

Abstract
The cyclic-AMP response element-binding protein (CREB) is a nuclear transcription factor activated by phosphorylation at Ser133 by multiple serine/threonine (Ser/Thr) kinases. Upon phosphorylation, CREB binds the transcriptional co-activator, CBP (CREB-binding protein), to initiate CREB-dependent gene transcription. CREB is a critical regulator of cell differentiation, proliferation and survival in the nervous system. Recent studies have shown that CREB is involved tumor initiation, progression and metastasis, supporting its role as a proto-oncogene. Overexpression and over-activation of CREB were observed in cancer tissues from patients with prostate cancer, breast cancer, non-small-cell lung cancer and acute leukemia while down-regulation of CREB in several distinct cancer cell lines resulted in inhibition of cell proliferation and induction of apoptosis, suggesting that CREB may be a promising target for cancer therapy. Although CREB, as a transcription factor, is a challenging target for small molecules, various small molecules have been discovered to inhibit CREB phosphorylation, CREB-DNA, or CREB-CBP interaction. These results suggest that CREB is a suitable transcription factor for drug targeting and therefore targeting CREB could represent a novel strategy for cancer therapy.
Keywords: Cancer, CBP, CREB, inhibitors, KID, KIX, naphthol AS-E, Ro 31-8220
INTRODUCTION
The cyclic AMP (cAMP) response element binding protein (CREB) was first identified in 1987 during an investigation for nuclear proteins that bind to a stretch of DNA containing the cAMP response element (CRE) 5′-TGACGTCA-3′ [1, 2]. This 43 KDa protein belongs to a large family of basic leucine zipper (bZIP)-containing transcription factors including c-Jun, c-Fos and c-Myc (Fig. 1) [3, 4]. A salient feature of this transcription factor is that its transcriptional activity is induced upon phosphorylation at Ser133 located in the KID (kinase-inducible domain) domain by many different serine/threonine (Ser/Thr) protein kinases to yield phosphorylated CREB (p-CREB) [4]. The kinases known to phosphorylate CREB include protein kinase A (PKA) [1], Akt/protein kinase B (PKB) [5], mitogen-activated protein kinases (MAPK) [6, 7], and p90 ribosomal S6 kinase (p90RSK) (Fig. 2) [8]. Phosphorylation at Ser133 is required for its binding to the mammalian transcriptional co-activator, CREB-binding protein (CBP), through the KID domain in CREB and KIX (KID-interacting) domain in CBP [9]. This binding event enables recruitment of other transcriptional machinery to the gene promoter to initiate CREB-dependent gene transcription (Fig. 2) [4]. In addition to Ser133, many other sites in CREB are also known to be phosphorylated by a variety of kinases [10]. Although the biological relevance of these additional phosphorylation events is less well understood, they could potentially regulate CREB’s transcription activity in a more delicate fashion [10]#.
Fig. 1.

Domain structure of rat CREB. The basic leucine zipper (bZIP) is located at the C-terminus for DNA binding and homodimerization or heterodimerization with other bZIP family members. The kinase-inducible domain (KID) is the inducible activation domain activated by phosphorylation at Ser133. The glutamine rich regions [Q1 and Q2 or constitutive activation domain (CAD)] are the basal transcriptional activation domains. Human CREB lacks the α domain, which forms a presumed amphipathic α-helix to regulate the transcription activity of CREB.
Fig. 2.

Multiple signaling pathways activate CREB by phosphorylation. Illustrated are potential points of intervention by chemical inhibitors: 1) inhibition of kinases; 2) inhibition of CREB-CRE interaction; and 3) inhibition of CREB-CBP interaction.
CREB serves a variety of biological functions including cellular proliferation, differentiation and adaptive responses in the neuronal system [4, 11]. Recently, accumulating evidence has revealed that CREB participates in the regulation of immortalization and transformation of cancer cells. Therefore it is hypothesized that CREB is directly involved in oncogenesis of a variety of cancers [12–14].
CREB IN HUMAN MALIGNANCIES
The first indication of CREB’s involvement in cancer stemmed from the identification of a chromosomal t(12;22)(q13;q12) translocation in clear cell sarcomas of soft tissue (CCSST) to give a fusion protein EWS-ATF1 [15]. In the resulting chimeric protein, the N-terminal region of EWS (Ewing’s Sarcoma) [16], an RNA-binding protein, is fused with the C-terminal region (bZIP) of ATF-1 (activating transcription factor 1), a CREB related transcription factor which binds CRE and heterodimerizes with CREB [17]. This fusion protein loses a consensus PKA phosphorylation site mediating regulation by PKA and other kinases for transcription activation. Instead, the activation domain of the N-terminal region of EWS renders this fusion as a constitutively active transcription activator to upregulate the expression of some CREB target genes [18, 19]. This gene translocation is present in nearly all the CCSSTs [15, 18].
Immunohistochemical analysis of primary and bone metastatic prostate cancer tissues from patients demonstrated that normal, benign prostate glands showed no detectable p-CREB [20]. On the other hand, positive p-CREB staining was detected in all the examined poorly-differentiated prostate cancers and bone metastatic tissue specimens [20]. This positive correlation between the level of p-CREB and the extent of tumor differentiation and metastasis suggests that CREB is critically involved in tumor progression and metastasis. In prostate cancer LNCaP cells, activation of CREB has been implicated in the promotion of neuroendocrine differentiation (NED) induced by ionizing radiation [21] and elevated intracellular cAMP level [22, 23]. The neuroendocrine transdifferentiation process appears to be associated with the development of cross-resistance to radiotherapy, chemotherapy and androgen-independence in prostate cancers [21, 24]. In addition to prostate cancer, increased mRNA levels of CREB were also consistently detected in breast cancer tissues compared to normal mammary tissues [25]. Notably, the level of CREB expression was correlated with disease progression and survival [25]. In non-small-cell lung cancer (NSCLC) never-smoking patients, the expression levels of CREB and p-CREB were distinctively elevated in tumor tissues compared to the adjacent normal tissues [26]. In these examined patient samples, there exists an inverse correlation between CREB overexpression and disease-free survival [26]. In an animal model of lung adenocarcinoma induced by tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), the expression level of p-CREB progressively increased along the development of lung adenocarcinoma [27]. Similar observations were made in human lung adenocarcinoma NCI-H322 cells, where NKK potently stimulated phosphorylation of CREB and DNA synthesis [28].
