effectors responding in different organs against IAV are discussed below.
Figure 6. Compartmentalization of transcriptional regulators in CD4 T cell effectors following IAV challenge.

Heat map, generated using Gene-E (Broad Institute), showing the signal strength of genes in the DAVID ontogeny pathway of transcriptional regulation differentially expressed by primary CD4 T cell effectors isolated from the spleen, draining lymph node (dLN), and lung at 7 days post infection.
Eomes
Eomesodermin, encoded by a T box gene eomes, is a member of the interferon regulatory factor family known to be essential for the development of CD8 T cell IFN-γ, granzyme, and perforin production (56, 57). Deletion of eomes in CD8 T cells reduces production of IFN-γ and reduces cytolytic activity (56). Ligation of CD134 (OX40) and CD137 (4-1BB) promotes eomes expression in CD4 T cells in vivo and in vitro and enhances the development of CD4 T cells capable of killing tumor cells (58). We see high levels of eomesodermin expression in cells generated by IAV infection with high ThCTL activity by intracellular staining (Marshall, unpublished) and others find eomesodermin deficient mice develop few cytolytic cells (58). Much like its functions in CD8 T cells, ectopic expression of this transcription factor in CD4 T cell clones can similarly drive increased expression of perforin and granzymes and additionally increases FasL expression (59). We found by microarray that CD4 T cell effectors from the lung expressed significantly higher levels of eomes compared to cells from the draining lymph node or spleen (5), further supporting that cells with the capacity to directly control virus are enriched in the lung. This data is consistent with the concept that eomesodermin promotes cytolytic activity in CD8 T cells and CD4 T cells, and thus contributes to a potent anti-viral effector phenotype in infected tissues such as the lung following IAV infection.
Id2
The inflammatory milieu can impact the expression of inhibitors of DNA binding proteins (Id) proteins, Id2 and Id3 (60). Id proteins negatively regulate the DNA binding activity of E-protein transcription factors, which are important in B and T cell development, cell survival, and proliferation (61). Inflammatory cytokines, such as IL-12, drive increased expression of Id2 in CD8 T cells and the subsequent development of more terminally differentiated ‘short-lived effector cells' (62). In contrast to Id2, Id3 is reciprocally regulated and its expression is decreased in the presence of inflammation. High levels of Id3 expression in CD8 T cells are associated with non-terminally differentiated ‘long-lived effector’ CD8 T cells (60). High levels of multiple pro-inflammatory cytokines and chemokines characterize the lung environment during influenza infection (6), and in agreement with the paradigm of Id protein expression in CD8 T cells, we found that CD4 T cell effectors isolated from the lung express high levels of mRNA for Id2, compared to cells in the secondary lymphoid organs. The reverse was found for Id3, with high expression in the spleen and draining lymph nodes and low expression in the lung, suggesting that CD4 T cells parallel CD8 T cells in the regulation of this transcriptional pathway and that it might underlie different patterns of CD4 T cell effector contraction in the lung and secondary lymphoid organs (63) and control important aspects of memory generation.
Hlx
H2.0-like homeobox is a transcription factor encoded by Hlx or Hlx1. This transcription factor is a downstream target of T-bet (64, 65). T-bet promotes Th1 cell differentiation by increasing expression of Hlx, and together with other transcription factors they reinforce IFN-γ expression and the Th1 development program (64, 65). In many models, T cells that secrete large amounts of IFN-γ at the sites of infection also co-produce IL-10. As mentioned earlier, more IL-10 producing cells that also co-produce IFN-γ are found in the lung following primary IAV infection (35). That we find the greatest Hlx expression where we find the greatest proportion of IFN/IL-10 double-producing effectors may suggest that this pathway is involved in regulating the production of IL-10 by effectors in the lung.
Runx2
Runt related transcription factors play key mechanistic links between cell fate, proliferation and growth control (66-69). Recently, in conjunction with an essential transcription factor RORγt, Runx1 and Runx2 have been implicated in promoting IL-17 production in CD4 T cells (70). Akin to IL-10 production by CD4 T cell effectors during IAV challenge, IL-17 producing cells are found primarily in the lung (35). We observed enhanced expression of the gene encoding Runx2 in the lung effectors versus those in the secondary lymphoid organs, suggesting that expression of this transcription factor may be involved in regulating Th17 responses during IAV challenge. It is interesting to speculate that differential expression of Runx2 could underlie the differential production of IL-17 by CD4 T cell effectors in the lung versus secondary lymphoid organs during IAV.
Runx2 can also promote the expression of the pro-apoptotic molecule Bim (71). Correlating with the enhanced inflammatory milieu in the lung versus secondary lymphoid organs, we observed the highest levels of Bim protein levels in lung-resident CD4 T cell effectors, suggesting that Runx2 may also influence the extent of death of the effectors in the lung that is coincident with viral clearance (unpublished data). Following primary IAV infection, CD4 effector T cells in the lung contract more dramatically than those in spleen and draining lymph node (63). Bim has been shown to play a key role in T cell effector contraction (72, 73). Thus, we are investigating whether there is a lung-specific pathway of CD4 effector contraction involving upregulation of Runx2.
Nfil3
Nuclear factor, interleukin-3 (IL-3), regulated protein or E4BP4 is expressed by many leukocytes, including dendritic cells, NK cells, B cells, and T cells. The protein encoded by this gene is a member of the basic region/leucine zipper transcription factor super family, and as implied by its name, is upregulated in response to IL-3 (74-76). Both Th1- and Th2-polarized CD4 T cells can express nfil3. In Th1 cells in particular, its expression is thought to contribute to effector cell cytokine production plasticity in terms of the ability to express cytokines other than IFN-γ (77). More specifically, expression of nfil3 is believed to maintain or promote expression of IL-10 and IL-13. Th1 cells deficient in nfil3 show impaired IL-10 and IL-13 production, whereas Th2 cells appear unaffected by its absence, and forced expression of nfil3 drives expression of IL-10 and IL-13 in CD4 T cell effectors (77). That nfil3 expression is higher in lung CD4 T cells versus CD4 T cell effectors isolated from the spleen and draining lymph nodes again correlates with the increased IL-10 production found in the lungs, suggesting another pathway that might be important for the development of the IFN-γ/IL-10 dual-producing subset.
