

Neurodegenerative diseases are the sixth leading cause of death in the United States,1 but unlike other major causes of mortality, no significant disease-modifying therapies exist. Moreover, the number of patients with neurodegenerative disease is expected to nearly triple by 2050.2 Without effective therapies, these disorders will devastate our health care systems, families, and millions of patients. The lack of treatments is not for lack of effort. In fiscal year 2011, the NIH spent $713 million on neurodegenerative disease research (http://report.nih.gov/index.aspx) and PubMed lists over 227,000 studies on neurodegeneration. So why are there no effective treatments?
While many challenges to drug development for neurodegenerative diseases have been suggested,3–6 we suspect that a fundamental problem in trial design remains a critical factor. Trials have largely recruited patients by clinical features, including symptoms and neurologic examination findings. Although this allows maximum study generalizability by testing patients according to their neurodegeneration syndrome, it assumes that most participants share the same underlying disease mechanism or common pathway, or that the trial drug will influence disease progression despite a wide variety of disease mechanisms. These are risky propositions because patients can share a syndrome that results from common alterations in brain function but have drastically different underlying mechanisms of neurodegeneration. For example, loss of substantia nigra neurons leading to parkinsonism may result from (1) mutations in genes that regulate protein degradation (e.g., PARK27), (2) mutations in genes controlling mitochondrial turnover (e.g., PINK18), (3) mutations in genes with as yet unclear function (e.g., LRRK29), (4) exposure to toxins, such as MPTP,10 and (5) unknown causes (sporadic disease). Just as boosting PARK2 levels would not prevent Parkinson disease in those without PARK2 mutations, an agent targeting only one pathway in a clinical trial may influence only a small subset of study participants. Because trials are powered with the assumption that all patients are potentially responsive, a drug affecting a subset of patients might be wrongly labeled as ineffective, killing future research. Such assumptions may delay the field for decades.
How can we select trial participants to maximize our chances of success? Recent trials have used CSF and neuroimaging biomarkers to study patients with specific neuropathologies. For example, many Aβ-neutralizing antibody studies are now only performed in patients with amyloid plaques.11 Although this trial design represents an exciting advance, these studies assume that plaques identify patients whose disease is caused by Aβ. Alternatively, as evidenced by Aβ deposits in unimpaired elderly adults,12 Aβ may be present but not mechanistically linked to disease in some patients. Trials that group patients by and target Aβ, tau, or other biomarkers risk failure if these proteins are coincident with—rather than causative of—disease.
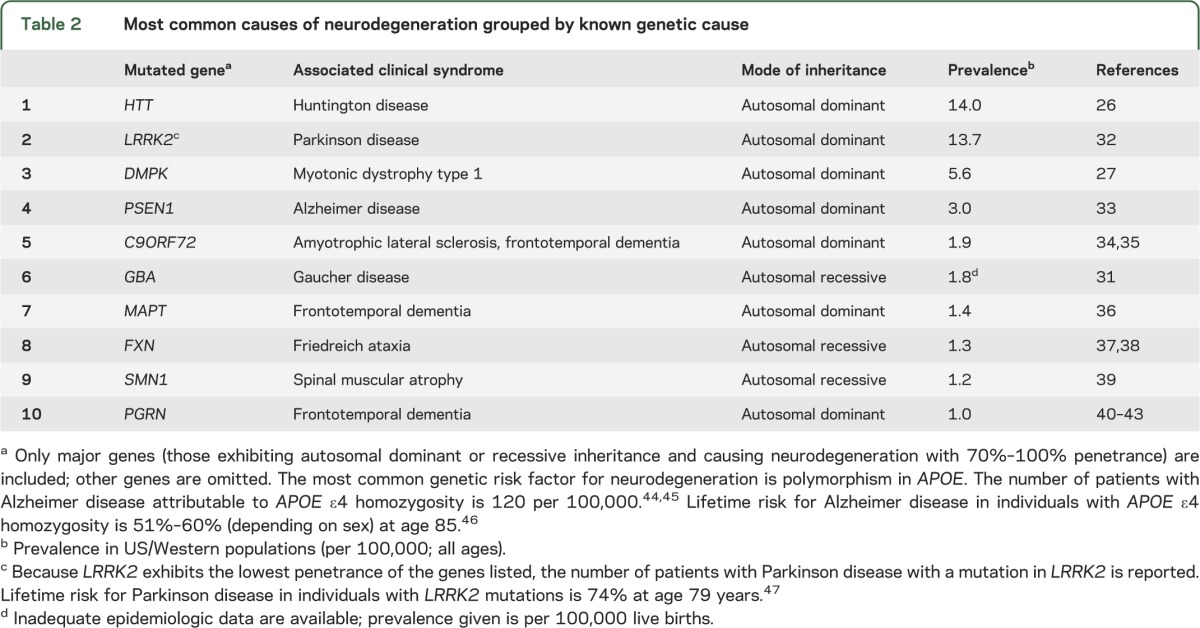
As an alternative to enrolling patients using only clinical features or biomarkers, we propose studying patients with a known mechanistic trigger—patients with a gene mutation that causes a well-defined clinical syndrome. In the past decade, many highly penetrant mutations have been identified.13,14 Many are rare compared to the total number of patients with a given neurodegenerative disease (table 1), but some are sufficiently prevalent to enroll patients on the basis of genotype (table 2).