Besides its role in the development of solid tumors, mounting evidence has shown that CREB may also play a role in the development of bone marrow neoplasms. The oncogenic virus human T-cell leukemia virus type 1 (HTLV-1) is strongly associated with aggressive cases of adult T-cell leukemia (ATL) [29, 30]. The oncogenic transformation caused by the HTLV-1 Tax oncoprotein requires intact CREB signaling [31, 32]. Furthermore, the role of CREB itself as a proto-oncogene in leukemogenesis is supported by studies of leukemia patient samples and leukemia cell cultures [33–36]. Higher CREB and p-CREB expression levels were detected in the bone marrow from patients with ALL (acute lymphoid leukemia) and AML (acute myeloid leukemia) compared to that from patients without neoplastic hematologic disorders [35, 36]. The degree of increased p-CREB level does not correlate with the cAMP levels in the leukemia patient samples [36], suggesting CREB kinases other than PKA are contributing to the enhanced phosphorylation of CREB. In addition, CREB expression clearly correlates with disease stage in the leukemia patients where high expression was observed at diagnosis and relapse, but low expression was seen upon remission from the same patients [35, 36]. Kaplan-Meier analysis showed that CREB overexpression is also associated with a decrease in time to relapse and a decrease in event-free survival [33]. On the flip side, the expression profiles of inhibitors of CREB, including ICER (inducible cyclic AMP early repressor) [36] and miR-34b (a microRNA targeting CREB) [37] are down-regulated in the bone marrow of AML patients compared to healthy individuals. These empirical expression data from human patient samples have been supported by animal models. The transgenic mice that overexpress CREB in the cells of the myeloid lineage developed increased monocytosis and myeloproliferative syndrome with splenomegaly [33]. The bone marrow cells from these transgenic mice also displayed transformed blast-cell phenotypes including increased proliferation, immortalization and growth factor-independence [33]. Overall, the data from both clinical samples and animal models indicate that CREB not only serves as a diagnostic marker, due to its role in induction and maintenance of malignancy, but also an oncogene of its own accord when inappropriately activated.
TARGETING CREB FOR CANCER THERAPY: PROOF-OF-CONCEPT
The observation that CREB is overexpressed and/or over-activated in a variety of different clinical cancer tissues argues that CREB may represent a promising target for cancer therapy. Although the exact mechanisms by which CREB contributes to cancer development are not clear, CREB, as a transcription factor, directly regulates a number of critical genes involved in cellular proliferation, anti-apoptosis and metastasis. These targets include cyclin A1 [33], cyclin D1 [38], bcl-2 (b-cell leukemia 2) [33], VEGF (vascular endothelial growth factor) [20], type IV collagenase MMP-2 (matrix metalloproteinase 2) and cell adhesion molecule MUC18/MCAM (melanoma cell adhesion molecule) [39]. In hematopoietic cells, overexpression of CREB may also block cellular differentiation by up-regulating a member of the hox gene family, Meis1 (myeloid ecotropic viral integration site 1) [40, 41]. As mentioned earlier, CREB is also implicated in promoting prostate cancer cell NED.
The biological function of CREB, as a transcription factor, entails protein-DNA and protein-protein interactions. Both of these binding interfaces are challenging targets for rational small molecule design even though the high-resolution structures of CREB-CRE and KIX-KID complexes have been determined [9, 42]. Therefore, initial studies to test the hypothesis of targeting CREB as a strategy for cancer therapy are focused on biological approaches to inhibit CREB’s function.
Dominant-negative CREB mutants were employed to inhibit CREB’s transcription activity. A melanoma cell line (MeWo) was transfected with a dominant-negative CREB, KCREB, which is a full-length CREB with R287L mutation [43] and does not bind CRE but does heterodimerize with wild-type CREB [39, 44]. Expression of KCREB in the highly metastatic MeWo cell line resulted in 1) decreased potential for metastasis in vitro and in vivo [39]; 2) decreased tumor growth in the mouse xenograft model [39]; 3) decreased capability to grow in soft agar, indicating decreased transforming capacity [44]; and 4) reduced resistance to radiation [44]. These results are possibly due to the decreased expression of CREB target genes involved in metastasis, including type IV collagenase (MMP-2) and cell adhesion molecule MUC18/MCAM [39]. Similar tumor growth inhibition with dominant-negative CREB was also observed in hepatocellular carcinoma BNL1ME cells [45] and non-small-cell lung cancer A549 cells [46]. Inducible expression of a dominant-negative CREB mutant in the mouse basal epidermis significantly reduced the incidence of skin papillomas induced by sequential treatment with 7,12-dimethyl-benz[a]anthracene (DMBA) and phorbol-12-myristate-13-acetate, suggesting potential cancer preventive value of CREB inhibitors [47].