Pou2af1 and Pou6f1
POU domain proteins are key regulators of gene expression during lymphocyte development and activation (78). One such protein, POU domain, class 2, associating factor, also known as BOB.1 or OBF-1, is a transcription factor encoded by pou2af1 that is expressed in B cells and T cells (43, 79). Mouse B cells deficient in BOB.1/OBF-1 respond to TLR stimulation by producing lower levels of IL-6 (43). Some evidence suggests that this transcription factor is also involved in regulating IL-6 production in human cells. In mouse T cells, expression of BOB.1/OBF-1 in Th0, Th1, and Th2 subsets is increased upon TcR and anti-CD28 stimulation (79). BOB.1/OBF.1 can enhance PU.1 transcription factor activity (79, 80), which participates in the Th2 development program (81), as well as in recruiting Bcl-6 to DNA (82). POU domain, class 6, transcription factor, also known as emb/brn5.0 in mouse or TCFB1 in humans (83) can interact with other POU domain proteins Pou4f2 and Pou6f2, as well as Stat3. Observations suggest that Pou6f2 is involved in cell proliferation, perhaps through its ability to activate the IL-2 promoter in an activation dependent manner (84). Studies employing human cells suggest that it also has a role in regulating TCR β gene expression (83). In our studies, Th1-like effector cells isolated from the secondary lymphoid organs express higher levels of pou2af1 and pou6f1 compared to the effectors isolated from the lung. A recent study also found pou6f1 to be expressed in early fate committed Tfh (85). Given that Tfh are enriched in the secondary lymphoid organs, it is tempting to speculate that these transcription factors contribute to the site-specific Tfh development program during IAV challenge.
Blimp-1 and Bcl-6
B lymphocyte induced maturation protein-1 (Blimp-1), is a transcriptional repressor involved in lymphocyte homeostasis, maturation, and function (86). Loss of Blimp-1 results in the accumulation of effector and memory phenotype T cells and culminates in severe inflammatory disease likely caused by the diminished IL-10 production in effector CD4 and CD8 T cells (87-89). IL-2 induces Blimp-1 expression that goes on to counter regulate IL-2 cytokine production (86) and prevent terminal differentiation. Blimp-1 is considered a counter-regulator of Bcl-6, a key factor necessary for Tfh development. This transcription factor regulatory network has recently been reviewed extensively (2, 90-92). Blimp-1, is also considered key to non-follicular CD4 development (93). In our microarray analysis of CD4 T cell effectors generated in response to IAV infection, message for the gene that encodes blimp-1, prdm1, is highest in CD4 T cell lung effectors (Figure 7) that are the highest IL-10 producers, and lowest in spleen and draining lymph node effectors that are enriched in Tfh phenotype cells. In agreement with this, following IAV infection, Blimp-1 is more intensely expressed in CD4 and CD8 T cell effectors in the lung compared to the secondary lymphoid organs when Blimp-1 expression is visualized with a GFP reporter (Marshall and Swain, unpublished data). Our preliminary results suggest that conditional Blimp-1 deficient mice develop fewer cells with a ThCTL phenotype, those with less cytolytic activity, and lower granzyme B and eomesodermin expression (Marshall, unpublished data), consistent with this transcriptional repressor playing a key role in controlling ThCTL differentiation.
Figure 7. Differential expression of Blimp-1 in CD4 T cell effectors following IAV challenge.
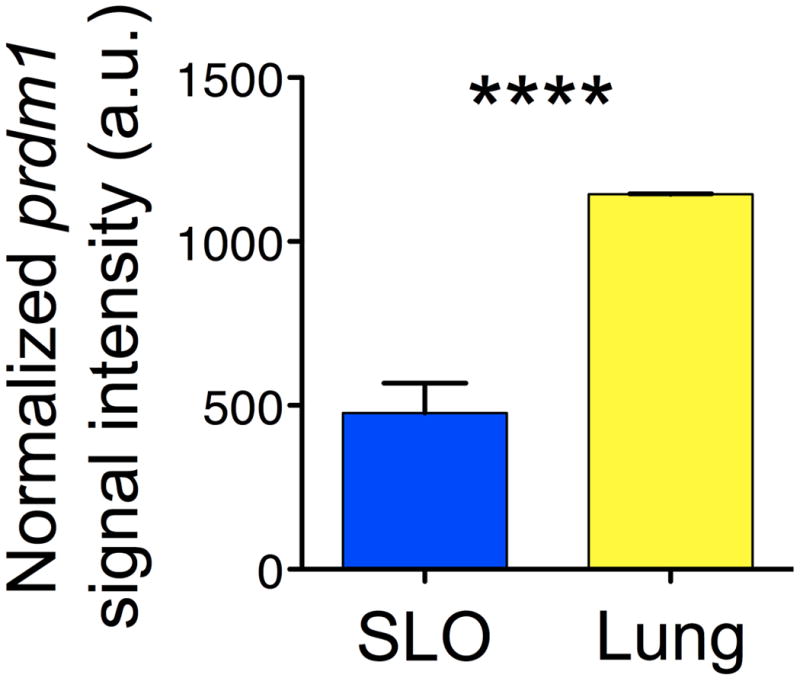
Relative signal of the gene encoding Blimp-1, prdm1, in CD4 T cell effectors isolated from the secondary lymphoid organs (SLO) versus the lung at 7 days post infection.
These data implicating differential expression of key transcription factors in different organs support the idea that many factors work together to dictate the eventual effector phenotype and suggest that the local environment of the site of infection (the lung for IAV infection) vs. secondary lymphoid organs may act on recent effector immigrants to further mold effector CD4 T cell functions several days after their initial activation.
Simultaneous Generation of Functional CD4 Effector Subsets
Studies of cytokine polarized-subsets often stress their counter-regulation. In certain strains of mice, and in response to particular infections, one subset will dominate and this can be ascribed to their signature cytokines that often suppress other fates. For instance, Th1 effector signature cytokine IFN-γ suppresses generation of Th2 polarized cells and the Th2 signature cytokine IL-4 suppresses Th1 generation (94). In vivo this plays out in responses that tend to be dominated by one or other of these responses.
The systemic regulation of the functionally defined subsets that segregate into different organs, or different defined regions within organs, such as Tfh and ThCTL, as well as Treg and perhaps Th17, seems to work on different principles. For instance in IAV infection, while a predominantly Th1 cytokine pattern develops, all of the above functionally defined subsets develop simultaneously. There is a vigorous generation of Tfh confined to the follicular areas of spleen and draining lymph node (5, 53), and generation of multiple other subsets in non-follicular sites including Th17 and ThCTL in the lung (22, 35) and Treg in multiple organs (55).