Table 1.
Most common causes of neurodegeneration grouped by clinical syndrome
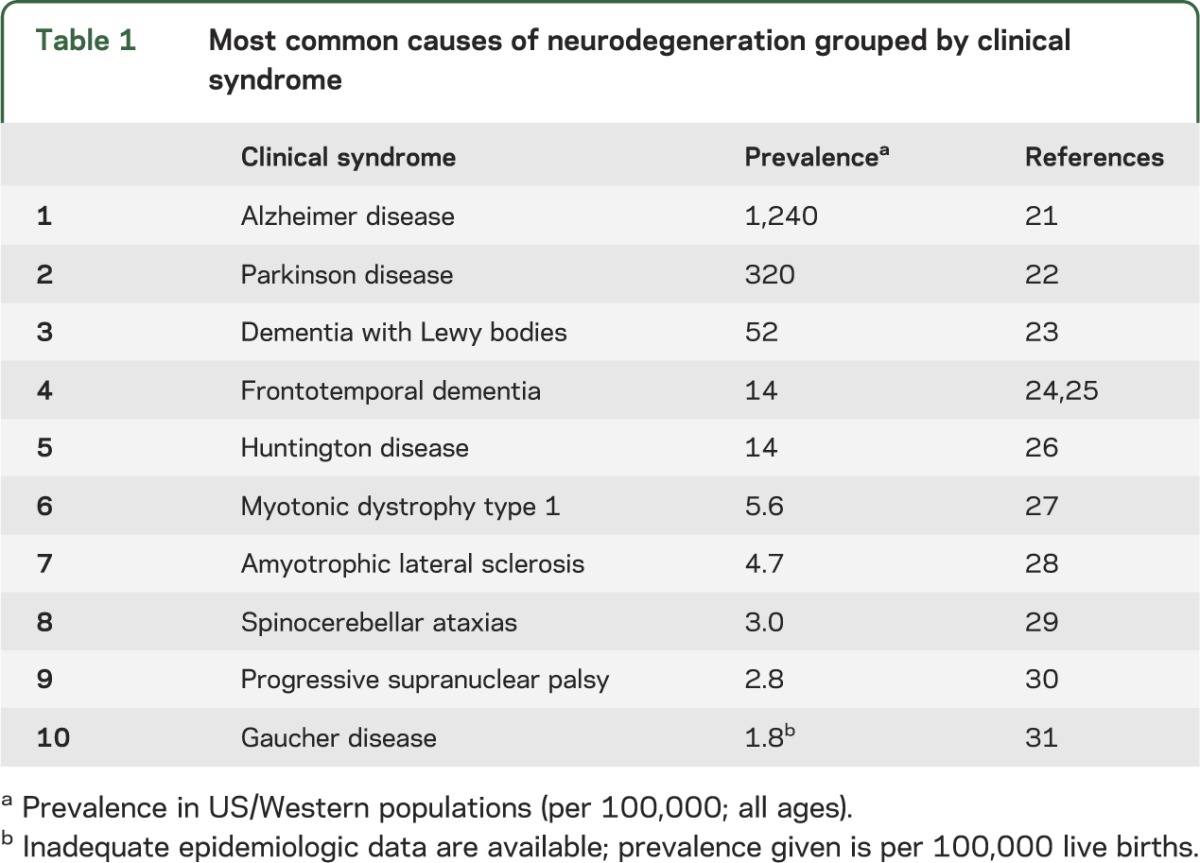
Table 2.
