

Abstract
Tumor suppressor p53 is inactivated in most cancers and the critical role of p53 in the suppression of carcinogenesis has been confirmed in many mouse models. The protein product of the tumor suppressor p53 gene works as a transcriptional regulator, activating expression of numerous genes involved in cell death, cell cycle arrest, senescence, DNA-repair and many other processes. In spite of the multiple efforts to characterize the functions of p53, the mechanisms of tumor suppression by p53 are still elusive. Recently, new activities of p53 such as regulation of reactive oxygen species (ROS) and metabolism have been described and the p53-regulated genes responsible for these functions have been identified. Metabolic derangements and accumulation of ROS are features of carcinogenesis, supporting the idea that many tumor suppressive effects of p53 can be mediated by regulation of metabolism and/or ROS. Mutations in the p53 gene can not only inactivate wild type function of p53 but also endow p53 with new functions such as activation of new metabolic pathways contributing to carcinogenesis. Understanding the metabolic and antioxidant functions of p53 allow us to develop approaches to restore p53 function in cancers, where p53 is inactivated, in other to ensure the best outcome of anti-cancer treatment.
1. Introduction
p53 was discovered more than three decades ago as a protein interacting with large T-antigen of human polyoma virus SV40, which is known to be responsible for malignant transformation of different human and mouse cells. This discovery paved the way for intensive studies of the role of p53 in transformation and carcinogenesis (Levine, 1997). A few years later, the tumor suppressor p53 (TSp53) gene was cloned by several groups and it was demonstrated that p53 is mutated in a vast majority of human cancers (Levine, 1997). Beyond point mutations, p53 can be inactivated via many other mechanisms including chromosome deletions, amplification of its natural negative regulator Mdm2 (mouse double minute 2) or expression of viral oncogenes such as papillomavirus E6 or adenovirus E1B proteins (Levine, 1997). The following studies found inactivation of one p53 allele in families with Li-Fraumeni syndrome, the disease characterized by the predisposition to many types of cancer at an early age (Li and Fraumeni, 1969a, b). The ultimate evidence that p53 is a critical tumor suppressor came with the advent of gene knockout technology. p53 knockout mice were normal, indicating that p53 does not play a role in embryogenesis and differentiation, but they developed cancers, mostly lymphomas and sarcomas with 100% penetrance and died from cancer by the age of 6 months (Donehower et al., 1992). Surprisingly, the pattern of cancers observed in the p53-knockout animals differed from the majority of human cancers, which are mostly carcinomas. This may be because human cancers mostly bear a mutant form of the proteins rather than total inactivation of p53 and the most common mutations in p53, called “hot-spot” mutations, give the mutant form of p53 an ability to stimulate invasiveness or cancer-associated metabolism in the process called “gain-of-function” (Donehower et al., 1992; Freed-Pastor and Prives, 2012; Levine, 1997). Accordingly, two mouse p53 knockin strains with mutations R172H and R270H (where arginines (R) are replaced to histidines (H), corresponding to hot-spot human p53 mutations in codons 175 and 273) were generated, and it was demonstrated that mice developed mostly highly-invasive carcinomas (Lang et al., 2004; Olive et al., 2004).
As it has been believed for many years, p53 protects from carcinogenesis via “crime and punishment” mechanisms by restricting proliferation of genetically damaged cells via activation of cell death or permanent cell cycle arrest or senescence (Vousden and Prives, 2009). This dogma has been significantly re-visited during recent years when it was shown that inactivation of major targets of p53 involved in regulation of cell death - Noxa and Puma, or senescence/cell cycle arrest gene p21 does not recapitulate the cancer-prone phenotype of p53-deficient mice (Deng et al., 1995; Jeffers et al., 2003; Villunger et al., 2003). In following studies it was also established that p53 triple mutant knockin mice (p533KR), where 3 lysines (117, 161 and 162), subjects of p53 acetylation, were replaced with arginines, lost the ability to activate cell death, cell cycle arrest or senescence but still maintained the ability to suppress carcinogenesis (Li et al., 2012). This data opens a new venue for the studies of the mechanisms of “good maintenance” applied by p53 to prevent accumulation of damage via regulation of reactive oxygen species and metabolism leaving the “punishment” as the last resort for the cells which go awry.
2. Tumor suppressor gene p53 encodes stress-responsive transcriptional factor
p53 works as a transcription factor involved in transcriptional regulation of multiple genes. p53 functions in the form of homo-tetramers, recognizing p53-responsive elements composed of two decamers separated by short spacer 5′-RRRCWWGYYYn0–14RRRCWWGYYY-3′ where R – purine, Y – pyrimidine, C – cytosine, G – guanine, W – adenine or thymine and N – nucleotide and activates or suppresses different promoters dependent on context (Freed-Pastor and Prives, 2012). It is well established that p53 is induced by DNA-damage via consequent activation of ATM/ATR and Chk1/Chk2 kinases which phosphorylate p53 on N-terminus, causing p53 stabilization and activation. As a result, p53 induces cell cycle arrest, stimulates DNA-repair and protects genomic stability earning p53 the name “guardian of the genome” (Lane, 1992). Other stimuli, including oxidative stress, hypoxia, nutrient deprivation or activation of oncogenes, induce p53 via mechanisms yet to be established and stimulate expression of p53-dependent genes, facilitating stress consequences (Vousden and Lane, 2007). Nevertheless, if the stress insult is too intense, p53 can stimulate pathways that lead to the elimination of cells via induction of cell death or senescence. The choice between a pro-survival or pro-death outcome is determined by the sensitivity of different promoters to p53 (Vousden and Lane, 2007). In response to weak or moderate stress insults, p53 stimulates expression of pro-survival genes which protect cells from damage. The promoters of these genes are very sensitive to even low p53 activation and usually activate very quickly after the stress is applied. On the contrary, pro-apoptotic genes are activated in response to intense stress and with a significant delay as compared to pro-survival genes (Sablina et al., 2005; Vousden and Prives, 2009) (Figure 1).
Figure 1. Yin and Yang activities of p53 in tumor suppression.

Many stress insults induce p53 which activates expression of multiple genes via interaction with p53-responsive elements on their promoters. The outcome of p53 activation can be cell protection or cell death depending on the nature and intensity of the stress. Both processes can be required to reach maximal protection of the organism against carcinogenesis.
Among the targets of p53 are its own negative regulators such as Mdm2, PIRH2 (p53-induced protein with a RING (Really Interesting New Gene)-H2 domain) and COP1 (constitutively photomorphogenic 1). They work as E3-ubiquitine ligases which stimulate p53 ubiquitination and degradation by proteasomes (Dornan et al., 2004; Honda et al., 1997; Leng et al., 2003). Mdm2 plays a major role in regulation of p53 stability and activity via constant control of p53 expression (Bond et al., 2005; Lahav et al., 2004). Inactivation of Mdm2 in mice leads to permanent p53 activation, causing embryonic lethality and the lethal phenotype was completely rescued by p53 inactivation supporting the critical role of Mdm2 in p53 regulation (Jones et al., 1995). Moreover, single nucleotide polymorphisms in SP1 site in regulatory area of the human Mdm2 gene causes elevated Mdm2 expression, which increases susceptibility of the organism to carcinogenesis (Bond et al., 2004).