A second biological approach to inhibit CREB’s function was the utilization of CRE “decoy” oligonucleotides. Once inside cells, these “decoy” oligonucleotides can bind CREB and sequester it away from the genomic CRE sequences and thus effectively inhibit CREB-mediated gene transcription. Introduction of a 24-mer CRE “decoy” into a variety of solid tumor cell lines was shown to suppress the growth of cancer cells both in culture and in the xenograft models [48, 49]. On the other hand, delivery of the same CRE “decoy” oligonucleotide into normal cells did not result in toxicity [49]. These results further suggest that pharmacological inhibition of the transcription activity of CREB could be a promising strategy to develop next-generation nontoxic, anticancer drugs.
Finally, a third approach that was investigated for inhibition of CREB was RNA interference (RNAi). Our laboratory has recently shown that knockdown of CREB in human myeloid leukemia cells (K562 and TF-1) resulted in decreased cellular proliferation and viability in vitro [33, 50]. In a murine myeloid leukemia model, NOD-SCID (non-obese diabetic, severe combined immuodeficicent) mice injected with Ba/F3 cells transduced with Bcr-Abl and shRNA (short hairpin RNA) against CREB exhibited decreased disease burden and increased median survival [50]. More importantly, this effect was also observed in imatinib-resistant Bcr-AblT315I-expressing cells [50]. Knockdown of CREB by siRNA has also been shown to enhance oxidant- and asbestos-induced apoptosis [51, 52].
TARGETING CREB FOR CANCER THERAPY: SMALL MOLECULES
The aforementioned studies of inhibiting the transcriptional activity of CREB by various genetic methods provide proof-of-concept evidence that CREB is a promising target for anticancer drug development. However, direct translation of these methods for potential cancer therapies would require gene therapy techniques, whose clinical application is still rather limited and controversial [53]. An alternative approach to inhibit CREB-mediated gene transcription is to utilize small organic molecules, which, in general, have better pharmacokinetic properties as cancer therapeutics. Three potential intervention points for small molecules as chemical inhibitors of CREB-mediated gene transcription are depicted in Fig. (2).
The first approach of pharmacological inhibition of CREB-mediated gene transcription in cells involves the use of kinase inhibitors to inhibit phosphorylation and therefore activation of CREB. As presented in Fig. (2), multiple Ser/Thr protein kinases could phosphorylate CREB. Therefore, effective inhibition of CREB phosphorylation in cancer cells would require simultaneous inhibition of multiple CREB kinases either by combining different specific kinase inhibitors or employing non-specific kinase inhibitors. This polypharmacology approach may elicit many off-target effects [54]. However, promising results were obtained from Aggarwal et al who reported that Ro 31-8220 (Chart 1), an analog of staurosporine [55] with medium kinase selectivity,$ inhibited both CREB upstream and downstream signaling components in non-small-cell lung cancer cell lines (NSCLC) [56]. Specifically, phosphorylation of CREB kinase p90RSK and CREB in H1734 cells was inhibited by Ro 31-8220. The expression of CREB target genes involved in anti-apoptosis, Bcl-2 and Bcl-xL, was down-regulated in H1734 cells treated with Ro 31-8220. Furthermore, NSCLC cells treated with Ro 31-8220 exhibited caspase-dependent apoptosis while similar treatment in normal human tracheo-bronchial epithelial (NHTBE) cells did not result in apoptosis [56], suggesting that cancer cells are more sensitive to CREB inhibition. While these results are intriguing, it remains to be determined whether the observed cellular phenotype is a result of inhibition of CREB’s activity because of the moderate specificity profile of Ro 31-8220 against different kinases in cells.
Chart 1.

Chemical inhibitors of CREB-mediated gene transcription [56–59].
The second approach to inhibit CREB’s activity is to inhibit CREB-CRE interaction. Unlike enzymes which have a well-defined and relatively small-sized binding pocket for small molecule binding, protein-DNA interactions are traditionally thought to be difficult to be targeted by small molecules. However, a fluorescent polarization–based high throughput screening assay was designed to identify potential inhibitors of CREB-CRE interaction from the NCI-diversity set of ~1900 compounds [57]. NSC 12155 and NSC 45576 (Chart 1) were identified as submicromolar and low micromolar inhibitors of CREB-CRE interaction. However, these compounds are not specific in inhibiting CREB-CRE interaction and their cellular activity remains to be determined [57]. Another potential approach to target CREB-CRE interaction is to employ the programmable N-methylpyrrole-N-methylimidazole polyamides [60], which can be designed to target specific DNA sequences and have been engineered to inhibit HIF-1α (hypoxia inducible factor 1α) and androgen receptor [61, 62].
The third approach to the inhibition of CREB-mediated gene transcription is to target CREB-CBP interaction, which necessarily precedes CREB-dependent gene transcriptional activation [63]. The binding interface between CREB and CBP is KID-KIX interaction and it is structurally well-characterized by NMR spectroscopy [9], but targeting protein-protein interactions by small molecules is still challenging. However, the recent successful examples of small molecule inhibitors of various protein-protein interactions suggest this is feasible provided there are binding “hotspots” on the protein surfaces [64]. Two distinct binding sites on KIX are utilized by natural transcription factors [65, 66], suggesting its potential to be targeted by small molecules. Therefore, a medium-throughput NMR screening assay was designed to identify potential small molecule binders of KIX that could disrupt the CREB-CBP interaction [58]. From a pre-selected drug-like library of 762 compounds, naphthol AS-E phosphate (Chart 1) was identified as a candidate compound to inhibit the KID-KIX interaction with IC50 of ~90 μM. However, this same compound displayed significantly more potent activity in inhibiting CREB-mediated gene transcription in living cells [58]. Further studies in the authors’ laboratory showed that naphthol AS-E phosphate is not stable in serum or cell-permeable and the actual active species in the cellular experiments is the dephosphorylated product, naphthol AS-E (Chart 1), which is a much more potent inhibitor of the KID-KIX interaction [59]. The identification of this cell-permeable KID-KIX interaction inhibitor will enable us to further investigate the hypothesis that KID-KIX interaction inhibitors are potential anticancer agents.& Furthermore, the modular nature of naphthol AS-E is amenable to further structure-activity relationship studies to uncover the structural manipulations that would enhance their biological activities. Some other KIX-binding small molecules were also reported in the literature [67–69], however, their capability to inhibit CREB-mediated gene transcription in cells is unknown.