Most common causes of neurodegeneration grouped by known genetic cause
A genetic approach to drug development and trial design has achieved impressive results for non-neurologic disorders. For example, ivacaftor is effective in a small subset of patients with cystic fibrosis (CF). Around 90% of patients with CF express little cell-surface CF transmembrane conductance regulator (CFTR); however, the CF-evoking mutation CFTR G551D results in a poorly conducting but correctly localized channel in 4% of patients. Ivacaftor enhances CFTR conductance and thus lung function in G551D patients.15,16 Trastuzumab, a monoclonal antibody against HER2, is another genetically targeted therapy. Trastuzumab significantly extends life in patients with HER2-positive tumors.17 Importantly, trastuzumab would not have shown efficacy in a clinical trial enrolling all patients with breast cancer as only 20% of breast cancer patients are HER2-positive.18
Developing and testing therapeutics based on mutation status is not without challenges. First, drug discovery is never straightforward, even in diseases with a known genetic target. Many causative genes in neurodegeneration are scaffolds, structural proteins, and other nonenzymes that are difficult to target directly. Even after a suitable target has been identified, extensive pharmacologic and chemical studies are required. However, several new technologies hold promise for genetic disease, including antisense oligonucleotides (ASO). For patients with known mutations, ASO bypass the challenging target identification process as they allow allele-specific knockdown of virtually any gene. With ASO, any gain-of-function neurodegenerative disease gene—and even some loss-of-function genes— is “druggable.” In 2014, a phase III clinical trial will assess the use of ASO to treat spinal muscular atrophy (http://quest.mda.org/news/sma-isis-smnrx-shows-benefit-infants-children), a disorder caused by loss-of-function mutations in SMN1; in this case, the ASO rescue loss of SMN1 by promoting alternative splicing and therefore full activity of a conserved gene, SMN2. More trials, including one in Huntington disease, are expected in 2015.
After drug discovery, clinical trials for genetic forms of neurodegeneration will present more challenges. While most neurodegenerative disorders have a familial form caused by known gene mutations, these inherited variants constitute a small fraction of total cases (tables 1 and 2). It is thus costly and time-consuming to recruit patients by genotype. One strategy, which could partially mitigate this problem, is to incorporate substudies of patients with genetic disease into larger trials including mostly sporadic patients. Another innovative trial design, the adaptive I-SPY 2 trial design, which assigns breast cancer patients to multiple different treatment arms according to the mutations found in their tumors,19 could be applied to neurodegeneration and may further improve trial outcomes. Another challenge is that studies or substudies of patients with genetic forms of neurodegeneration will necessarily be small; therefore, these studies will be powered to detect only relatively large effects. We do not see this as a disadvantage; the identification of therapies with large effects should be a priority in neurodegenerative disease research. Finally, the relatively modest financial reward for targeting uncommon genetic diseases may dissuade investment. However, many large biotechnology companies are increasing their focus on rare diseases, and some companies have successful drug portfolios solely targeting rare diseases. The potential efficacy and profitability of targeting genetically at-risk populations have already encouraged biotechnology leaders to start grouping patients by genotype. For example, Genentech's upcoming trial of crenezumab will include 100 Colombian patients with mutations in PSEN1 (http://www.clinicaltrials.gov/ct2/show/NCT01998841). Additionally, the Dominantly Inherited Alzheimer Network is recruiting patients with known genetic causes of Alzheimer disease to participate in trials and tissue banking. An important trial examining an anti-Aβ antibody (solanezumab) in patients with mutations leading to increased Aβ fragments (APP and PSEN1 mutations) is underway (http://www.clinicaltrials.gov/show/NCT01760005). Both of these trials are enrolling very young patients who are either asymptomatic or mildly symptomatic; one advantage of studying patients with genetic disease is the ability to identify those who will develop neurodegeneration before symptoms appear, as early intervention may be crucial in halting the disease process.
Identifying a disease-modifying therapy—in any population regardless of size—is the most pressing issue in neurodegeneration research. Prior trials have prioritized study designs that would impact a large number of patients, but it may be time to retarget the lowest hanging fruit, no matter how modest the size. We do not advocate ceasing clinical trials in patients with sporadic forms of neurodegeneration—trials in genetic and sporadic disease are not mutually exclusive—but we do advocate devoting more resources and time to trials of patients with genetic disease. If the first effective treatment for neurodegeneration is identified in patients with genetic disease, it may prove efficacious in a broader population of patients who share the same underlying disease mechanism as the genetic patients. This has previously been seen with familial hypercholesterolemia and statin treatment.20 A successful therapy will also catalyze renewed academic and industry investment in translational neurodegeneration research. Thus, we are optimistic that effective therapies for neurodegeneration can be identified through clinical trials designed with the best chance for success.