Although p53 protein is suppressed by Mdm2 under non-stressed conditions and has a lifespan around 20 min, it can be easily activated by multiple stress insults (Levine, 1997). Mild stress can be induced by mistakes in DNA-replication, ROS accumulation or decrease in ATP levels. It causes modification of p53 via phosphorylation and some other mechanisms such as acetylation, methylation, ubiquitylation, neddylation or summoylation (Vousden and Lane, 2007). p53 can be also activated via redox-dependent mechanisms involving redox factor Ref1 (Seo et al., 2002). As a result, it leads to stimulation of p53-dependent genes involved in suppression of ROS and tuning up the metabolism. In contrast, activation of p53 by severe stress leads to cell death or permanent cell cycle arrest (Budanov, 2011; Vousden and Lane, 2007).
3. P53 regulates redox balance in cells
3.1 Pro-oxidant function of p53
p53 is the major regulator of programmed cell death or apoptosis, and for many years this activity was considered to be the major mechanism of p53-controlled tumor suppression (Selivanova, 2004). In an attempt to explain the mechanisms of the pro-apoptotic activity of p53, several targets named PIG1–13 (p53-inducible genes 1–13) with strong pro-oxidant properties were described (Polyak et al., 1997). Some examples include: PIG1 - member of galectin family involved in superoxide production; PIG3 - homolog of NADPH–quinone oxidoreductase, a potent ROS generator; PIG8 - human homolog of mouse E-24 gene, a quinone known to regulate ROS (Polyak et al., 1997). Accordingly, activation of p53 in different cancer cell lines via overexpression or strong genotoxic stress causes accumulation of reactive oxygen species (ROS) and oxidative stress contributing to induction of cell death (Polyak et al., 1997; Sablina et al., 2005). Overexpression of BH3-only protein Puma, a critical mediator of p53-activated cell death, also stimulates ROS production which most likely is associated with mitochondria disintegration (Sablina et al., 2005). In accordance with these data, overexpression of p53 stimulates ROS production mostly in cells with intact mitochondria, which are susceptible to p53-induced cell death (Sablina et al., 2005). Besides its effects on mitochondria, the parallel mechanism of ROS production by p53 can involve transcriptional activation of a component of NADPH oxidase, the critical enzyme responsible for O2− production, NCF2/p67phox (neutrophil cytosol factor 2) (Italiano et al., 2012). Although pro-oxidant activity of p53 can be beneficial for elimination of cancer cells, it might have some disadvantages for the organism under conditions of ischemia, neurodegeneration or aging (Vousden and Prives, 2009); consequently tight p53 regulation is critical for proper control of many physiological processes associated with p53 activation (Figure 2).
Figure 2. Regulation of anti-oxidant and pro-oxidant genes by p53.

Low-intensities of stress stimulate expression of highly sensitive p53-dependent pro- survival genes involved in ROS suppression, metabolism, mitochondrial function and autophagy, which protect cell viability. Highly intense stress insults activate cell death via the induction of pro-apoptotic and pro-oxidant genes. In response to low stress, PGC1α binds p53 and stimulates expression of pro-survival genes, although its degradation induced by chronic prolonged stress may be responsible for the activation of genes responsible for cell death.
3.2 Antioxidant function of p53
The paradigm that p53 is a bona fide pro-oxidant factor changed since it had been found that under physiological or low stress conditions p53 suppresses ROS accumulation (Sablina et al., 2005). ROS cause oxidative DNA-damage which increase the rate of mutagenesis and chromosomal instability. Being widely recognized as the protector of genomic stability, p53 inhibits DNA-oxidation and mutagenesis via suppression of ROS (Budanov, 2011). ROS are also involved in the activation of signaling pathways controlling cell growth, proliferation, viability and transformation such as PI3K-AKT, JAK-STAT, PLC-PKC, MAPK cascade or IKK-NF-κB (Martindale and Holbrook, 2002). They also play an important role in stimulation of angiogenesis and epithelial-mesenchymal transition, both critical steps of cancer progression (Cui, 2012). Thus, ROS accumulation, often observed in cancer cells (Behrend et al., 2003), can be responsible for the high rate of mutagenesis in oncogenes and tumor suppressor genes, stimulation of cell proliferation and malignant transformation, and eventually for angiogenesis, invasiveness and metastasis (Behrend et al., 2003; Martindale and Holbrook, 2002).
These considerations are supported by animal studies. The lifespans of the p53-deficient mice, characterized by increased ROS levels, are shortened by 5–6 fold due to accelerated carcinogenesis (Donehower et al., 1992; Sablina et al., 2005). Increased levels of oxidative stress is also observed in patients with Li-Fraumeni cancer predisposition syndrome characterized by inactivation of one copy of the p53 gene (Wang et al., 2013). Inactivation of ATM, an upstream p53 regulator, also leads to oxidative stress, DNA oxidation, mutagenesis and carcinogenesis (Reliene et al., 2004; Xu et al., 1996). Tumor phenotypes of p53 and ATM knockout mice were significantly suppressed in animals supplemented with an antioxidant N-acetyl-cysteine (NAC) (Sablina et al., 2005; Schubert et al., 2004). Additionally xenograft studies on lung adenocarcinoma cells demonstrated that p53 silencing accelerates tumor growth, while these effects were strongly suppressed by NAC treatment (Sablina et al., 2005).
In another set of experiments, a mouse strain carrying extra-genomic copies of p53 and its upstream regulator Arf1 (alternative reading frame 1) was generated (Matheu et al., 2007). These mice, designated s-p53/Arf1 (super-p53/Arf1) are characterized by slightly elevated p53 expression and a continuous activation of p53-dependent antioxidant genes. s-p53/Arf1 mice have an extended lifespan as compared to wild type control and are highly resistant to carcinogenesis. Interestingly, the cells isolated from the s-p53/Arf1 mice are also well protected against transformation by cooperating oncogenes, providing a protecting mechanism against carcinogenesis (Matheu et al., 2007).
Since characterization of the antioxidant function of p53, several groups of p53-inducible antioxidant proteins were identified.
a) Antioxidant enzymes
MnSOD (Manganese superoxide dismutase) is an enzyme responsible for decomposition of superoxide (O2−) converting it to less toxic H2O2 form in the reaction 2O2−+2H+=H2O2+O2 (Balaban et al., 2005). Superoxide is produced as a by-product of mitochondrial oxidative phosphorylation or via activation of NADPH oxidases. Mitochondria are the major source of superoxide and other ROS, and as estimated approximately 2% of oxygen consumed by healthy mitochondria is converted into the O2− form (Finkel and Holbrook, 2000). Moreover, mitochondrial dysfunction induced by stress or improper control of mitochondrial integrity causes increased leakage of electrons from the respiratory chain. Superoxide is extremely reactive and, consequently, very unstable. Nevertheless, it can damage different macromolecules in mitochondria including lipids, proteins and DNA, affecting mitochondrial function and stimulating extensive electron leakage and ROS production (Balaban et al., 2005; Finkel and Holbrook, 2000). MnSOD, residing in the mitochondrial matrix, plays a critical role in the detoxification of O2−, producing less reactive H2O2. p53 activates expression of the MnSOD via direct recognition of MnSOD promoter in -2032 - -2009 position of the human gene (Hussain et al., 2004).