TARGETING CREB: GOOD OR BAD
mRNA expression data showed that CREB was expressed in all the adult tissues examined [70] and CREB is a focal point of many different signaling pathways (see Fig. 2) [4, 11]. Thus, specific and efficacious inhibition of CREB might invoke non-specific adverse effects in the normal cells. In fact, CREB-null mice are lethal at birth due to post-natal lung defects [71]. However, the centrality of CREB might be advantageous for anticancer drug design because cancer cells often have multiple lesions in different signaling pathways [72, 73] and targeting CREB could potentially block a number of these aberrantly activated pathways (see Fig. 2). Although genetic knockout data are critical in inferring the biological function of a gene of interest, one has to realize that the key differences between genetic knockouts and chemical inhibitors are that chemical inhibition of CREB is reversible, transient and does not change the endogenous CREB protein level. The residual CREB activity after chemical inhibition may be sufficient to maintain the normal cell homeostasis, but below the threshold to maintain the cancer cell phenotype. As a matter of fact, the hypomorphic CREB mice are viable without developmental defects [74].
Studies with CRE-decoy oligonucleotides demonstrated that normal cells are not sensitive to CREB inhibition [49]. The exact mechanisms underlying this selectivity remain to be determined, but one can conceptualize the selective toxicity to cancer cells over normal cells by CREB inhibition as in Fig. (3). Cancer cells acquire an incredible amount of apoptotic stress during malignant transformation by aberrant activation of oncogenes [72]. To prevent cell death, cancer cells are able to block apoptosis by up-regulating anti-apoptotic signals [72]. Since cancer is believed to be a product of evolutionary selection [72], there must be a reason for over-activation of CREB consistently seen in different tumor specimens. This may be that activated CREB transcribes its target genes required for anti-apoptosis to maintain the cancer phenotype. Endogenous genes are often regulated by more than one transcription factor. The contribution to a given transcript from CREB might be bigger in cancer cells with over-activated CREB than that in normal cells. Therefore, inhibitors of CREB-mediated gene transcription will have a more profound effect in cancer cells than normal cells, which will be sufficient to induce apoptosis in cancer cells. But the effect in normal cells is too small to induce an adverse effect due to redundancy (Fig. 3). Alternatively, promoter usage for a given gene in cancer cells may be different from that in normal cells in that CREB is predominantly driving transcription of a particular target gene that is required to maintain the cancer phenotype. A good example of this latter point is illustrated in the transcription of aromatase in different tissues, where CREB is major contributor in breast cancer cells but not in other cells [75]. Therefore, chemical inhibitors of CREB-mediated gene transcription could display anticancer activity with no or acceptable minimal toxicity.
Fig. 3.
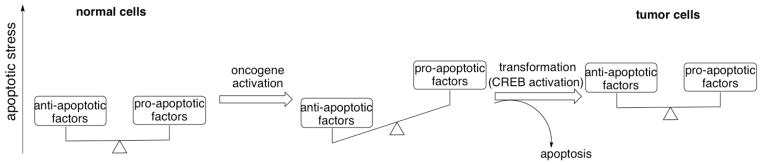
Cancer cells bear higher apoptotic stress than normal cells. Normal cells (left) maintain a homeostasis with low levels of pro-apoptotic factors and anti-apoptotic factors. Upon aberrant activation of oncogenes, the cells start to accumulate apoptotic stress (middle) and most of the cells will die. However, a small percentage of the cells will survive through activation or inactivation of other cell signaling components (e.g. activation of CREB) resulting in up-regulation of anti-apoptotic factors. These transformed cancer cells (right) will maintain this new cellular homeostasis with overall higher levels of both pro-apoptotic factors and anti-apoptotic factors than their normal counterparts.
CONCLUSIONS AND OUTLOOK
In conclusion, there is a great deal of evidence to implicate CREB as an important regulator of tumor initiation, progression and metastasis. Proof-of-principle studies using various strategies have demonstrated therapeutic potential of inhibitors of CREB-mediated gene transcription. In the coming years, we anticipate that more chemical inhibitors will be developed to allow investigators to further test the hypothesis of CREB inhibitors for cancer therapy in preclinical and clinical settings. However, the following challenges need to be addressed in order to fully appreciate the clinical utility of any chemical inhibitor of CREB-mediated gene transcription: 1) What are the mechanisms responsible for the anti-cancer activity/selectivity of CREB inhibitors? 2) How specific are the small molecule inhibitors in inhibiting CREB-mediated gene transcription? 3) What patients are appropriate for the trials of CREB inhibitors? 4) How and whether these inhibitors should be used in combination with other existing cancer therapies? 5) What are the endpoint markers to evaluate the efficacy and on-target effects? The discovery of various inhibitors presented in Chart 1 represents just the opening chapter of an entire book of a novel strategy for cancer therapy requiring more extensive investigations.
Acknowledgments
X.X. wants to thank financial supports from Oregon Health & Science University and Medical Research Foundation of Oregon. K.M.S. is supported by the Leukemia and Lymphoma Society of America, National Institutes of Health (NIH) HL75826 and HL83077, William Lawrence and Blanche Hughes Foundation, and the St. Baldrick’s Foundation. A.I. is supported by NIH HD034610-12/UCLA Child Health Research Career Development Award and the Couples Against Leukemia/Ronald McDonald House Children’s Cancer Research Fund 20091869. B.M. is a recipient of the American Academy of Pediatrics Resident Research Award.