So how is this simultaneous generation of distinct subsets regulated? Existing data indicate the development into follicular vs. non follicular pathways occurs early on (85), and that higher levels of IL-6 vs. IL-2 (95, 96) is a key factor in up-regulating Bcl-6 and Blimp-1 transcription factors associated with follicular vs. non-follicular fate (Figure 8). The most likely source of these key cytokines is the antigen-presenting cell (APC) or T cell itself during the initial cognate interaction. The differentiation of Tfh is particularly revealing, because the remaining steps in their differentiation require repeated interactions with follicular B cells and signals to the costimulatory ligand ICOS (97). We recently have dissected some of the signals favoring generation of ThCTL in the lung. We find they require IL-2 (98), are enhanced by induction of eomesodermin and signals through CD134 (OX40) and CD137 (4-1BB) (58) (Swain and Vong, unpublished) and they are dependent on Blimp-1 (Swain and Marshall, unpublished). Thus, we suggest that the fate of the developing effectors is determined by multiple interactions with APC in distinct environments that are specialized to support distinct Th subset differentiation, as illustrated in Figure 8.
Figure 8. CD4 T cell effector subset generation.
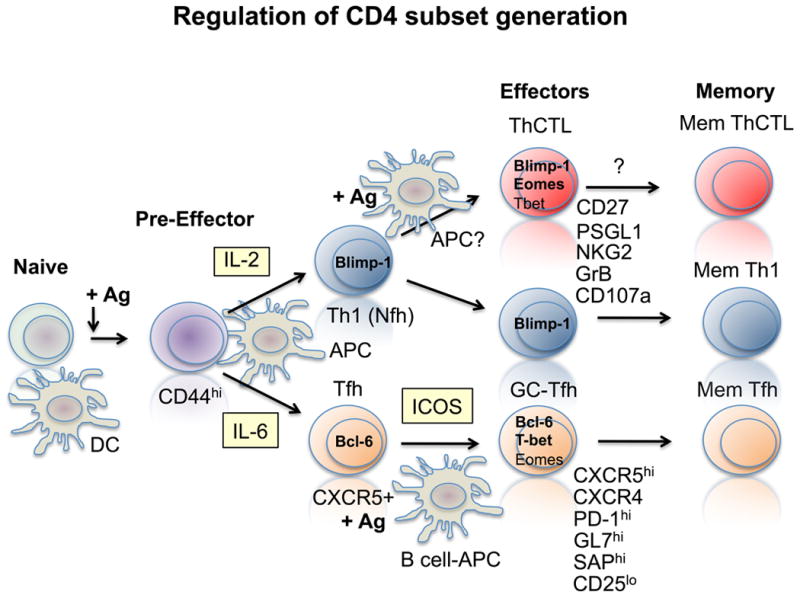
Following initial activation Bcl-6 and Blimp-1 reciprocally regulate the generation of follicular Th (Tfh and GC-Tfh) vs. non-follicular (Nfh), which include the unique ThCTL subset.
Memory CD4 T cell protection against IAV – synergy and redundancy of protective mechanisms
The above results suggest that distinct CD4 T cell functions, mediated by specialized subsets of differentiated effector cells, coordinately act to effect clearance of IAV. To directly test this hypothesis, we systematically deconstructed the protective memory CD4 T cell response against IAV by employing host animals deficient for lymphocyte subsets that memory CD4 T cells might interact with to optimally clear virus (namely CD8 T cells and B cells), and by employing memory cells deficient in crucial effector molecules that could be instrumental in viral clearance (IFN-γ and perforin) (4). We observed that while a set number of memory CD4 T cells could fully protect WT mice against a high doses of virus (2 LD50 and higher), B- and T-cell deficient hosts receiving the same number of memory CD4 T cells were not able to survive these challenge doses, but were protected against a lower, but still lethal dose for these hosts (corresponding to 0.5 LD50 for WT mice). When the same number of memory cells was transferred to hosts lacking both T cells and B cells, their protective impact was further diminished, with only partial protection observed against a dose of IAV corresponding to 0.1 LD50 for WT mice. The memory cells did, however, initially control viral titers and promote recovery during the first two weeks following infection, but failed to clear virus, resulting in a chronic and ultimately fatal infection (4). These results suggest that protection against IAV by memory CD4 T cells is dramatically enhanced by synergy with either CD8 T cells or B cells, and that optimal protection requires both (Figure 9).
Figure 9. Layers of CD4 T cell-mediated protection.
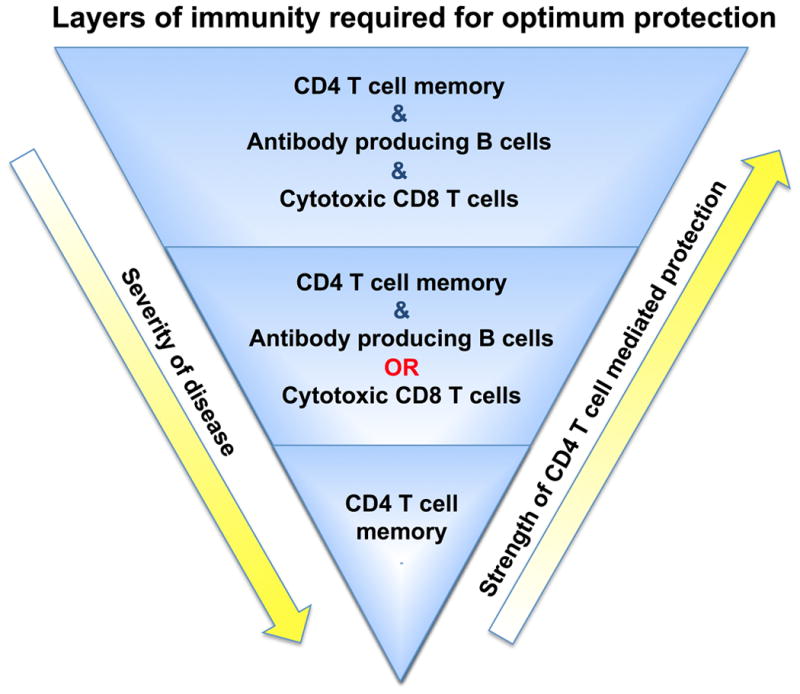
Optimal protection against IAV provided by memory CD4 T cells requires memory CD4 T cells to synergize with B cells and CD8 T cells (either through synergies with the cells themselves, or alternatively, synergies of anti-viral memory CD4 T cell functions with the distinct anti-viral functions of B cells and CD8 T cells). Protection is similarly and substantially reduced if either B cells or CD8 T cells are absent, correlating with delayed viral clearance and enhanced disease. When memory CD4 T cells respond against the same dose of virus in mice lacking both CD8 T cells and B cells, protection and viral clearance are greatly compromised.