Another critical antioxidant enzyme regulated by p53 is GPx1 (glutathione peroxidase 1), an enzyme responsible for decomposition of H2O2 via the reaction 2GSH+H2O2=GS-SG+H2O (Balaban et al., 2005), where GSH and GS-SG are reduced and oxidized forms of glutathione, a major cellular antioxidant (Finkel and Holbrook, 2000). Selenoprotein GPx1 plays a major role in decomposition of H2O2, a highly diffusible molecule which reacts with different macromolecules including membrane lipids, proteins and nuclear DNA. p53 activates expression of the GPx1 gene through binding to the -694 - -720 region of its promoter, stimulating an antioxidant response (Hussain et al., 2004). In particular cell types, such as developing rat retina and retinal ganglion cells, p53 can stimulate expression of catalase, another enzyme involved in H2O2 decomposition (O’Connor et al., 2008). It was also reported that p53 regulates activity of catalase via direct protein-protein interactions (Kang et al., 2013). Interestingly, p53-responsive p53R2 (p53-inducible ribonucleotide reductase) was found in a p53-catalase complex under physiological conditions and stimulates catalase activity, while another p53 target PIG3 was found in the p53-catalase complex under genotoxic stress conditions and inhibits catalase activity (Kang et al., 2013).
b) Sestrins
Another group of antioxidant genes, Sestrins, play a major role in regulation of ROS in response to p53 activation (Budanov et al., 2004). Sestrins are a family of genes highly conserved in evolution from protists to mammals (Budanov et al., 2010). The mammalian Sestrin gene family is composed of 3 members, Sesn1, Sesn2 and Sesn3, while the invertebrate genome contains only one Sestrin gene (Budanov et al., 2010; Lee et al., 2008). The first family member, Sesn1, was identified as a p53-inducible gene activated by DNA-damage (Velasco-Miguel et al., 1999). Several potential p53-responive elements were described within intron 1 and 2 (Velasco-Miguel et al., 1999; Wei et al., 2006). Sesn1 gene is transcribed in three mRNA forms using alternative promoters and produces three different proteins with molecular weights of 46,55 and 68 kDa (Velasco-Miguel et al., 1999). Among these proteins, only the 46 and 55 kDa forms are regulated by p53, while the 68kDa form is constantly expressed (Velasco-Miguel et al., 1999). The second member of the family, Sesn2, was isolated as hypoxia inducible gene (Budanov et al., 2002). In following studies it was established that the Sesn2 gene is also activated by many other stimuli including oxidative stress, nutrient deficiency and DNA-damage (Ben-Sahra et al., 2013a; Budanov et al., 2004; Budanov et al., 2002). p53 is responsible for Sesn2 activation by DNA-damage, and contributes to Sesn2 activation by oxidative stress (Budanov et al., 2004; Budanov et al., 2002). The p53-responsive element was identified 9.7 kb downstream of the Sesn2 gene by PET (paired-end ditag) sequencing (Wei et al., 2006). In different study, another p53-responsive element was found in the 1st exon of the Sesn2 gene (Lee et al., 2011). The Sesn2 gene encodes a protein with a molecular weight of 60 kDa showing close similarity with 55 kDa protein product of the Sesn1 gene (Budanov et al., 2002).
Sesn3, the third family member, was identified in silico as a close homolog of both Sesn1 and Sesn2 genes (Budanov et al., 2002; Peeters et al., 2003). Interestingly, while two of three members of the Sestrin family - Sesn1 and Sesn2 - are direct targets of p53, Sesn1 and Sesn3 are activated by the FoxO family of transcription factors (Budanov et al., 2010; Nogueira et al., 2008). Similar to p53, FoxO controls both pro-survival and pro-apoptotic processes via the activation of different sets of genes involved in regulation of cell death and cell cycle arrest as well as metabolism and ROS (Hagenbuchner et al., 2012). Regulation of Sestrins by p53 and FoxO is very conserved in evolution, as demonstrated by the Drosophila Sestrin gene (Lee et al., 2009).
Although Sestrins do not have similarity with other proteins, detailed sequence analysis has shown that part of Sestrin protein (corresponding to 100–175 aa of Sesn2) shares homology with the Mycobacterium Tuberculosis antioxidant protein AhpD, which is a critical regulator of bacterial thiol peroxidase AhpC (Bryk et al., 2002; Budanov et al., 2004). During the catalytic cycle AhpC is oxidized and then restored by AhpD. Mammalian peroxiredoxins are AhpC homologs, which work via similar mechanisms (D’Autreaux and Toledano, 2007). In contrast to bacterial proteins, mammalian peroxiredoxins can easily be inactivated via overoxidation of catalytical cysteine (Wood et al., 2003). Sestrins contribute to regeneration of peroxiredoxins potentially through activation of sulfinilreductase for mammalian peroxiredoxins – Srx (sulfiredoxin) (Budanov et al., 2010). Sestrins activate expression of Srx and some other antioxidant genes stimulating autophagic degradation of protein Keap1 (Kelch-like ECH-associated protein 1), an inhibitor of antioxidant transcription factor NRF2 (nuclear factor (erythroid-derived 2)-like 2) (Bae et al., 2013). The regulation of antioxidant response by Sestrins via regeneration of peroxiredoxins can play an important role in protection of neurons and macrophages from oxidative stress (Essler et al., 2009; Papadia et al., 2008). Sestrin-mediated autophagy also plays a critical role in support of integrity of the mitochondria and other organelles responsible for ROS production (Budanov and Karin, 2008; Lee et al., 2009; Lee et al., 2012). Inactivation of Sestrins via p53 mutation or other mechanism might play a critical role in carcinogenesis, facilitating cell transformation. Accordingly, Sesn2-deficient cells are much more susceptible to transformation than their wild-type counterparts (Budanov and Karin, 2008). Moreover, cell transformation by Ras oncogenes requires ROS production and Ras stimulates ROS via suppression of Sesn1 and Sesn3 gene expression (Kopnin et al., 2007).
c) TIGAR
TIGAR (p53-induced glycolysis and apoptotic regulator) was discovered as a p53-inducible gene by microarray analysis. It is a direct p53 target with kinetics of activation similar with other genes involved in pro-survival activities involved in ROS regulation, cell cycle control and DNA-repair, which are activated quickly after p53 induction (Bensaad et al., 2006). Two p53-responsive elements were found in the promoter and first intron of the human TIGAR gene (Bensaad et al., 2006). TIGAR shares similarity with proteins of the PGM (phosphoglycerate mutase) family, but the highest degree homology is observed with the biphosphatase domain of PFK-2/FBPase-2 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase) (Bensaad et al., 2006). Ectopic expression of TIGAR decreases the levels of Fru-2,6-P2 (fructose-2,6-biphosphate). As expected, it leads to a decrease in glycolytic rates, and silencing TIGAR stimulates Fru-2,6-P2 production and glycolysis. Being an inhibitor of glycolysis, TIGAR can re-direct glycolytic intermediates into the pentose phosphate pathway (PPP), which is involved in the production of the reducing agent NADPH. NADPH is used by glutathione reductase to convert GSSG to GSH. The GSH/GSSG ratio is the major indicator of redox balance in the cell, which affects cell metabolism, viability and antioxidant defense. Accordingly, activation of TIGAR in response to p53 decreases the levels of ROS and suppresses p53-induced cell death (Bensaad et al., 2006). TIGAR protein was also found in mitochondria where it interacts with mitochondrial HK2 (hexokinase 2) and stimulates its activity. HK2 arguably helps to couple glycolysis with oxidative phosphorylation and limits ROS production in mitochondria via control of ADP levels (Cheung et al., 2012).