ABBREVIATIONS
- Abl
Abelson murine leukemia viral oncogene homolog
- ALL
Acute lymphoid leukemia
- AML
Acute myeloid leukemia
- ATF-1
Activating transcription factor 1
- ATL
Adult T-cell leukemia
- Bcr
Breakpoint cluster region
- bcl-2
b-cell leukemia 2
- bZIP
Basic leucine zipper
- CBP
CREB binding protein
- CCSST
Clear cell sarcomas of soft tissue
- CRE
Cyclic-AMP response element
- CREB
CRE-binding protein
- DMBA
7,12-dimethylbenz[a]anthracene
- EWS
Ewing’s Sarcoma
- HIF-1α
Hypoxia inducible factor 1α
- HTLV-1
Human T-cell leukemia virus type 1
- ICER
Inducible cyclic AMP early repressor
- KID
Kinase-inducible domain
- KIX
KID-interacting
- MAPK
Mitogen-activated protein kinase
- MCAM/MUC18
Melanoma cell adhesion molecule
- Meis1
Myeloid ecotropic viral integration site 1
- MMP-2
Matrix metalloproteinase 2
- NCI
National Cancer Institute
- NED
Neuroendocrine differentiation
- NMR
Nuclear magnetic resonance
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NSCLC
Non-small-cell lung cancer
- p-CREB
Phosphorylated CREB
- PKA
Protein kinase A
- PKB/Akt
Protein kinase B
- p90RSK
p90 ribosomal S6 kinase
- VEGF
Vascular endothelial growth factor
Footnotes
The remaining discussion of phosphorylation of CREB in this article refers to Ser133.
see technical article of Ro 31-8220 at www.sigma-aldrich.com
Li, B.X., Bryant, D., Yamanaka, K., Dorsa, D., Xiao, X. In 238th ACS National Meeting: Washington, DC, United States, 2009.
References
- 1.Montminy MR, Bilezikjian LM. Binding of a nuclear-protein to the cyclic-AMP response element of the somatostatin gene. Nature. 1987;328:175–178. doi: 10.1038/328175a0. [DOI] [PubMed] [Google Scholar]
- 2.Montminy MR, Sevarino KA, Wagner JA, Mandel G, Goodman RH. Identification of a cyclic-AMP-responsive element within the rat somatostatin gene. Proc Natl Acad Sci USA. 1986;83:6682–6686. doi: 10.1073/pnas.83.18.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vinson CR, Sigler PB, McKnight SL. Scissors-grip model for DNA recognition by a family of leucine zipper proteins. Science. 1989;246:911–916. doi: 10.1126/science.2683088. [DOI] [PubMed] [Google Scholar]
- 4.Shaywitz AJ, Greenberg ME. CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 5.Du KY, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- 6.Tan Y, Rouse J, Zhang AH, Cariati S, Cohen P, Comb MJ. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996;15:4629–4642. [PMC free article] [PubMed] [Google Scholar]
- 7.Ginty DD, Bonni A, Greenberg ME. Nerve growth factor activates a Ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell. 1994;77:713–725. doi: 10.1016/0092-8674(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 8.Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 9.Radhakrishnan I, PerezAlvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator:Coactivator interactions. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 10.Johannessen M, Moens U. Multisite phosphorylation of the cAMP response element-binding protein (CREB) by a diversity of protein kinases. Front Biosci. 2007;12:1814–1832. doi: 10.2741/2190. [DOI] [PubMed] [Google Scholar]
- 11.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 12.Conkright MD, Montminy M. CREB: the unindicted cancer co-conspirator. Trends Cell Biol. 2005;15:457–459. doi: 10.1016/j.tcb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 13.Sakamoto KM, Frank DA. CREB in the pathophysiology of cancer: implications for targeting transcription factors for cancer therapy. Clin Cancer Res. 2009;15:2583–2587. doi: 10.1158/1078-0432.CCR-08-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandoval S, Pigazzi M, Sakamoto KM. CREB: A Key Regulator of Normal and Neoplastic Hematopoiesis. Adv Hematol. 2009;2009:634292. doi: 10.1155/2009/634292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zucman J, Delattre O, Desmaze C, Epstein AL, Stenman G, Speleman F, Fletchers CDM, Aurias A, Thomas G. EWS and ATF-1 gene fusion induced by T(12,22) translocation in malignant melanoma of soft parts. Nat Genet. 1993;4:341–345. doi: 10.1038/ng0893-341. [DOI] [PubMed] [Google Scholar]
- 16.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, Dejong P, Rouleau G, Aurias A, Thomas G. Gene fusion with an ETS DNA-binding domain caused by chromosone-translocation in human tumors. Nature. 1992;359:162–165. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- 17.Hai TW, Liu F, Coukos WJ, Green MR. Transcription factor ATF cDNA clones--an extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes Dev. 1989;3:2083–2090. doi: 10.1101/gad.3.12b.2083. [DOI] [PubMed] [Google Scholar]
- 18.Schaefer KL, Brachwitz K, Wai DH, Braun Y, Diallo R, Korsching E, Eisenacher M, Voss R, van Valen F, Baer C, Selle B, Spahn L, Liao SK, Lee KAW, Hogendoorn PCW, Reifenberger G, Gabbert HE, Poremba C. Expression profiling of t(12;22) positive clear cell sarcoma of soft tissue cell lines reveals characteristic up-regulation of potential new marker genes including ERBB3. Cancer Res. 2004;64:3395–3405. doi: 10.1158/0008-5472.CAN-03-0809. [DOI] [PubMed] [Google Scholar]