Acknowledgments
Acknowledgment: A. Mason, A. Ziemann, and S. Finkbeiner thank R. Albin, G. Block, L. Bruijn, J. Dunlop, S. Freedman, S. Landis, R. Mahley, G. Seabrook, M. Sutherland, R.S. Williams, and members of the Finkbeiner Laboratory for discussions or comments on this article, G. Howard for editorial assistance, and K. Nelson for administrative assistance.
Footnotes
Author contributions: A. Mason wrote the manuscript and tables. A. Ziemann helped revise the manuscript and provided comments. S. Finkbeiner had the idea for the manuscript's argument and provided comments.
Study funding: CIRM TG2-01153, CIRM RB4-06079, NIH T32HD007470, NIH T32 GM007618, the Huntington's Disease Society of America, the Hereditary Disease Foundation, the Michael J. Fox Foundation, the ALS Association, the Packard Center, Target ALS, the Taube/Koret Center for Neurodegenerative Disease Research, the Hellman Foundation Program in Alzheimer's Disease Research, and the J. David Gladstone Institutes.
Disclosure: A. Mason is supported by CIRM TG2-01153, NIH T32HD007470, and NIH T32 GM007618. A. Ziemann is currently employed by Lundbeck, Deerfield, IL. S. Finkbeiner's funding includes CIRM RB4-06079, the Huntington's Disease Society of America, the Hereditary Disease Foundation, the Michael J. Fox Foundation, the ALS Association, the Packard Center, Target ALS, the Taube/Koret Center for Neurodegenerative Disease Research, the Hellman Foundation Program in Alzheimer's Disease, and the J. David Gladstone Institutes. Go to Neurology.org for full disclosures.
References
- 1.Heron M. Deaths: leading causes for 2009. Natl Vital Stat Rep 2012;61:1–94 [PubMed] [Google Scholar]
- 2.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Archi Neurol 2003;60:1119–1122 [DOI] [PubMed] [Google Scholar]
- 3.Finkbeiner S. Bridging the valley of death of therapeutics for neurodegeneration. Nat Med 2010;16:1227–1232 [DOI] [PubMed] [Google Scholar]
- 4.Smith Y, Wichmann T, Factor SA, DeLong MR. Parkinson's disease therapeutics: new developments and challenges since the introduction of levodopa. Neuropsychopharmacology 2012;37:213–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iqbal K, Grundke-Iqbal I. Opportunities and challenges in developing Alzheimer disease therapeutics. Acta Neuropathol 2011;122:543–549 [DOI] [PubMed] [Google Scholar]
- 6.Huey ED, Armstrong N, Momeni P, Grafman J. Challenges and new opportunities in the investigation of new drug therapies to treat frontotemporal dementia. Expert Opin Ther Targets 2008;12:1367–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605–608 [DOI] [PubMed] [Google Scholar]
- 8.Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004;304:1158–1160 [DOI] [PubMed] [Google Scholar]
- 9.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004;44:601–607 [DOI] [PubMed] [Google Scholar]
- 10.Ballard PA, Tetrud JW, Langston JW. Permanent human parkinsonism due to 1-methy 1-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): seven cases. Neurology 1985;35:949–949 [DOI] [PubMed] [Google Scholar]
- 11.Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol 2010;9:363–372 [DOI] [PubMed] [Google Scholar]
- 12.Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol 2008;65:1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol 2013;9:445–454 [DOI] [PubMed] [Google Scholar]
- 14.Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 2012;8:423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramsey B, Davies J, McElvaney G, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. NEJM 2011;365:1663–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ledford H. Drug bests cystic-fibrosis mutation. Nature 2012;482:145. [DOI] [PubMed] [Google Scholar]
- 17.Norum J, Risberg T, Olsen J. A monoclonal antibody against HER-2 (trastuzumab) for metastatic breast cancer: a model-based cost-effectiveness analysis. Ann Oncol 2005;16:909–914 [DOI] [PubMed] [Google Scholar]
- 18.Slamon DJ, Godolphin W, Jones L, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989;244:707–712 [DOI] [PubMed] [Google Scholar]
- 19.Printz C. I-SPY 2 may change how clinical trials are conducted. Cancer 2013;119:1925–1927 [DOI] [PubMed] [Google Scholar]
- 20.Vega GL, Scott M. Treatment of hypercholesterolemia with lovastatin (mevinolin) and colestipol. JAMA 1987;257:33–38 [PubMed] [Google Scholar]