d) GLS2
The GLS2 (Glutaminase 2) gene encodes a mitochondrial glutaminase which catalyzes the hydrolysis of glutamine to glutamate. Activation of GLS2 by p53 leads to increased production of glutamate and α-ketoglutarate, last one is the critical substrate for the tricarboxylic acid (TCA) cycle. As a result, GLS2 stimulates mitochondrial respiration and ATP production. Moreover, GSL2 induction causes GSH accumulation and ROS downregulation, providing protection against oxidative stress (Hu et al., 2010). Thus, GSL2 regulates ROS supporting mitochondrial function and preventing ROS generation by dysfunctional mitochondria. Activation of GSL2 also protects cells against oxidative DNA-damage and supports genomic stability (Suzuki et al., 2010).
e) Other p53 targets involved in redox regulation
Several other p53-inducible proteins are involved in redox regulation, although the mechanisms are yet to be established. p53INP1 (p53-inducible nuclear protein 1) when overexpressed facilitates cell death. However, p53INP1-deficient MEFs and spleenocytes accumulate ROS, and P53INP1-null thymocytes are highly susceptible to cell death linked with enhanced ROS production (Cano et al., 2009; N’Guessan et al., 2011). p53INP1-null cells are characterized by decreased levels of autophagy which might be responsible for p53INP1-regulated antioxidant protection (N’Guessan et al., 2011). Another p53 target, ALDH4, is a mitochondrial-matrix NAD+-dependent enzyme which catalyzes the second step in the proline degradation pathway. ALDH4 downregulates ROS levels via a yet to be defined mechanism and its silencing enhances cell death in response to p53 overexpression (Yoon et al., 2004). Many effects of p53 on ROS can be explained via support of mitochondrial function. P53 is involved in the maintenance of mitochondrial DNA copy number and mitochondrial mass, and p53 inactivation can lead to improper control of mitochondrial integrity and an increased ROS production (Lebedeva et al., 2009). p53 can also support stability of mitochondrial DNA interacting with mitochondrial DNA-polymerase γ, ensuring integrity and proper functioning of mitochondria (Achanta et al., 2005). As an additional mechanism of control of mitochondrial function, p53 activates expression of Mieap which induces the formation of a lysosome-like vacuole structure in mitochondria and is involved in mitochondria quality control, suppressing ROS production (Kitamura et al., 2011) (Figure 2).
3.3 P53 and autophagy
Macroautophagy (therein in the text - autophagy) is the mechanism of two-membrane encapsulation and lysosomal degradation of cellular constituents such as organelles, protein aggregates and bulk of cytoplasm (Kroemer et al., 2010). Autophagy is responsible for the control of the integrity of organelles, such as mitochondria and peroxisomes, when damaged produce excessive amounts of ROS. Thus, impaired autophagy can be responsible for oxidative stress and genomic instability (Mathew et al., 2007). p53 regulates autophagy in a positive and negative manner dependent on context (Feng et al., 2005; Tasdemir et al., 2008). The nuclear form of p53 transcriptionally activates expression of multiple genes involved in autophagy such as DRAM, Sesn2, ULK1, ULK2, Atg4a, Atg4c, Atg7, and Uvrag in response to DNA-damage or oncogene activation (Crighton et al., 2006; Feng et al., 2005; Gao et al., 2011; Kenzelmann Broz et al., 2013; Maiuri et al., 2009). In contrast, the cytoplasmic form of p53 inhibits autophagy via an interaction with autophagy proteins Atg17 and RB1CC1/FLP200 (Morselli et al., 2011; Tasdemir et al., 2008). This promiscuous effect of p53 on autophagy regulation can be explained by the link of autophagy with cell death. Autophagy can suppress cell death in response to stress supporting integrity and function of organelles, suppressing ROS production and providing ATP for the repair processes (Kroemer et al., 2010). When p53 functions in a pro-survival fashion, it might support autophagy via transcriptional activation of autophagy genes. On the contrary, when cell death is the more desirable scenario p53 can suppress autophagy via protein-protein interactions and ensure completion of cell death program. Interestingly, non-nuclear p53 plays an important role in activation of cell death via an interaction with the proteins of the Bcl2-family, so these p53 forms can also be responsible for regulation of autophagy (Green and Kroemer, 2009) (Figure 2).
4. Role p53 in regulation of metabolism
4.1 AMPK-mTOR pathway
Carcinogenesis involves metabolic changes required for tumor growth and an adaptation to the environment. Constant growth and proliferation of tumor cells requires constant synthesis of proteins, lipids and nucleotides and all of these processes are regulated by mTOR (mammalian target of rapamycin) kinase (Ben-Sahra et al., 2013b; Peterson et al., 2011; Wullschleger et al., 2006). mTOR is a highly conserved protein kinase which belongs to the PIKK (phosphatidylinositol kinase related kinase) family found in all eukaryotic organisms (Wullschleger et al., 2006). mTOR forms two complexes mTORC1 (mTOR complex 1) and mTORC2 (mTOR complex 2) (Wullschleger et al., 2006). mTORC1 regulates protein synthesis via phosphorylation of p70S6K (p70S6 kinase) and 4EBP1 (eIF4E binding protein 1). While p70S6K phosphorylates ribosomal S6 protein potentially modulating its activity, 4EBP1 binds and inactivates a factor of initiation of Cap-dependent transcription eIF4E. In its hypophosphorylated forms, 4EBP1 has high avidity to eIF4E causing inhibition of translation of many mRNA with structured 5′UTR, encoding proteins critical for cell growth and metabolism. Phosphorylation of 4EBP1 on multiple sites by mTORC1 leads to disintegration of the 4EBP1-eIF4E protein complex, leading to eIF4E release and the activation of translation (Wullschleger et al., 2006). mTORC1 is activated by Rheb and RagA,B,C, and D small GTPases in a cooperative manner. While Rag proteins are activated by amino acids, Rheb is tightly controlled by growth factors or stress insults via tuberoses scleroses 1 and 2 protein complex (TSC1:TSC2) where TSC2 is GAP (GTP activating protein) for Rheb (Laplante and Sabatini, 2012). Being activated by upstream signals, the TSC1:TSC2 complex causes conversion of Rheb bound GTP to GDP form leading to Rheb inhibition. Insulin and growth factors inhibit TSC1:TSC2 complex via phosphorylation by AKT kinase, causing activation of Rheb-mTORC1 axis and stimulation of cell growth and proliferation. Among the negative regulators of mTORC1, AMPK (AMP-activated protein kinase) plays a major role due to its susceptibility to many stress factors, including nutrient/energy deficiency, accumulation of Ca2+, oxidative stress and DNA-damage. AMPK directly phosphorylates TSC2 and Raptor proteins, which leads to inhibition of TORC1 (Laplante and Sabatini, 2012; Mihaylova and Shaw, 2011).
p53 inhibits mTORC1 in response to genotoxic stress and some other stimuli via the activation of expression of mTORC1 inhibitors such as IF-BP3, TSC2, PTEN and AMPKβ1 (Budanov, 2011; Levine et al., 2006). p53 also stimulates phosphorylation of AMPKα subunits on T172, required for mTORC1 inhibition (Feng et al., 2005). p53-activated Sesn1 and Sesn2 play a major role in activation of AMPK phosphorylation in response to p53 (Budanov and Karin, 2008). As a result, mTORC1 inhibition leads to suppression of protein synthesis and activation of autophagy (Budanov, 2011; Loayza-Puch et al., 2013), preventing accumulation of protein aggregates and damaged mitochondria, which is detrimental for cell viability. Inhibition of protein synthesis can also re-direct ATP from energy-consuming anabolism toward repair processes to protect cell homeostasis. In parallel, autophagy supplies cells with demanded ATP via digestion of cellular constituents (Kroemer et al., 2010). Inactivation of p53 in cancer cells has long-lasting consequences on metabolism and angiogenesis instigated by hyperactivation of mTORC1. One of the critical targets of mTORC1 is the transcription factor hypoxia-inducible factor 1 (HIF1) (Laplante and Sabatini, 2012). HIF1 is composed from stable HIF1β and inducible HIF1α subunits (Semenza, 2012). mTORC1 stimulates the translation of HIF1α, activating expression of many genes involved in adaptation to metabolic derangements and hypoxia such as glycolytic enzymes and angiogenic factors supporting the viability of cancer cells in conditions of nutrient and oxygen deprivation (Semenza, 2012). mTORC1 is also a critical activator of transcriptional factor SREBP (sterol regulatory element-binding protein), that induces genes of lipid biosynthesis required for the growth of cancer cells (Zoncu et al., 2011)(Figure 3).