What functions of CD4 T cells might promote these protective synergies? Memory CD4 T cells induce a faster, earlier Ab response against IAV by day 8 post-infection (5), consistent with the idea that one component of synergy between memory CD4 and B cells is help for this early Ab. Indeed we see synergy between Ab collected from wild-type convalescent mice and transfer of CD4+ effector (21) or memory cells recognizing virus (4). Surprisingly, we found that canonical Tfh help was not required for memory cell protection of T cell-deficient mice as SAP-deficient memory cells, that cannot become Tfh, protected equally as well as WT cells (4). However, consistent with previous observations (99), SAP-deficient memory cell transfer did not lead to the formation of long-lived IAV-specific IgG (4). These results do not rule out that non-follicular help provided by memory CD4 T cells is a crucial component on the protective synergy observed. The signals involved in this process remain poorly understood. As mentioned above, transfer of memory CD4 T cells, both WT and SAP-deficient (unpublished observations), markedly enhance early titers of IAV-specific IgG present at 8 dpi in otherwise unmanipulated mice versus naïve CD4 T cell transfer (5), consistent with other observations of enhanced B cell help provided by memory versus naïve CD4 T cells in other models (100), suggesting that even slightly earlier Ab production can have a tremendous impact in protection. Another possibility could be local Ab production in the lung is enhanced preferentially by memory CD4 T cells resident in the lung.
Whether memory CD4 T cell synergy with CD8 T cells is due to memory cells providing help for CD8 T cells is unclear. After primary IAV challenge, there is little evidence to suggest that naïve CD4 T cells help shape the magnitude nor kinetics of the CD8 T cell effector responses (101), although optimal CD8 T cell memory seems to require CD4 T cell help (102). It is, however, possible, that the memory cells, as opposed to naïve CD4 T cells, can enhance the kinetics of, or functional capacity of primary CD8 T cell responses perhaps through activating APC quicker, or through production of IL-2. Indeed, recent studies have found impaired CD8 T cell responses against IAV in the absence of CD28 costimulation (101), and we have shown that memory CD4 T cells rapidly upregulate CD80 on dendritic cells within 2 days of IAV challenge, well before CD80 upregulation is observed on dendritic cells in recipients of naïve CD4 T cells and challenged with IAV (6). In this regard, it is tempting to speculate that certain memory CD4 T cells may differentiate to become specialized helpers of CD8 T cells.
We investigated what direct functions might be involved in CD4 T cell protection by utilizing memory populations deficient for either IFN-γ or perforin. We tested IFN-γ-deficient memory cells and found that they protected WT hosts or hosts lacking B cells or CD8 T cells equally well as WT memory cells (4). However, when IFN-γ-deficient memory CD4 T cells were transferred to hosts deficient in T and B cells, protection was lost. This was not due to aberrant differentiation of IFN-γ-deficient cells as treating recipients of WT cells with IFN-γ neutralizing Abs also abrogated protection (4). These results suggest that the importance of IFN-γ production to CD4 T cell-mediated protection is largely dependent on the relative strength of other arms of the immune response, namely CD8 T cells and Ab. When CD8 T cells and/or neutralizing Ab responses are optimal, mechanisms of viral clearance are present that supersede IFN-γ-mediated control. However, in their absence, the IFN-γ-mediated control is critical. As introduced earlier, it is not exactly how IFN-γ-mediated control against IAV operates, and further experiments are required to test the many possibilities.
We also tested the impact of removing perforin, which is necessary for ThCTL-mediated killing of targets. Perforin-deficient memory CD4 T cells protected WT mice equally well from lethal challenges as WT memory cells (4), consistent with results utilizing perforin-deficient effector transfer (21), demonstrating a negligible impact on protection. Even IFN-γ and perforin double-deficient memory cells protected unprimed WT hosts as well as corresponding WT cells (unpublished observations). However, when perforin-deficient memory cells were transferred to SCID hosts, protection was significantly reduced as evidenced by one log higher viral titers in the lungs compared to recipients of WT memory cells (4). These results confirm that perforin expression, presumably by ThCTL, can contribute to protection against IAV, but also stress that this mechanism, and others, are often redundant with CD8 T cell and B cell mediated-mechanisms of viral control present in intact mice.
Interestingly, we found that WT memory CD4 T cells responding in SCID hosts drove the emergence of TcR epitope-specific viral escape mutants (4). The outgrowth of such mutants was virtually eliminated in recipients of perforin-deficient memory cells, implicating ThCTL as necessary for this selection. These results thus further reinforce that perforin-dependent killing of infected target cells contributes to memory CD4 T cell-mediated protection against IAV, and that this mechanism can efficiently select for escape mutants by monoclonal CD4 T cells responding in the absence of CD8 T cell and B cells. These results are similar to studies ascribing a dominant role to perforin in CD8 T cell-mediated IAV escape utilizing a similar model (103, 104). Amino acid substitutions in hemagglutinin epitopes associated with escape from a characterized human CD4 T cell epitope have been described (105), suggesting that this mechanism might play a role in determining the severity of disease in some individual cases of IAV clinically.
We observed two distinct patterns of the emergence of viral escape mutants using two different viruses. Using the more virulent A/PuertoRico/8/34 (PR8) strain, all SCID mice challenged with a 0.1 LD50 dose of virus died, but escape mutants were only observed in about 1/3 of animals. However, when mice were challenged with the less-virulent PR8-OVAII virus, only about half of the animals receiving memory CD4 T cells died, while the other half cleared virus. Interestingly, all PR8-OVAII infected mice that were not protected by memory CD4 T cells harbored TcR-specific escape mutants, suggesting that the death of animals infected with the weaker virus was due to the lack of control of IAV due to outgrowth of mutants not recognized by the TcR, while death of mice challenged with the more virulent PR8 was largely due to unchecked inflammatory/healing responses driven by chronic infection irrespective of whether escape mutants were present or not.
What are the most protective CD4 T cells?
Overall, a picture emerges of memory CD4 T cells contributing to protection through multiple synergizing as well as redundant mechanisms that result in timely and efficient clearance of IAV. Is there a subset of memory cells with more protective capacity than others? Recent observations suggest that memory CD4 T cells that are resident in the lung provide stronger protection against IAV than memory cells isolated from secondary lymphoid organs upon adoptive transfer to naïve animals then challenged with virus (5, 8). Enhanced protection mediated by lung-resident memory cells correlated with preferential presence in the lung versus in secondary lymphoid organs following transfer (8), suggesting that improved homing to the site of infection may contribute to their superior protective capacity. We also directly transferred primary effectors isolated from the lung of IAV-infected animals or isolated from the secondary lymphoid organs and similarly observed significantly improved protection from effector cells isolated from the lung. Studies of the phenotype of cells in the lung, indicate they are unique CD4 T cell effectors (5, 18, 98) and Table I, and include cells with cytotoxic capacity, ThCTL (22), as well as populations defined by different cytokine production (5) indicating that lung CD4 effectors, and probably resident lung memory cells, are functionally diverse.