- 19.Fujimura Y, Ohno T, Siddique H, Lee L, Rao VN, Reddy ESP. The EWS-ATF-1 gene involved in malignant melanoma of soft parts with t(12;22) chromosome translocation, encodes a constitutive transcriptional activator. Oncogene. 1996;12:159–167. [PubMed] [Google Scholar]
- 20.Wu W, Zhau HE, Huang WC, Iqbal S, Habib FK, Sartor O, Cvitanovic L, Marshall FF, Xu Z, Chung LWK. cAMP-responsive element-binding protein regulates vascular endothelial growth factor expression: implication in human prostate cancer bone metastasis. Oncogene. 2007;26:5070–5077. doi: 10.1038/sj.onc.1210316. [DOI] [PubMed] [Google Scholar]
- 21.Deng X, Liu H, Huang J, Cheng L, Keller ET, Parsons SJ, Hu CD. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Cancer Res. 2008;68:9663–9670. doi: 10.1158/0008-5472.CAN-08-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bang YJ, Pirnia F, Fang WG, Kang WK, Sartor O, Whitesell L, Ha MJ, Tsokos M, Sheahan MD, Nguyen P, Niklinski WT, Myers CE, Trepel JB. Terminal neuroendocrine differentiation of human prostate carcinoma cells in response to increased intracellular cyclic AMP. Proc Natl Acad Sci USA. 1994;91:5330–5334. doi: 10.1073/pnas.91.12.5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan TC, Veeramani S, Lin MF. Neuroendocrine-like prostate cancer cells: neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocr Relat Cancer. 2007;14:531–547. doi: 10.1677/ERC-07-0061. [DOI] [PubMed] [Google Scholar]
- 24.Deeble PD, Cox ME, Frierson HF, Jr, Sikes RA, Palmer JB, Davidson RJ, Casarez EV, Amorino GP, Parsons SJ. Androgen-independent growth and tumorigenesis of prostate cancer cells are enhanced by the presence of PKA-differentiated neuroendocrine cells. Cancer Res. 2007;67:3663–3672. doi: 10.1158/0008-5472.CAN-06-2616. [DOI] [PubMed] [Google Scholar]
- 25.Chhabra A, Fernando H, Watkins G, Mansel RE, Jiang WG. Expression of transcription factor CREB1 in human breast cancer and its correlation with prognosis. Oncol Rep. 2007;18:953–958. [PubMed] [Google Scholar]
- 26.Seo HS, Liu DD, Bekele BN, Kim MK, Pisters K, Lippman SM, Wistuba II, Koo JS. Cyclic AMP response element-binding protein overexpression: A feature associated with negative prognosis in never smokers with non-small cell lung cancer. Cancer Res. 2008;68:6065–6073. doi: 10.1158/0008-5472.CAN-07-5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cekanova M, Majidy M, Masi T, Al-Wadei HA, Schuller HM. Overexpressed Raf-1 and phosphorylated cyclic adenosine 3′-5′-monophosphatate response element-binding protein are early markers for lung adenocarcinoma. Cancer. 2007;109:1164–1173. doi: 10.1002/cncr.22520. [DOI] [PubMed] [Google Scholar]
- 28.Laag E, Majidi M, Cekanova M, Masi T, Takahashi T, Schuller HM. NNK activates ERK1/2 and CREB/ATF-1 via beta-1-AR and EGFR signaling in human lung adenocarcinoma and small airway epithelial cells. Int J Cancer. 2006;119:1547–1552. doi: 10.1002/ijc.21987. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida M, Seiki M, Yamaguchi K, Takatsuki K. Monoclonal integration of human T-cell leukemia provirus in all primary tumors of adult T-cell leukemia suggests causative role of human T-cell leukemia-virus in the disease. Proc Natl Acad Sci USA. 1984;81:2534–2537. doi: 10.1073/pnas.81.8.2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA. 1980;77:7415–7419. doi: 10.1073/pnas.77.12.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akagi T, Ono H, Nyunoya H, Shimotohno K. Characterization of peripheral blood T-lymphocytes transduced with HTLV-I Tax mutants with different transactivating phenotypes. Oncogene. 1997;14:2071–2078. doi: 10.1038/sj.onc.1201045. [DOI] [PubMed] [Google Scholar]
- 32.Smith MR, Greene WC. Type-I human T-cell leukemia-virus Tax protein transforms rat fibroblasts through the cyclic adenosine-monophosphate response element binding-protein/activating transcription factor pathway. J Clin Invest. 1991;88:1038–1042. doi: 10.1172/JCI115364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shankar DB, Cheng JC, Kinjo K, Federman N, Moore TB, Gill A, Rao NP, Landaw EM, Sakamoto KM. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell. 2005;7:351–362. doi: 10.1016/j.ccr.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 34.Kinjo K, Sandoval S, Sakamoto KM, Shankar DB. The role of CREB as a proto-oncogene in hematopoiesis. Cell Cycle. 2005;4:1134–1135. doi: 10.4161/cc.4.9.1991. [DOI] [PubMed] [Google Scholar]
- 35.Crans-Vargas HN, Landaw EM, Bhatia S, Sandusky G, Moore TB, Sakamoto KM. Expression of cyclic adenosine monophosphate response-element binding protein in acute leukemia. Blood. 2002;99:2617–2619. doi: 10.1182/blood.v99.7.2617. [DOI] [PubMed] [Google Scholar]