- 21.WHO. Dementia: a public health priority [Internet]. 2012. Available at: http://apps.who.int/iris/bitstream/10665/75263/1/9789241564458_eng.pdf?ua=1. Accessed June 26, 2014
- 22.De Lau L, Breteler M. Epidemiology of Parkinson's disease. Lancet Neurol 2006;5:525–535 [DOI] [PubMed] [Google Scholar]
- 23.Zaccai J, McCracken C, Brayne C. A systematic review of prevalence and incidence studies of dementia with Lewy bodies. Age Ageing 2005;34:561–566 [DOI] [PubMed] [Google Scholar]
- 24.Onyike C, Diehl-Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry 2013;25:130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knopman DS, Roberts RO. Estimating the number of persons with frontotemporal lobar degeneration in the US population. J Mol Neurosci 2011;45:330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fisher ER, Hayden MR. Multisource ascertainment of Huntington disease in Canada: prevalence and population at risk. Mov Disord 2014;29:105–114 [DOI] [PubMed] [Google Scholar]
- 27.Monckton G, Hoskin V, Warren S. Prevalence and incidence of muscular dystrophy in Alberta, Canada. Clin Genet 1982;21:19–24 [DOI] [PubMed] [Google Scholar]
- 28.Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Incidence and prevalence of ALS in Ireland, 1995–1997: a population-based study. Neurology 1999;52:504–509 [DOI] [PubMed] [Google Scholar]
- 29.Van de Warrenburg BPC, Sinke RJ, Verschuuren-Bemelmans CC, et al. Spinocerebellar ataxias in the Netherlands: prevalence and age at onset variance analysis. Neurology 2002;58:702–708 [DOI] [PubMed] [Google Scholar]
- 30.Nath U, Ben-Shlomo Y, Thomson RG, et al. The prevalence of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) in the UK. Brain 2001;124:1438–1449 [DOI] [PubMed] [Google Scholar]
- 31.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281:249–254 [DOI] [PubMed] [Google Scholar]
- 32.Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 2009;18:R48–R59 [DOI] [PubMed] [Google Scholar]
- 33.Campion D, Dumanchin C, Hannequin D, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet 1999;65:664–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11:323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van der Zee J, Gijselinck I, Dillen L, et al. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat 2013;34:363–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat 2004;24:277–295 [DOI] [PubMed] [Google Scholar]
- 37.Leone M, Brignolio F, Rosso MG, et al. Friedreich's ataxia: a descriptive epidemiological study in an Italian population. Clin Genet 2008;38:161–169 [DOI] [PubMed] [Google Scholar]
- 38.López-Arlandis JM, Vilchez JJ, Palau F, Sevilla T. Friedreich's ataxia: an epidemiological study in Valencia, Spain, based on consanguinity analysis. Neuroepidemiology 1995;14:14–19 [DOI] [PubMed] [Google Scholar]
- 39.Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet 1978;15:409–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu C-E, Bird TD, Bekris LM, et al. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol 2010;67:161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 2006;15:2988–3001 [DOI] [PubMed] [Google Scholar]
- 42.Le Ber I, van der Zee J, Hannequin D, et al. Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat 2007;28:846–855 [DOI] [PubMed] [Google Scholar]
- 43.Pickering-Brown S, Rollinson S, Du Plessis D, et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 2008;131:721–731 [DOI] [PubMed] [Google Scholar]
- 44.Ward A, Crean S, Mercaldi CJ, et al. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer's disease: a systematic review and meta-analysis. Neuroepidemiology 2012;38:1–17 [DOI] [PubMed] [Google Scholar]
- 45.Tang MX, Maestre G, Tsai WY, et al. Relative risk of Alzheimer disease and age-at-onset distributions, based on APOE genotypes among elderly African Americans, Caucasians, and Hispanics in New York City. Am J Hum Genet 1996;58:574–584 [PMC free article] [PubMed] [Google Scholar]
- 46.Genin E, Hannequin D, Wallon D, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry 2011;16:903–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Healy DG, Falchi M, O'Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol 2008;7:583–590 [DOI] [PMC free article] [PubMed] [Google Scholar]