Many studies have found that CD4 T cells are not required for clearance of primary IAV challenge (17). However, we found that SAP-deficient animals, that do not develop GC-Tfh were not protected against secondary homotypic IAV challenge, and that this poor protection correlated with very low levels of long-lived neutralizing Ab (99). Similar results were seen when we transferred SAP-deficient or WT CD4 T cells to CD4-deficient hosts (99). Since SAP-dependent GC-Tfh are required for the generation of long-lived memory B cells and neutralizing serum Ab, and such cells are virtually absent from the lung during primary challenge, these results argue that CD4 T cells responding in secondary lymphoid organs also make an important contribution to immunity against IAV, especially against homotypic challenge.
We conclude that CD4 T cells act by many mechanisms to combat IAV and likely other viruses, and that together while each mechanism may be effective at low doses of virus, there is synergy among them and with B and CD8 T cell protective mechanisms that can provide the most highly effective immunity. We also suggest that these same mechanisms will be relevant to other respiratory viruses and to multiple types of infections.
How to generate the most protective memory CD4 T cells?
The observations discussed here suggest that vaccines that aim to generate the most protective CD4 T cell responses against IAV should induce multiple, distinct memory subsets. Given the complexity of diverse T cell responses generated by infection, it is likely that vaccination with a live-attenuated virus can most readily replicate the spectrum of subsets generated by wild-type virus in contrast to vaccines based on non-replicating platforms. In addition, live attenuated IAV induces a significantly larger number of T cell effectors than killed vaccines, ultimately giving rise to more memory cells (106). As the subsets of memory CD4 T cells continue to expand, it seems that it is time to re-evaluate the ability of different vaccine strategies to generate effector and memory cells of the different subsets, especially those defined by function such as Tfh and ThCTL, which have not yet been widely used to assess vaccine efficacy.
For protection against influenza, it appears especially important to generate strong and lasting T cell memory in the lung, the site of infection. Studies demonstrate that intranasal immunization with live, attenuated IAV (unpublished observations) or other vaccines (107) is superior in generating strong T cell responses in the lung compared to other routes of vaccination. Important cues for the generation, and maintenance, of tissue-resident memory populations remain unclear, but we suggest a better understanding of this subset could significantly improve vaccines aiming to generate protective, and lasting T cell memory against IAV.
Summary and closing thoughts
Textbook paradigms suggest that the T cell effectors generated in secondary lymphoid organs following pathogen encounter must traffic to sites of infection in order to carryout protective functions. However, it is becoming increasingly clear that multiple subsets of specialized effectors are simultaneously generated in response to pathogen challenge, and that signals in the tissue-environment delivered by both costimulatory molecules and soluble molecules can act on already differentiated effector cells to further tailor their function. By better understanding these tissue-specific cues we will gain more insight into how T cell effectors coordinate and contribute to protective responses against pathogens, and develop strategies to better shape effector responses, as required.
Determining robust correlates of protection is key to determining what subsets of T cells are the most desirable to generate via vaccination in order to provide long lasting immunity. Recent results highlighting functional differences between CD4 T cells responding in different organs complicate this analysis since distinct protective correlates will be needed to characterize cells specialized for different roles that are present at different sites. Furthermore, since CD4 T cells mediate distinct protective functions at different phases of an immune response, correlates must be determined at several time points during the effector response. Since generation of memory is under separate regulation, it is also critical to evaluate memory months after vaccine administration. These considerations are especially problematical given that peripheral blood is the most commonly assessed clinical sample and it is often collected from patients only at one time after initial infection or vaccination. Strategies to overcome these limitations need to be developed to identify vaccines that will be most effective in generating the most protective memory CD4 T cells.
References
- 1.Powell TJ, et al. Priming with cold-adapted influenza A does not prevent infection but elicits long-lived protection against supralethal challenge with heterosubtypic virus. J Immunol. 2007;178:1030–8. doi: 10.4049/jimmunol.178.2.1030. [DOI] [PubMed] [Google Scholar]
- 2.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–63. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 3.Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol. 2012;12:136–48. doi: 10.1038/nri3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKinstry KK, et al. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J Clin Invest. 2012;122:2847–56. doi: 10.1172/JCI63689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strutt TM, McKinstry KK, Kuang Y, Bradley LM, Swain SL. Memory CD4+ T-cell-mediated protection depends on secondary effectors that are distinct from and superior to primary effectors. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1205894109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strutt TM, et al. Memory CD4+ T cells induce innate responses independently of pathogen. Nat Med. 2010;16:558–64. doi: 10.1038/nm.2142. 1p following 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teijaro JR, Verhoeven D, Page CA, Turner D, Farber DL. Memory CD4 T cells direct protective responses to influenza virus in the lungs through helper-independent mechanisms. J Virol. 2010;84:9217–26. doi: 10.1128/JVI.01069-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol. 2011;187:5510–4. doi: 10.4049/jimmunol.1102243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang S, Mozdzanowska K, Palladino G, Gerhard W. Heterosubtypic immunity to influenza type A virus in mice. Effector mechanisms and their longevity. J Immunol. 1994;152:1653–61. [PubMed] [Google Scholar]
- 10.Epstein SL, et al. Mechanisms of heterosubtypic immunity to lethal influenza A virus infection in fully immunocompetent, T cell-depleted, beta2-microglobulin-deficient, and J chain-deficient mice. J Immunol. 1997;158:1222–30. [PubMed] [Google Scholar]
- 11.Sun K, Ye J, Perez DR, Metzger DW. Seasonal FluMist Vaccination Induces Cross-Reactive T Cell Immunity against H1N1 (2009) Influenza and Secondary Bacterial Infections. J Immunol. 2011;186:987–93. doi: 10.4049/jimmunol.1002664. [DOI] [PubMed] [Google Scholar]