Figure 3. p53 plays an important role in adaptation to metabolic derangements.

A shortage in metabolites such as glucose, glutamine and serine activate p53 which tune up metabolism to suppress undesired anabolic processes via the inhibition of mTORC1, stimulate ATP production via mitochondrial respiration rather than glycolysis, and use alternative fatty acid oxidation as other sources of ATP production. Dysregulation of these processes due to p53 inactivation in cancer cells can be beneficial for tumor growth.
4.2 Regulation of glycolysis and oxidative phosphorylation by p53
a) Glycolysis
Cancer cells have higher glycolytic rates and rely more on glycolysis for ATP production than normal cells (DeBerardinis and Thompson, 2012). This observation has been made by Otto Warburg in the 1930s and was called the “Warburg effect”. Although the glycolytic pathway is much less efficient for ATP production, under abundant glucose supply, ATP production through glycolysis is faster than via oxidative phosphorylation. Moreover, glycolysis allows cells to generate many intermediates of biosynthetic pathways involved in the synthesis of carbohydrates, DNA, proteins and lipids (DeBerardinis and Thompson, 2012). p53 inhibits the Warburg effect by suppressing glycolysis and stimulating mitochondrial respiration (Maddocks and Vousden, 2011). As discussed earlier, p53-inducible protein TIGAR acts as a PFK-2/FBPase-2 biphosphatase (Bensaad et al., 2006). Consequently, via induction of TIGAR, p53 redirects glucose derivatives to the pentose phosphate pathway (PPP) (Bensaad et al., 2006). This leads to the production of NADPH, the important component required for the biosynthesis of ribose-6-phosphate (Maddocks and Vousden, 2011). p53 also inhibits glycolysis via ubiquitination and degradation of the PGM protein (Liang et al., 2013). Besides its direct effects on glycolytic enzymes, p53 also suppresses glycolysis by restricting glucose transport by inhibiting glucose transporters such as GLUT1, GLUT4 and GLUT3 (Liang et al., 2013).
p53 and mitochondrial respiration
While having a negative impact on glycolysis, p53 also stimulates mitochondrial respiration, the most efficient process for ATP production (DeBerardinis and Thompson, 2012). It can be regulated through several mechanisms. p53 directly activates several genes which support oxidative phosphorylation in mitochondria such as SCO2 (synthesis of cytochrome C oxidase 2), apoptosis-inducing factor (AIF), glutaminase 2 (GLS2), Parkin and p53R2 (Budanov, 2011; Liang et al., 2013). SCO2 is involved in the formation of the cytochrome c oxidase complex (complex IV) (Matoba et al., 2006), while AIF supports the stability of the mitochondrial complex I (Vahsen et al., 2004). Additionally, GLS2, the enzyme that controls the production of glutamate and α-ketoglutarate, supports mitochondrial oxidative phosphorylation and ATP production (Hu et al., 2010). Moreover, glutamate is a precursor of antioxidant GSH, thus GLS2 stimulates GSH production and the antioxidant response (Suzuki et al., 2010). Parkin also supports mitochondrial oxidative phosphorylation (Zhang et al., 2011). The potential mechanism involves stimulation of the expression of PDHA1 (pyruvate dehydrogenase E1α1), a component of the pyruvate dehydrogenase complex (PDH), which converts pyruvate into acetyl-CoA, the primary substrate for the TCA cycle. In parallel, p53 stimulates PDH through the repression of PDK2 (pyruvate dehydrogenase kinase 2), which phosphorylates and inhibits PDHA1. As a result p53 stimulates acetyl-CoA production and mitochondrial respiration, preventing the conversion of pyruvate to lactate (Zhang et al., 2011). p53 can also enhance mitochondrial respiration through the control of mitochondrial integrity via regulation of autophagy, DNA-stability and antioxidant protection (Figure 3).
5. p53 controls of cell viability in response to nutrient deprivation
Metabolic stress induced by glucose deprivation leads to a significant decrease in ATP production and induces p53 via activation of AMPK and ATM. Both kinases directly and indirectly phosphorylate human p53 on Ser15, leading to the activation of inhibitor Cyclin-CDK complexes p21 and reversible cell cycle arrest in G1 (Assaily et al., 2011; Jones et al., 2005). As a result, p53 inhibits cell proliferation to protect cells against cell death induced by glucose deprivation (Jones et al., 2005). Interestingly, pharmacological activators of AMPK such as metformin and Aicar suppress growth of p53-deficient tumors, but has no effect on p53-positive tumors in xenograft based studies, indicating the important role of p53 in protection of cell viability in response to AMPK activation. Consequently, metformin stimulates cell death in p53-deficient, but not in p53-proficient cells deprived with glucose (Buzzai et al., 2007). The protective effect of p53 against cell death can be explained by several mechanisms including the activation of autophagy and fatty acid β-oxidation, which supply cells with ATP. Besides glucose deprivation, serine starvation is another type of metabolic stress involved in p53 activation. Similar to its effects on the viability of glucose-starved cells, p53 activates p21 and induces cell cycle arrest preventing cell death in response to serine withdrawal. Moreover, p53-proficient cells show suppressed glycolysis and higher rates of oxidative phosphorylation stimulating higher outcome of ATP production. Serine starved p53-deficient cells fail to recover from serine depletion, resulting in oxidative stress and cell death (Maddocks et al., 2013). Depletion of another amino acid – glutamine also activates p53, that supports cell viability under glutamine-depleted conditions (Reid et al., 2013). Glutamine induces p53 through transcriptional upregulation of B55α (Ppp2r2a), a regulatory subunit of PP2A (protein phosphatase 2A). PP2A activates p53 via dephosphorylation and inactivation of EDD (E3 identified by differential display), HECT domain-containing E3 ubiquitin-ligase which negatively regulates p53 (Reid et al., 2013). Nevertheless, in the conditions of genotoxic stress, metabolic regulation by p53 can play pro-apoptotic role. In response to DNA-damage p53 directly activates expression of GAMT (guanidinoacetate methyltransferase), an enzyme involved in creatine synthesis (Ide et al., 2009). Creatine metabolism helps to maintain the proper levels of ATP, which is critical for apoptosis. GAMT also stimulates fatty acid β-oxidation, the source of energy in nutrient-depleted conditions. Accordingly GAMT silencing resulted in a significant decrease of ATP production and suppression of p53-induced cell death (Ide et al., 2009). Lpin1 is another factor that stimulates fatty acid β-oxidation in a cell-type specific manner in response to p53 activation. It interacts with PPARα (peroxisome proliferator-activated receptor α) and PGC1α (PPARγ coactivator 1α) stimulating transcription of genes involved in fatty acid βoxidation. As a result, it potentiates the effects of p53 on the regulation of metabolism, ROS and cell viability (Assaily et al., 2011) (Figure 3).