- 36.Pigazzi M, Ricotti E, Germano G, Faggian D, Arico M, Basso G. cAMP response element binding protein (CREB) over-expression CREB has been described as critical for leukemia progression. Haematologica. 2007;92:1435–1437. doi: 10.3324/haematol.11122. [DOI] [PubMed] [Google Scholar]
- 37.Pigazzi M, Manara E, Baron E, Basso G. miR-34b targets cyclic AMP-responsive element binding protein in acute myeloid leukemia. Cancer Res. 2009;69:2471–2478. doi: 10.1158/0008-5472.CAN-08-3404. [DOI] [PubMed] [Google Scholar]
- 38.Kumar AP, Bhaskaran S, Ganapathy M, Crosby K, Davis MD, Kochunov P, Schoolfield J, Yeh IT, Troyer DA, Ghosh R. Akt/cAMP-responsive element binding protein/cyclin D1 network: A novel target for prostate cancer inhibition in transgenic adenocarcinoma of mouse prostate model mediated by Nexrutine, a Phellodendron amurense bark extract. Clin Cancer Res. 2007;13:2784–2794. doi: 10.1158/1078-0432.CCR-06-2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie SH, Price JE, Luca M, Jean D, Ronai Z, BarEli M. Dominant-negative CREB inhibits tumor growth and metastasis of human melanoma cells. Oncogene. 1997;15:2069–2075. doi: 10.1038/sj.onc.1201358. [DOI] [PubMed] [Google Scholar]
- 40.Abramovich C, Pineault N, Ohta H, Humphries RK. Hox genes: from leukemia to hematopoietic stem cell expansion. Ann N Y Acad Sci. 2005;1044:109–116. doi: 10.1196/annals.1349.014. [DOI] [PubMed] [Google Scholar]
- 41.Esparza SD, Chang J, Shankar DB, Zhang B, Nelson SF, Sakamoto KM. CREB regulates Meis1 expression in normal and malignant hematopoietic cells. Leukemia. 2008;22:665–667. doi: 10.1038/sj.leu.2404933. [DOI] [PubMed] [Google Scholar]
- 42.Schumacher MA, Goodman RH, Brennan RG. The structure of a CREB bZIP.somatostatin CRE complex reveals the basis for selective dimerization and divalent cation-enhanced DNA binding. J Biol Chem. 2000;275:35242–35247. doi: 10.1074/jbc.M007293200. [DOI] [PubMed] [Google Scholar]
- 43.Walton KM, Rehfuss RP, Chrivia JC, Lochner JE, Goodman RH. A dominant repessor of cyclic adenosin-3′,5′-monphosphate (cAMP)-regulated enhancer-binding protein activity inhibits the cAMP-mediated induction of the somatostatin promoter in vivo. Mol Endocrinol. 1992;6:647–655. doi: 10.1210/mend.6.4.1350057. [DOI] [PubMed] [Google Scholar]
- 44.Yang YM, Dolan LR, Ronai Z. Expression of dominant negative CREB reduces resistance to radiation of human melanoma cells. Oncogene. 1996;12:2223–2233. [PubMed] [Google Scholar]
- 45.Abramovitch R, Tavor E, Jacob-Hirsch J, Zeira E, Amariglio N, Pappo O, Rechavi G, Galun E, Honigman A. A pivotal role of cyclic AMP-responsive element binding protein in tumor progression. Cancer Res. 2004;64:1338–1346. doi: 10.1158/0008-5472.can-03-2089. [DOI] [PubMed] [Google Scholar]
- 46.Linnerth NM, Baldwin M, Campbell C, Brown M, McGowan H, Moorehead RA. IGF-II induces CREB phosphorylation and cell survival in human lung cancer cells. Oncogene. 2005;24:7310–7319. doi: 10.1038/sj.onc.1208882. [DOI] [PubMed] [Google Scholar]
- 47.Rozenberg J, Rishi V, Orosz A, Moitra J, Glick A, Vinson C. Inhibition of CREB function in mouse epidermis reduces papilloma formation. Mol Cancer Res. 2009;7:654–664. doi: 10.1158/1541-7786.MCR-08-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho YS, Kim MK, Cheadle C, Neary C, Park YG, Becker KG, Cho-Chung YS. A genomic-scale view of the cAMP response element-enhancer decoy: A tumor target-based genetic tool. Proc Natl Acad Sci USA. 2002;99:15626–15631. doi: 10.1073/pnas.242617799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park YG, Nesterova M, Agrawal S, Cho-Chung YS. Dual blockade of cyclic AMP response element-(CRE) and AP-1-directed transcription by CRE-transcription factor decoy oligonucleotide - Gene-specific inhibition of tumor growth. J Biol Chem. 1999;274:1573–1580. doi: 10.1074/jbc.274.3.1573. [DOI] [PubMed] [Google Scholar]
- 50.Cheng JC, Kinjo K, Judelson DR, Chang J, Wu WS, Schmid I, Shankar DB, Kasahara N, Stripecke R, Bhatia R, Landaw EM, Sakamoto KM. CREB is a critical regulator of normal hematopoiesis and leukemogenesis. Blood. 2008;111:1182–1192. doi: 10.1182/blood-2007-04-083600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barlow CA, Kitiphongspattana K, Siddiqui N, Roe MW, Mossman BT, Lounsbury KM. Protein kinase A-mediated CREB phosphorylation is an oxidant-induced survival pathway in alveolar type II cells. Apoptosis. 2008;13:681–692. doi: 10.1007/s10495-008-0203-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shukla A, Bosenberg MW, MacPherson MB, Butnor KJ, Heintz NH, Pass HI, Carbone M, Testa JR, Mossman BT. Activated cAMP response element binding protein is overexpressed in human mesotheliomas and inhibits apoptosis. Am J Pathol. 2009;175:2197–2206. doi: 10.2353/ajpath.2009.090400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verma IM, Somia N. Gene therapy - promises, problems and prospects. Nature. 1997;389:239–242. doi: 10.1038/38410. [DOI] [PubMed] [Google Scholar]