- 12.Guo H, Santiago F, Lambert K, Takimoto T, Topham DJ. T Cell-Mediated Protection against Lethal 2009 Pandemic H1N1 Influenza Virus Infection in a Mouse Model. J Virol. 2011;85:448–55. doi: 10.1128/JVI.01812-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bentebibel SE, et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med. 2013;5:176ra32. doi: 10.1126/scitranslmed.3005191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilkinson TM, et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med. 2012;18:274–80. doi: 10.1038/nm.2612. [DOI] [PubMed] [Google Scholar]
- 15.Swain SL, et al. CD4+ T-cell memory: generation and multi-faceted roles for CD4+ T cells in protective immunity to influenza. Immunol Rev. 2006;211:8–22. doi: 10.1111/j.0105-2896.2006.00388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boonnak K, Subbarao K. Memory CD4+ T cells: beyond “helper” functions. J Clin Invest. 2012;122:2768–70. doi: 10.1172/JCI65208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKinstry KK, Strutt TM, Swain SL. Hallmarks of CD4 T cell immunity against influenza. J Intern Med. 2011;269:507–18. doi: 10.1111/j.1365-2796.2011.02367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roman E, et al. CD4 effector T cell subsets in the response to influenza: heterogeneity, migration, and function. J Exp Med. 2002;196:957–68. doi: 10.1084/jem.20021052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bingaman AW, et al. Novel phenotypes and migratory properties distinguish memory CD4 T cell subsets in lymphoid and lung tissue. Eur J Immunol. 2005;35:3173–86. doi: 10.1002/eji.200526004. [DOI] [PubMed] [Google Scholar]
- 20.Graham MB, Braciale VL, Braciale TJ. Influenza virus-specific CD4+ T helper type 2 T lymphocytes do not promote recovery from experimental virus infection. J Exp Med. 1994;180:1273–82. doi: 10.1084/jem.180.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown DM, Dilzer AM, Meents DL, Swain SL. CD4 T cell-mediated protection from lethal influenza: perforin and antibody-mediated mechanisms give a one-two punch. J Immunol. 2006;177:2888–98. doi: 10.4049/jimmunol.177.5.2888. [DOI] [PubMed] [Google Scholar]
- 22.Brown DM, Lee S, Garcia-Hernandez MD, Swain SL. Multi-functional CD4 cells expressing IFN-gamma and perforin mediate protection against lethal influenza infection. J Virol. 2012 doi: 10.1128/JVI.07172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moran TM, Isobe H, Fernandez-Sesma A, Schulman JL. Interleukin-4 causes delayed virus clearance in influenza virus-infected mice. J Virol. 1996;70:5230–5. doi: 10.1128/jvi.70.8.5230-5235.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karupiah G, Chen JH, Mahalingam S, Nathan CF, MacMicking JD. Rapid interferon gamma-dependent clearance of influenza A virus and protection from consolidating pneumonitis in nitric oxide synthase 2-deficient mice. J Exp Med. 1998;188:1541–6. doi: 10.1084/jem.188.8.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tate MD, Pickett DL, van Rooijen N, Brooks AG, Reading PC. Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J Virol. 2010;84:7569–80. doi: 10.1128/JVI.00291-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weiss ID, et al. IFN-gamma treatment at early stages of influenza virus infection protects mice from death in a NK cell-dependent manner. J Interferon Cytokine Res. 2010;30:439–49. doi: 10.1089/jir.2009.0084. [DOI] [PubMed] [Google Scholar]
- 27.Tate MD, Deng YM, Jones JE, Anderson GP, Brooks AG, Reading PC. Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J Immunol. 2009;183:7441–50. doi: 10.4049/jimmunol.0902497. [DOI] [PubMed] [Google Scholar]
- 28.Dienz O, et al. Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol. 2012;5:258–66. doi: 10.1038/mi.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiley JA, Cerwenka A, Harkema JR, Dutton RW, Harmsen AG. Production of interferon-gamma by influenza hemagglutinin-specific CD8 effector T cells influences the development of pulmonary immunopathology. Am J Pathol. 2001;158:119–30. doi: 10.1016/s0002-9440(10)63950-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baumgarth N, Kelso A. In vivo blockade of gamma interferon affects the influenza virus-induced humoral and the local cellular immune response in lung tissue. J Virol. 1996;70:4411–8. doi: 10.1128/jvi.70.7.4411-4418.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huber VC, et al. Distinct contributions of vaccine-induced immunoglobulin G1 (IgG1) and IgG2a antibodies to protective immunity against influenza. Clin Vaccine Immunol. 2006;13:981–90. doi: 10.1128/CVI.00156-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Damjanovic D, et al. Negative regulation of lung inflammation and immunopathology by TNF-alpha during acute influenza infection. Am J Pathol. 2011;179:2963–76. doi: 10.1016/j.ajpath.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hussell T, Pennycook A, Openshaw PJ. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur J Immunol. 2001;31:2566–73. doi: 10.1002/1521-4141(200109)31:9<2566::aid-immu2566>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 34.Belisle SE, et al. Genomic profiling of tumor necrosis factor alpha (TNF-alpha) receptor and interleukin-1 receptor knockout mice reveals a link between TNF-alpha signaling and increased severity of 1918 pandemic influenza virus infection. J Virol. 2010;84:12576–88. doi: 10.1128/JVI.01310-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKinstry KK, et al. IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high-dose challenge. J Immunol. 2009;182:7353–63. doi: 10.4049/jimmunol.0900657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamada H, et al. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J Immunol. 2009;182:3469–81. doi: 10.4049/jimmunol.0801814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crowe CR, et al. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol. 2009;183:5301–10. doi: 10.4049/jimmunol.0900995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamada H, et al. Multiple redundant effector mechanisms of CD8+ T cells protect against influenza infection. J Immunol. 2013;190:296–306. doi: 10.4049/jimmunol.1200571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pociask DA, et al. IL-22 Is Essential for Lung Epithelial Repair following Influenza Infection. Am J Pathol. 2013;182:1286–96. doi: 10.1016/j.ajpath.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paget C, et al. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: potential role in protection against lung epithelial damages. J Biol Chem. 2012;287:8816–29. doi: 10.1074/jbc.M111.304758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar P, Thakar MS, Ouyang W, Malarkannan S. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol. 2013;6:69–82. doi: 10.1038/mi.