Among the factors, which modulate pro-survival or pro-apoptotic functions of p53 via the activation of pro-survival genes involved in regulation of ROS, metabolism, and cell cycle arrest, a critical role is played by p53-co-activator PGC1α. Under starvation conditions PGC1α binds p53 and stimulates activation of pro-survival genes including TIGAR, GADD45, SCO2, Sesn2 and p21, but not pro-apoptotic Puma and Bax (Sen et al., 2011). Prolonged starvation causes PGC1α degradation via ubiquitin-proteosomal pathway. This process is controlled by binding of PGC1α with RNF2, a polycomb group protein that possesses E3 ubiquitin ligase activity. Activation of RNF2 by prolonged stress causes inactivation of PGC1α and a transcriptional switch toward p53-regulated proapoptotic genes (Sen et al., 2011).
6. Mutant p53 and regulation of metabolism
p53 is inactivated in many human tumors through point mutations, deletions, overexpression of Mdm2 or some viral proteins leading to the loss-of-function phenotype (Levine, 1997). Inactivation of p53 leads to metabolic derangements, uncontrolled ROS production and genomic instability (Budanov, 2011). In addition, many tumors express a mutant form of p53 which although is unable to normally regulate the expression of many p53-regulated genes, still might have residual activity of the wild-type protein or regulate expression of a new set of genes via the gain-of-function mechanism. It was demonstrated that mutant p53 forms contribute to viability, invasiveness, migration, and metastasis of cancer cells (Muller and Vousden, 2013). Mutant p53 can also stimulate persistent inflammation by activating the NF-κB transcription factor. Inflammation is a critical promoter of carcinogenesis, involved in the production of ROS and RNS, which can fuel mutagenesis (Cooks et al., 2013).
In contrast to the wild-type form, a hot-spot p53 R273H mutant stimulates production of reactive oxygen species via inhibition of the NRF2 transcription factor causing a decrease in expression of antioxidant and detoxifying enzymes such as NQO1 and HO1 (Kalo et al., 2012). Mutant p53 can also inhibit p73, another member of the p53 family, via direct protein-protein interactions (Di Como et al., 1999). As reported, the transcriptionally active form of p73 (TAp73) regulates mitochondrial activity and prevents ROS accumulation by via transcriptional activation of the mitochondrial complex IV cytochrome C oxidase subunit 4 (Cox4i1). Accordingly, deficiency in TAp73 suppresses ATP production, mitochondrial complex IV activity, and oxygen consumption causing accumulation of ROS (Rufini et al., 2012).
Additionally, mutant p53 stimulates the expression of genes involved in biosynthesis of sterols (mevalonate pathway) and other lipids (Freed-Pastor et al., 2012). The mevalonate pathway is responsible for the synthesis of cholesterol, a critical component of the cell membrane. Lipids are a necessary component required for cell growth and proliferation. Moreover mevalonate pathway is critical for production of farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), the indispensable components of posttranslational modification of Ras or RhoA, correspondingly. Both proteins play a major role in carcinogenesis, supporting proliferation, invasiveness and metastatic ability of cancer cells. Mutant p53 regulates the mevalonate pathway via interaction and activation of transcriptional factor SREBP (Freed-Pastor et al., 2012). Mutant p53 can also affect pyrimidine metabolism providing resistance against anticancer fluoropyrimidine drugs such as 5FU (5-fluorouracil) (Pugacheva et al., 2002). The p53H175 hot-spot mutant activates the expression of the enzyme dUTPase, which exerts conversion of dUTP to dUMP. The enzyme thymidylate synthase uses dUMP for synthesis of dTMP. Uracil can be mistakenly incorporated into DNA and its mis-incorporation is responsible for the cytotoxic activity of 5FU; consequently, inhibitors of thymidylate synthase cause increases in dUTP and increased uracil incorporation into DNA. As a result, it leads to massive DNA-damage, which cause DNA-strain breaks and cell death. On the contrary, dUTPase diminishes dUTP levels preventing uracil mis-incorporation and protects the cell from DNA-damage induced by fluoropyrimidine drugs (Pugacheva et al., 2002)(Figure 4).
Figure 4. Mutant p53 can contribute to carcinogenesis via gain-of-function mechanism.
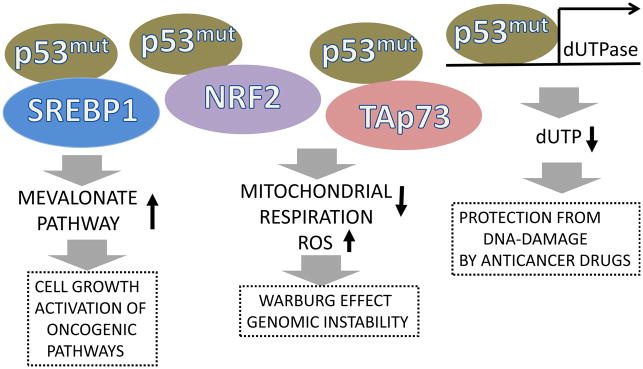
Hot-spot p53 mutants can regulate metabolism, suppress mitochondrial respiration, stimulate ROS production and protect against anti-cancer therapy. These activities are mediated by an interaction with different transcription factors such as: SREBP, critical for activation of mevalonate pathway, as well as NRF2 and Tap73, involved in control of mitochondrial respiration and antioxidant defense. Mutant p53 can also activate the promoter of the dUTPase gene, inducing the expression of enzyme dUTPase which suppresses therapeutic effects of fluoropyrimidine anticancer drugs.
7. Conclusion
While in previous years the p53 research was focused on its role in restraining and eliminating cancer cells as a major mechanism of tumor suppression, now the paradigm is being revisited. The mechanism of “good maintenance” involved in the control of metabolism and ROS seems to be indispensable for the tumor suppressive function of p53 and their inefficiency and perversion can cause numerous defects leading to the attainment of carcinogenic properties. Interestingly, some hot-spot p53 mutants can contribute to carcinogenesis through activation of metabolic pathways not exerted by wild-type p53, which play a role in cell growth and proliferation. Finally, mutant p53 can also decrease sensitivity to anticancer drugs through metabolic modulation, which can make tumors more resistant to anticancer therapy. Thus, restoration of the wild-type function of p53 or inactivation of the mutant p53 form is a highly desirable approach for the future of anticancer treatments.
Acknowledgments
This work is supported by NIH NCI 1RO1CA172660 to Andrei Budanov. The author thanks Brittany Binion for the help in preparation of the manuscript. The author is especially grateful to Nadulya Pryadilova for everyday support.