- 54.Bilanges B, Torbett N, Vanhaesebroeck B. Killing two kinase families with one stone. Nat Chem Biol. 2008;4:648–649. doi: 10.1038/nchembio1108-648. [DOI] [PubMed] [Google Scholar]
- 55.Davis PD, Hill CH, Keech E, Lawton G, Nixon JS, Sedgwick AD, Wadsworth J, Westmacott D, Wilkinson SE. Potent selective inhibitors of protein kinase C. FEBS Lett. 1989;259:61–63. doi: 10.1016/0014-5793(89)81494-2. [DOI] [PubMed] [Google Scholar]
- 56.Aggarwal S, Kim SW, Ryu SH, Chung WC, Koo JS. Growth suppression of lung cancer cells by targeting cyclic AMP response element-binding protein. Cancer Res. 2008;68:981–988. doi: 10.1158/0008-5472.CAN-06-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rishi V, Potter T, Laudeman J, Reinhart R, Silvers T, Selby M, Stevenson T, Krosky P, Stephen AG, Acharya A, Moll J, Oh WJ, Scudiero D, Shoemaker RH, Vinson C. A high-throughput fluorescence-anisotropy screen that identifies small molecule inhibitors of the DNA binding of B-ZIP transcription factors. Anal Biochem. 2005;340:259–271. doi: 10.1016/j.ab.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 58.Best JL, Amezcua CA, Mayr B, Flechner L, Murawsky CM, Emerson B, Zor T, Gardner KH, Montminy M. Identification of small-molecule antagonists that inhibit an activator: coactivator interaction. Proc Natl Acad Sci USA. 2004;101:17622–17627. doi: 10.1073/pnas.0406374101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li BX, Xiao X. Discovery of a Small-Molecule Inhibitor of the KIX-KID Interaction. Chem Bio Chem. 2009;10:2721–2724. doi: 10.1002/cbic.200900552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wemmer DE, Dervan PB. Targeting the minor groove of DNA. Curr Opin Struct Biol. 1997;7:355–361. doi: 10.1016/s0959-440x(97)80051-6. [DOI] [PubMed] [Google Scholar]
- 61.Nickols NG, Dervan PB. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc Natl Acad Sci USA. 2007;104:10418–10423. doi: 10.1073/pnas.0704217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Olenyuk BZ, Zhang GJ, Klco JM, Nickols NG, Kaelin WG, Jr, Dervan PB. Inhibition of vascular endothelial growth factor with a sequence-specific hypoxia response element antagonist. Proc Natl Acad Sci USA. 2004;101:16768–16773. doi: 10.1073/pnas.0407617101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cardinaux JR, Notis JC, Zhang Q, Vo N, Craig JC, Fass DM, Brennan RG, Goodman RH. Recruitment of CREB binding protein is sufficient for CREB-mediated gene activation. Mol Cell Biol. 2000;20:1546–1552. doi: 10.1128/mcb.20.5.1546-1552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 65.De Guzman RN, Goto NK, Dyson HJ, Wright PE. Structural basis for cooperative transcription factor binding to the CBP coactivator. J Mol Biol. 2006;355:1005–1013. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 66.Goto NK, Zor T, Martinez-Yamout M, Dyson HJ, Wright PE. Cooperativity in transcription factor binding to the coactivator CREB-binding protein (CBP). The mixed lineage leukemia protein (MLL) activation domain binds to an allosteric site on the KIX domain. J Biol Chem. 2002;277:43168–43174. doi: 10.1074/jbc.M207660200. [DOI] [PubMed] [Google Scholar]
- 67.Buhrlage SJ, Bates CA, Rowe SP, Minter AR, Brennan BB, Majmudar CY, Wemmer DE, Al-Hashimi H, Mapp AK. Amphipathic small molecules mimic the binding mode and function of endogenous transcription factors. ACS Chem Biol. 2009;4:335–344. doi: 10.1021/cb900028j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xiao X, Yu P, Lim HS, Sikder D, Kodadek T. Design and synthesis of a cell-permeable synthetic transcription factor mimic. J Comb Chem. 2007;9:592–600. doi: 10.1021/cc070023a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiao X, Yu P, Lim HS, Sikder D, Kodadek T. A cell-permeable synthetic transcription factor mimic. Angew Chem Int Ed. 2007;46:2865–2868. doi: 10.1002/anie.200604485. [DOI] [PubMed] [Google Scholar]
- 70.Ruppert S, Cole TJ, Boshart M, Schmid E, Schutz G. Multiple mRNA isoforms of the transcription activator protein CREB: generation by alternative splicing and specific expression in primary spermatocytes. EMBO J. 1992;11:1503–1512. doi: 10.1002/j.1460-2075.1992.tb05195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rudolph D, Tafuri A, Gass P, Hammerling GJ, Arnold B, Schutz G. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc Natl Acad Sci USA. 1998;95:4481–4486. doi: 10.1073/pnas.95.8.4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 73.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 74.Hummler E, Cole TJ, Blendy JA, Ganss R, Aguzzi A, Schmid W, Beermann F, Schutz G. Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proc Natl Acad Sci USA. 1994;91:5647–5651. doi: 10.1073/pnas.91.12.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bulun SE, Lin Z, Zhao H, Lu M, Amin S, Reierstad S, Chen D. Regulation of aromatase expression in breast cancer tissue. Ann N Y Acad Sci. 2009;1155:121–131. doi: 10.1111/j.1749-6632.2009.03705.x. [DOI] [PubMed] [Google Scholar]