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dienz O, et al. The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. J Exp Med. 2009;206:69–78. doi: 10.1084/jem.20081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karnowski A, et al. B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J Exp Med. 2012;209:2049–64. doi: 10.1084/jem.20111504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun K, Torres L, Metzger DW. A detrimental effect of interleukin-10 on protective pulmonary humoral immunity during primary influenza A virus infection. J Virol. 2010;84:5007–14. doi: 10.1128/JVI.02408-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schaefer JS, Montufar-Solis D, Vigneswaran N, Klein JR. ICOS promotes IL-17 synthesis in colonic intraepithelial lymphocytes in IL-10-/- mice. J Leukoc Biol. 2010;87:301–8. doi: 10.1189/jlb.0409238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pitt JM, et al. Blockade of IL-10 signaling during bacillus Calmette-Guerin vaccination enhances and sustains Th1, Th17, and innate lymphoid IFN-gamma and IL-17 responses and increases protection to Mycobacterium tuberculosis infection. J Immunol. 2012;189:4079–87. doi: 10.4049/jimmunol.1201061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang L, Yuan S, Cheng G, Guo B. Type I IFN promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS One. 2011;6:e28432. doi: 10.1371/journal.pone.0028432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huber S, et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3(-) and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity. 2011;34:554–65. doi: 10.1016/j.immuni.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun J, Madan R, Karp CL, Braciale TJ. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med. 2009;15:277–84. doi: 10.1038/nm.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ivanov, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–49. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McKinstry KK, Golech S, Lee WH, Huston G, Weng NP, Swain SL. Rapid default transition of CD4 T cell effectors to functional memory cells. J Exp Med. 2007;204:2199–211. doi: 10.1084/jem.20070041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marshall NB, Swain SL. Cytotoxic CD4 T cells in antiviral immunity. J Biomed Biotechnol. 2011;2011:954602. doi: 10.1155/2011/954602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyden AW, Legge KL, Waldschmidt TJ. Pulmonary infection with influenza A virus induces site-specific germinal center and T follicular helper cell responses. PLoS One. 2012;7:e40733. doi: 10.1371/journal.pone.0040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoo JK, Fish EN, Braciale TJ. LAPCs promote follicular helper T cell differentiation of Ag-primed CD4+ T cells during respiratory virus infection. J Exp Med. 2012;209:1853–67. doi: 10.1084/jem.20112256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brincks EL, et al. Antigen-Specific Memory Regulatory CD4+Foxp3+ T Cells Control Memory Responses to Influenza Virus Infection. J Immunol. 2013 doi: 10.4049/jimmunol.1203140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pearce EL, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–3. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 57.Glimcher LH, Townsend MJ, Sullivan BM, Lord GM. Recent developments in the transcriptional regulation of cytolytic effector cells. Nat Rev Immunol. 2004;4:900–11. doi: 10.1038/nri1490. [DOI] [PubMed] [Google Scholar]
- 58.Qui HZ, et al. CD134 plus CD137 dual costimulation induces Eomesodermin in CD4 T cells to program cytotoxic Th1 differentiation. J Immunol. 2011;187:3555–64. doi: 10.4049/jimmunol.1101244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eshima K, et al. Ectopic expression of a T-box transcription factor, eomesodermin, renders CD4(+) Th cells cytotoxic by activating both perforin- and FasL-pathways. Immunol Lett. 2012;144:7–15. doi: 10.1016/j.imlet.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 60.Yang CY, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. 2011;12:1221–9. doi: 10.1038/ni.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murre C. Helix-loop-helix proteins and lymphocyte development. Nat Immunol. 2005;6:1079–86. doi: 10.1038/ni1260. [DOI] [PubMed] [Google Scholar]
- 62.Cannarile MA, et al. Transcriptional regulator Id2 mediates CD8+ T cell immunity. Nat Immunol. 2006;7:1317–25. doi: 10.1038/ni1403. [DOI] [PubMed] [Google Scholar]
- 63.McKinstry KK, Strutt TM, Swain SL. Regulation of CD4+ T-cell contraction during pathogen challenge. Immunol Rev. 2010;236:110–24. doi: 10.1111/j.1600-065X.2010.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martins GA, Hutchins AS, Reiner SL. Transcriptional activators of helper T cell fate are required for establishment but not maintenance of signature cytokine expression. J Immunol. 2005;175:5981–5. doi: 10.4049/jimmunol.175.9.5981. [DOI] [PubMed] [Google Scholar]
- 65.Schulz EG, Mariani L, Radbruch A, Hofer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity. 2009;30:673–83. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 66.Taniuchi I, Littman DR. Epigenetic gene silencing by Runx proteins. Oncogene. 2004;23:4341–5. doi: 10.1038/sj.onc.1207671. [DOI] [PubMed] [Google Scholar]
- 67.Hu H, Djuretic I, Sundrud MS, Rao A. Transcriptional partners in regulatory T cells: Foxp3, Runx and NFAT. Trends Immunol. 2007;28:329–32. doi: 10.1016/j.it.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 68.Taniuchi I, et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–33. doi: 10.1016/s0092-8674(02)01111-x. [DOI] [PubMed] [Google Scholar]
- 69.Levanon D, Groner Y. Structure and regulated expression of mammalian RUNX genes. Oncogene. 2004;23:4211–9. doi: 10.1038/sj.onc.1207670. [DOI] [PubMed] [Google Scholar]
- 70.Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol. 2008;9:1297–306. doi: 10.1038/ni.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heidari N, Miller AV, Hicks MA, Marking CB, Harada H. Glucocorticoid-mediated BIM induction and apoptosis are regulated by Runx2 and c-Jun in leukemia cells. Cell Death Dis. 2012;3:e349. doi: 10.1038/cddis.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pellegrini M, Belz G, Bouillet P, Strasser A. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc Natl Acad Sci U S A. 2003;100:14175–80. doi: 10.1073/pnas.2336198100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wojciechowski S, Jordan MB, Zhu Y, White J, Zajac AJ, Hildeman DA. Bim mediates apoptosis of CD127(lo) effector T cells and limits T cell memory. Eur J Immunol. 2006;36:1694–706. doi: 10.1002/eji.200635897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang W, Zhang J, Kornuc M, Kwan K, Frank R, Nimer SD. Molecular cloning and characterization of NF-IL3A, a transcriptional activator of the human interleukin-3 promoter. Mol Cell Biol. 1995;15:6055–63. doi: 10.1128/mcb.15.11.6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ikushima S, Inukai T, Inaba T, Nimer SD, Cleveland JL, Look AT. Pivotal role for the NFIL3/E4BP4 transcription factor in interleukin 3-mediated survival of pro-B lymphocytes. Proc Natl Acad Sci U S A. 1997;94:2609–14. doi: 10.1073/pnas.94.6.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kamizono S, et al. Nfil3/E4bp4 is required for the development and maturation of NK cells in vivo. J Exp Med. 2009;206:2977–86. doi: 10.1084/jem.20092176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Motomura Y, et al. The transcription factor E4BP4 regulates the production of IL-10 and IL-13 in CD4+ T cells. Nat Immunol. 2011;12:450–9. doi: 10.1038/ni.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Phillips K, Luisi B. The virtuoso of versatility: POU proteins that flex to fit. J Mol Biol. 2000;302:1023–39. doi: 10.1006/jmbi.2000.4107. [DOI] [PubMed] [Google Scholar]