Abbreviations
- 4EBP1
eIF-4E binding protein 1
- Atg1–17
autophagy gene 1–17
- AIF
apoptosis-inducing factor
- AMPK
AMP activated protein kinase
- Arf1
alternative reading frame 1
- ATP
adenosine triphosphate
- ATM
ataxia-telangiectasia mutated kinase
- ATR
ATM related kinase
- Chk1/Chk2
checkpoint kinase 1/2
- COP1
constitutively photomorphogenic 1
- eIF-4E
eukaryotic translation initiation factor 4E
- FPP
farnesyl pyrophosphate
- FoxO
forkhead box O transcription factors
- GAMT
(guanidinoacetate methyltransferase
- GGPP
geranylgeranyl pyrophosphate
- GLS2
glutaminase 2
- GPX1
glutathione peroxidase 1
- GSH
glutathione
- GSSG
oxidized form of glutathione
- HIF1
hypoxia-inducible factor 1
- HK2
mitochondrial hexokinase 2
- Keap1
Kelch-like ECH-associated protein 1
- MnSOD
manganese superoxide dismutase
- Mdm2
mouse double minute
- NAC
N-acetyl cysteine
- NCF2/p67phox
neutrophil cytosol factor 2
- NRF2
nuclear factor (erythroid-derived 2)-like 2
- mTOR
mammalian target of rapamycin kinase
- mTORC1/2
mTOR complex 1/2
- p53INP1
p53-inducible nuclear protein 1
- PET
paired-end ditag
- PGC1α
PPARγ coactivator 1α
- PGM
phosphoglycerate mutase
- PFK-2/FBPase-2
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase
- PIG1–13
p53-inducible genes 1–13
- PIRH2
p53-induced protein with a RING (Really Interesting New Gene)-H2 domain
- PPARα/γ
peroxisome proliferator-activated receptor α/γ
- RagA,B,C,D
Ras –related GTP-binding protein A,B,C,D
- Rheb
Ras homolog enriched in brain
- ROS
reactive oxygen species
- SCO2
synthesis of cytochrome C oxidase 2
- SREBP1
sterol-regulatory element binding protein 1
- Tap73
transcriptionally active p73
- TIGAR
p53-induced glycolysis and apoptotic regulator
- TSC1/2
tuberoses sclerosis complex protein 1/2
References
- Achanta G, Sasaki R, Feng L, Carew JS, Lu W, Pelicano H, Keating MJ, Huang P. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol gamma. The EMBO journal. 2005;24:3482–3492. doi: 10.1038/sj.emboj.7600819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assaily W, Rubinger DA, Wheaton K, Lin Y, Ma W, Xuan W, Brown-Endres L, Tsuchihara K, Mak TW, Benchimol S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Molecular cell. 2011;44:491–501. doi: 10.1016/j.molcel.2011.08.038. [DOI] [PubMed] [Google Scholar]
- Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, Lee HE, Kang D, Rhee SG. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell metabolism. 2013;17:73–84. doi: 10.1016/j.cmet.2012.12.002. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans. 2003;31:1441–1444. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- Ben-Sahra I, Dirat B, Laurent K, Puissant A, Auberger P, Budanov A, Tanti JF, Bost F. Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death. Cell death and differentiation. 2013a;20:611–619. doi: 10.1038/cdd.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013b;339:1323–1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Bond GL, Hu W, Levine AJ. MDM2 is a central node in the p53 pathway: 12 years and counting. Current cancer drug targets. 2005;5:3–8. doi: 10.2174/1568009053332627. [DOI] [PubMed] [Google Scholar]
- Bryk R, Lima CD, Erdjument-Bromage H, Tempst P, Nathan C. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science. 2002;295:1073–1077. doi: 10.1126/science.1067798. [DOI] [PubMed] [Google Scholar]
- Budanov AV. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxidants & redox signaling. 2011;15:1679–1690. doi: 10.1089/ars.2010.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Lee JH, Karin M. Stressin’ Sestrins take an aging fight. EMBO Mol Med. 2010;2:388–400. doi: 10.1002/emmm.201000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene. 2002;21:6017–6031. doi: 10.1038/sj.onc.1205877. [DOI] [PubMed] [Google Scholar]
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer research. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- Cano CE, Gommeaux J, Pietri S, Culcasi M, Garcia S, Seux M, Barelier S, Vasseur S, Spoto RP, Pebusque MJ, et al. Tumor protein 53-induced nuclear protein 1 is a major mediator of p53 antioxidant function. Cancer research. 2009;69:219–226. doi: 10.1158/0008-5472.CAN-08-2320. [DOI] [PubMed] [Google Scholar]
- Cheung EC, Ludwig RL, Vousden KH. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:20491–20496. doi: 10.1073/pnas.1206530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S, Lozano G, Pikarsky E, Forshew T, Rozenfeld N, et al. Mutant p53 Prolongs NF-kappaB Activation and Promotes Chronic Inflammation and Inflammation-Associated Colorectal Cancer. Cancer cell. 2013;23:634–646. doi: 10.1016/j.ccr.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Cui X. Reactive oxygen species: the achilles’ heel of cancer cells? Antioxidants & redox signaling. 2012;16:1212–1214. doi: 10.1089/ars.2012.4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature reviews Molecular cell biology. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148:1132–1144. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- Di Como CJ, Gaiddon C, Prives C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Molecular and cellular biology. 1999;19:1438–1449. doi: 10.1128/mcb.19.2.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O’Rourke K, Koeppen H, Dixit VM. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429:86–92. doi: 10.1038/nature02514. [DOI] [PubMed] [Google Scholar]
- Essler S, Dehne N, Brune B. Role of sestrin2 in peroxide signaling in macrophages. FEBS letters. 2009;583:3531–3535. doi: 10.1016/j.febslet.2009.10.017. [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–258. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes & development. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell death and differentiation. 2011;18:1598–1607. doi: 10.1038/cdd.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–1130. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenbuchner J, Kuznetsov A, Hermann M, Hausott B, Obexer P, Ausserlechner MJ. FOXO3-induced reactive oxygen species are regulated by BCL2L11 (Bim) and SESN3. Journal of cell science. 2012 doi: 10.1242/jcs.092098. [DOI] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS letters. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7455–7460. doi: 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SP, Amstad P, He P, Robles A, Lupold S, Kaneko I, Ichimiya M, Sengupta S, Mechanic L, Okamura S, et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer research. 2004;64:2350–2356. doi: 10.1158/0008-5472.can-2287-2. [DOI] [PubMed] [Google Scholar]
- Ide T, Brown-Endres L, Chu K, Ongusaha PP, Ohtsuka T, El-Deiry WS, Aaronson SA, Lee SW. GAMT, a p53-inducible modulator of apoptosis, is critical for the adaptive response to nutrient stress. Molecular cell. 2009;36:379–392. doi: 10.1016/j.molcel.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Italiano D, Lena AM, Melino G, Candi E. Identification of NCF2/p67phox as a novel p53 target gene. Cell cycle. 2012;11:4589–4596. doi: 10.4161/cc.22853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Molecular cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- Kalo E, Kogan-Sakin I, Solomon H, Bar-Nathan E, Shay M, Shetzer Y, Dekel E, Goldfinger N, Buganim Y, Stambolsky P, et al. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. Journal of cell science. 2012;125:5578–5586. doi: 10.1242/jcs.106815. [DOI] [PubMed] [Google Scholar]
- Kang MY, Kim HB, Piao C, Lee KH, Hyun JW, Chang IY, You HJ. The critical role of catalase in prooxidant and antioxidant function of p53. Cell death and differentiation. 2013;20:117–129. doi: 10.1038/cdd.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes & development. 2013;27:1016–1031. doi: 10.1101/gad.212282.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura N, Nakamura Y, Miyamoto Y, Miyamoto T, Kabu K, Yoshida M, Futamura M, Ichinose S, Arakawa H. Mieap, a p53-inducible protein, controls mitochondrial quality by repairing or eliminating unhealthy mitochondria. PloS one. 2011;6:e16060. doi: 10.1371/journal.pone.0016060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopnin PB, Agapova LS, Kopnin BP, Chumakov PM. Repression of sestrin family genes contributes to oncogenic Ras-induced reactive oxygen species up-regulation and genetic instability. Cancer research. 2007;67:4671–4678. doi: 10.1158/0008-5472.CAN-06-2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Molecular cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, Alon U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nature genetics. 2004;36:147–150. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR Signaling in Growth Control and Disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva MA, Eaton JS, Shadel GS. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochimica et biophysica acta. 2009;1787:328–334. doi: 10.1016/j.bbabio.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Cho JS, Lambacher N, Lee J, Lee SJ, Lee TH, Gartner A, Koo HS. The Caenorhabditis elegans AMP-activated protein kinase AAK-2 is phosphorylated by LKB1 and is required for resistance to oxidative stress and for normal motility and foraging behavior. J Biol Chem. 2008;283:14988–14993. doi: 10.1074/jbc.M709115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, Ocorr K, Ellisman MH, Bodmer R, Bier E, et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. 2009;327:1223–1228. doi: 10.1126/science.1182228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, Bandyopadhyay G, Li N, Aghajan M, Jang I, et al. Maintenance of metabolic homeostasis by sestrin2 and sestrin3. Cell metabolism. 2012;16:311–321. doi: 10.1016/j.cmet.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SO, Andey T, Jin UH, Kim K, Sachdeva M, Safe S. The nuclear receptor TR3 regulates mTORC1 signaling in lung cancer cells expressing wild-type p53. Oncogene. 2011 doi: 10.1038/onc.2011.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–791. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Feng Z, Mak TW, You H, Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes & development. 2006;20:267–275. doi: 10.1101/gad.1363206. [DOI] [PubMed] [Google Scholar]
- Li FP, Fraumeni JF., Jr Rhabdomyosarcoma in children: epidemiologic study and identification of a familial cancer syndrome. Journal of the National Cancer Institute. 1969a;43:1365–1373. [PubMed] [Google Scholar]
- Li FP, Fraumeni JF., Jr Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Annals of internal medicine. 1969b;71:747–752. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Liu J, Feng Z. The regulation of cellular metabolism by tumor suppressor p53. Cell & bioscience. 2013;3:9. doi: 10.1186/2045-3701-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza-Puch F, Drost J, Rooijers K, Lopes R, Elkon R, Agami R. p53 induces transcriptional and translational programs to suppress cell proliferation and growth. Genome biology. 2013;14:R32. doi: 10.1186/gb-2013-14-4-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013;493:542–546. doi: 10.1038/nature11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks OD, Vousden KH. Metabolic regulation by p53. J Mol Med (Berl) 2011;89:237–245. doi: 10.1007/s00109-011-0735-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, Carnuccio R, Kroemer G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell cycle. 2009;8:1571–1576. doi: 10.4161/cc.8.10.8498. [DOI] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Vina J, Blasco MA, Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature cell biology. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli E, Shen S, Ruckenstuhl C, Bauer MA, Marino G, Galluzzi L, Criollo A, Michaud M, Maiuri MC, Chano T, et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell cycle. 2011;10:2763–2769. doi: 10.4161/cc.10.16.16868. [DOI] [PubMed] [Google Scholar]
- Muller PA, Vousden KH. p53 mutations in cancer. Nature cell biology. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- N’Guessan P, Pouyet L, Gosset G, Hamlaoui S, Seillier M, Cano CE, Seux M, Stocker P, Culcasi M, Iovanna JL, et al. Absence of tumor suppressor tumor protein 53-induced nuclear protein 1 (TP53INP1) sensitizes mouse thymocytes and embryonic fibroblasts to redox-driven apoptosis. Antioxidants & redox signaling. 2011;15:1639–1653. doi: 10.1089/ars.2010.3553. [DOI] [PubMed] [Google Scholar]
- Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer cell. 2008;14:458–470. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JC, Wallace DM, O’Brien CJ, Cotter TG. A novel antioxidant function for the tumor-suppressor gene p53 in the retinal ganglion cell. Invest Ophthalmol Vis Sci. 2008;49:4237–4244. doi: 10.1167/iovs.08-1963. [DOI] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters H, Debeer P, Bairoch A, Wilquet V, Huysmans C, Parthoens E, Fryns JP, Gewillig M, Nakamura Y, Niikawa N, et al. PA26 is a candidate gene for heterotaxia in humans: identification of a novel PA26-related gene family in human and mouse. Hum Genet. 2003;112:573–580. doi: 10.1007/s00439-003-0917-5. [DOI] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- Pugacheva EN, Ivanov AV, Kravchenko JE, Kopnin BP, Levine AJ, Chumakov PM. Novel gain of function activity of p53 mutants: activation of the dUTPase gene expression leading to resistance to 5-fluorouracil. Oncogene. 2002;21:4595–4600. doi: 10.1038/sj.onc.1205704. [DOI] [PubMed] [Google Scholar]
- Reid MA, Wang WI, Rosales KR, Welliver MX, Pan M, Kong M. The B55alpha Subunit of PP2A Drives a p53-Dependent Metabolic Adaptation to Glutamine Deprivation. Molecular cell. 2013;50:200–211. doi: 10.1016/j.molcel.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Reliene R, Fischer E, Schiestl RH. Effect of N-acetyl cysteine on oxidative DNA damage and the frequency of DNA deletions in atm-deficient mice. Cancer research. 2004;64:5148–5153. doi: 10.1158/0008-5472.CAN-04-0442. [DOI] [PubMed] [Google Scholar]
- Rufini A, Niklison-Chirou MV, Inoue S, Tomasini R, Harris IS, Marino A, Federici M, Dinsdale D, Knight RA, Melino G, et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes & development. 2012;26:2009–2014. doi: 10.1101/gad.197640.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert R, Erker L, Barlow C, Yakushiji H, Larson D, Russo A, Mitchell JB, Wynshaw-Boris A. Cancer chemoprevention by the antioxidant tempol in Atm-deficient mice. Human molecular genetics. 2004;13:1793–1802. doi: 10.1093/hmg/ddh189. [DOI] [PubMed] [Google Scholar]
- Selivanova G. p53: fighting cancer. Current cancer drug targets. 2004;4:385–402. doi: 10.2174/1568009043332934. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Satija YK, Das S. PGC-1alpha, a key modulator of p53, promotes cell survival upon metabolic stress. Molecular cell. 2011;44:621–634. doi: 10.1016/j.molcel.2011.08.044. [DOI] [PubMed] [Google Scholar]
- Seo YR, Kelley MR, Smith ML. Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14548–14553. doi: 10.1073/pnas.212319799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7461–7466. doi: 10.1073/pnas.1002459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al. Regulation of autophagy by cytoplasmic p53. Nature cell biology. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, et al. AIF deficiency compromises oxidative phosphorylation. The EMBO journal. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-Miguel S, Buckbinder L, Jean P, Gelbert L, Talbott R, Laidlaw J, Seizinger B, Kley N. PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene. 1999;18:127–137. doi: 10.1038/sj.onc.1202274. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. p53 in health and disease. Nature reviews Molecular cell biology. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wang PY, Ma W, Park JY, Celi FS, Arena R, Choi JW, Ali QA, Tripodi DJ, Zhuang J, Lago CU, et al. Increased oxidative metabolism in the Li-Fraumeni syndrome. The New England journal of medicine. 2013;368:1027–1032. doi: 10.1056/NEJMoa1214091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes & development. 1996;10:2411–2422. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]
- Yoon KA, Nakamura Y, Arakawa H. Identification of ALDH4 as a p53-inducible gene and its protective role in cellular stresses. Journal of human genetics. 2004;49:134–140. doi: 10.1007/s10038-003-0122-3. [DOI] [PubMed] [Google Scholar]
- Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16259–16264. doi: 10.1073/pnas.1113884108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nature reviews Molecular cell biology. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]