- 79.Brunner C, Sindrilaru A, Girkontaite I, Fischer KD, Sunderkotter C, Wirth T. BOB.1/OBF.1 controls the balance of TH1 and TH2 immune responses. EMBO J. 2007;26:3191–202. doi: 10.1038/sj.emboj.7601742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brunner C, Wirth T. Btk expression is controlled by Oct and BOB.1/OBF.1. Nucleic Acids Res. 2006;34:1807–15. doi: 10.1093/nar/gkl131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chang HC, et al. PU.1 expression delineates heterogeneity in primary Th2 cells. Immunity. 2005;22:693–703. doi: 10.1016/j.immuni.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 82.Wei F, Zaprazna K, Wang J, Atchison ML. PU.1 can recruit BCL6 to DNA to repress gene expression in germinal center B cells. Mol Cell Biol. 2009;29:4612–22. doi: 10.1128/MCB.00234-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Messier H, Brickner H, Gaikwad J, Fotedar A. A novel POU domain protein which binds to the T-cell receptor beta enhancer. Mol Cell Biol. 1993;13:5450–60. doi: 10.1128/mcb.13.9.5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kasibhatla S, Tailor P, Bonefoy-Berard N, Mustelin T, Altman A, Fotedar A. Jun kinase phosphorylates and regulates the DNA binding activity of an octamer binding protein, T-cell factor beta1. Mol Cell Biol. 1999;19:2021–31. doi: 10.1128/mcb.19.3.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Choi YS, et al. Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol. 2013;190:4014–26. doi: 10.4049/jimmunol.1202963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martins G, Calame K. Regulation and functions of Blimp-1 in T and B lymphocytes. Annu Rev Immunol. 2008;26:133–69. doi: 10.1146/annurev.immunol.26.021607.090241. [DOI] [PubMed] [Google Scholar]
- 87.Kallies A, et al. Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nat Immunol. 2006;7:466–74. doi: 10.1038/ni1321. [DOI] [PubMed] [Google Scholar]
- 88.Martins GA, et al. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat Immunol. 2006;7:457–65. doi: 10.1038/ni1320. [DOI] [PubMed] [Google Scholar]
- 89.Sun J, Dodd H, Moser EK, Sharma R, Braciale TJ. CD4+ T cell help and innate-derived IL-27 induce Blimp-1-dependent IL-10 production by antiviral CTLs. Nat Immunol. 2011;12:327–34. doi: 10.1038/ni.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Linterman MA, Liston A, Vinuesa CG. T-follicular helper cell differentiation and the co-option of this pathway by non-helper cells. Immunol Rev. 2012;247:143–59. doi: 10.1111/j.1600-065X.2012.01121.x. [DOI] [PubMed] [Google Scholar]
- 91.Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. 2010;11:114–20. doi: 10.1038/ni.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Okada T, Moriyama S, Kitano M. Differentiation of germinal center B cells and follicular helper T cells as viewed by tracking Bcl6 expression dynamics. Immunol Rev. 2012;247:120–32. doi: 10.1111/j.1600-065X.2012.01120.x. [DOI] [PubMed] [Google Scholar]
- 93.Cannons JL, Lu KT, Schwartzberg PL. T follicular helper cell diversity and plasticity. Trends Immunol. 2013 doi: 10.1016/j.it.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Seder RA, Paul WE. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu Rev Immunol. 1994;12:635–73. doi: 10.1146/annurev.iy.12.040194.003223. [DOI] [PubMed] [Google Scholar]
- 95.Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. 2012;209:243–50. doi: 10.1084/jem.20111174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ballesteros-Tato A, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity. 2012;36:847–56. doi: 10.1016/j.immuni.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Choi YS, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34:932–46. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brown DM, Kamperschroer C, Dilzer AM, Roberts DM, Swain SL. IL-2 and antigen dose differentially regulate perforin- and FasL-mediated cytolytic activity in antigen specific CD4+ T cells. Cell Immunol. 2009;257:69–79. doi: 10.1016/j.cellimm.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kamperschroer C, Dibble JP, Meents DL, Schwartzberg PL, Swain SL. SAP is required for Th cell function and for immunity to influenza. J Immunol. 2006;177:5317–27. doi: 10.4049/jimmunol.177.8.5317. [DOI] [PubMed] [Google Scholar]
- 100.MacLeod MK, David A, McKee AS, Crawford F, Kappler JW, Marrack P. Memory CD4 T cells that express CXCR5 provide accelerated help to B cells. J Immunol. 2011;186:2889–96. doi: 10.4049/jimmunol.1002955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Seah SG, et al. Unlike CD4+ T-cell help, CD28 costimulation is necessary for effective primary CD8+ T-cell influenza-specific immunity. Eur J Immunol. 2012;42:1744–54. doi: 10.1002/eji.201142211. [DOI] [PubMed] [Google Scholar]
- 102.Belz GT, Wodarz D, Diaz G, Nowak MA, Doherty PC. Compromised influenza virus-specific CD8(+)-T-cell memory in CD4(+)-T-cell-deficient mice. J Virol. 2002;76:12388–93. doi: 10.1128/JVI.76.23.12388-12393.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Price GE, Ou R, Jiang H, Huang L, Moskophidis D. Viral escape by selection of cytotoxic T cell-resistant variants in influenza A virus pneumonia. J Exp Med. 2000;191:1853–67. doi: 10.1084/jem.191.11.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Price GE, Huang L, Ou R, Zhang M, Moskophidis D. Perforin and Fas cytolytic pathways coordinately shape the selection and diversity of CD8+-T-cell escape variants of influenza virus. J Virol. 2005;79:8545–59. doi: 10.1128/JVI.79.13.8545-8559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Berkhoff EG, et al. An amino acid substitution in the influenza A virus hemagglutinin associated with escape from recognition by human virus-specific CD4+ T-cells. Virus Res. 2007;126:282–7. doi: 10.1016/j.virusres.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 106.Yang J, et al. CD4+ T cells recognize unique and conserved 2009 H1N1 influenza hemagglutinin epitopes after natural infection and vaccination. Int Immunol. 2013 doi: 10.1093/intimm/dxt005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Price GE, et al. Single-dose mucosal immunization with a candidate universal influenza vaccine provides rapid protection from virulent H5N1, H3N2 and H1N1 viruses. PLoS One. 2010;5:e13162. doi: 10.1371/journal.pone.0013162. [DOI] [PMC free article] [PubMed] [Google Scholar]