

Abstract
Recently, it has been suggested that pectinases could be used to hydrolyze pectin in biorefineries based on pectin-rich agro-industrial wastes. However, for this to be viable, the cost of their production would need to be lowered significantly. In fact, over the last few decades, there have been many attempts to improve pectinase production by existing strains or to screen for new strains from environmental isolates. In these studies, it is necessary to measure pectinase activities. Many researchers use single-time-point assays that involve incubation of pectinolytic extracts with pectic substrates for a fixed time, followed by determination of the liberated reducing sugars. However, different researchers use quite different conditions for this assay. Furthermore, no attention has been given to the reaction profile during the assay. In the current work, we show, for the first time, that a significant deceleration of the rate of liberation of reducing sugars occurs over the first ten minutes of the reaction. As a consequence, the incubation time used in a single-time-point assay has a large effect on the value obtained for the activity. In fact, we demonstrate that, depending on the particular combination of incubation time, pectin concentration and reaction temperature, the same extract could be reported to have activities that differ by an order of magnitude. In addition, we show that the relative activities obtained with polygalacturonic acid do not correlate with those obtained with pectin. We conclude that it is currently impossible to make meaningful comparisons between pectinase activities reported in the literature by workers who have used different assay conditions. Therefore there is an urgent need for the development of a standardized assay for evaluating the saccharification potential of pectinase complexes.
Introduction
The term “pectinases” refers to a group of enzymes that act together to solubilize, de-esterify and depolymerize the complex structure of native pectin [1]. These enzymes have varied applications in the food and beverage industries, including the extraction and clarification of juices, the improvement of the organoleptic properties of wine, the increasing of the efficiency of extraction of vegetable oils and the acceleration of coffee and tea fermentations [2], [3]. Pectinases are also used as additives in animal feeds, mainly for poultry and ruminants, to improve digestibility and nutritional value [4]. Pectinases could also be used for the saccharification of pectin-rich agricultural residues, with the resulting sugars being used for the production of ethanol or platform chemicals [5]–[7]. In this manner, they would play an important role in the recently proposed citrus waste biorefineries [8], [9].
The saccharification of pectin involves a number of enzymes working in conjunction (Table 1) [10], [11]. In the current paper, we use the term “pectinolytic activity” to denote the liberation of reducing sugars from pectin by this mixture of enzymes, which we refer to as a “pectinase complex”. The production of pectinase complexes by submerged-liquid fermentation and solid-state fermentation of various microorganisms has been studied over the last two decades or so, with many researchers having isolated new strains and undertaken medium optimization and strain improvement programs [12], [13]. In these programs, it is necessary to assay pectinolytic activity. When the interest is to produce pectinases for the food industry, it is common to determine pectinolytic activity based on measurements of the reduction in viscosity of a pectin solution [13]. Specific assays based on ruthenium red have also been developed for endopolygalacturoanases [14], [15]. However, the progress towards complete saccharification is best characterized by measuring the liberation of reducing sugars from pectin. The most frequently used method for doing this involves a “single-time-point” assay that requires two steps. The first step is a hydrolysis step, in which the pectinase-containing extract is incubated for a fixed time under selected conditions of pH, temperature and initial pectin concentration. In the second step, the reducing sugars liberated in the first step are quantified, either by the DNS method [16] or by the Somogyi-Nelson method [17], [18]. The advantage of this assay is that it is simple, uses relatively cheap reagents and can be used to process a relatively large number of samples simultaneously. It is performed manually in most laboratories, although a fully automated assay would be possible [19]. Activities are expressed in terms of “µmol of D-galacturonic acid equivalents produced per minute” and reported “per mL of fermentation broth”, in the case of submerged-liquid fermentation, or “per gram of dry solids”, in the case of solid-state fermentation.
Table 1. Enzymes that contribute to the degradation of pectin*.
| Name | EC number | Mechanism |
| Pectin lyase | 4.2.2.10 | Eliminative cleavage of (1→4)-α-d-galacturonan methyl ester to give oligosaccharides with 4-deoxy-6-O-methyl-α-d-galact-4-enuronosyl groups at their non-reducing ends |
| Pectate lyase | 4.2.2.2 | Eliminative cleavage of (1→4)-α-d-galacturonan to give oligosaccharides with 4-deoxy-α-d-galact-4-enuronosyl groups at their non-reducing ends |
| Pectate disaccharide lyase | 4.2.2.9 | Eliminative cleavage of 4-(4-deoxy-α-d-galact-4-enuronosyl)-d-galacturonate from the reducing end of pectate (i.e. de-esterified pectin) |
| Pectate trisaccharide-lyase | 4.2.2.22 | Eliminative cleavage of unsaturated trigalacturonate as the major product from the reducing ends of polygalacturonic acid/pectate |
| Rhamnogalacturonan endolyase | 4.2.2.23 | Endotype eliminative cleavage of l-α-rhamnopyranosyl-(1→4)-α-d-galactopyranosyluronic acid bonds in rhamnogalacturonan I domains of hairy regions of pectin |
| Endo-polygalacturonases | 3.2.1.15 | Random hydrolysis of (1→4)-α-d-galactosiduronic linkages in pectate and other galacturonans |
| Exo-polygalacturonases | 3.2.1.67 | Hydrolysis of d-galacturonic acid residues from the reducing ends of polygalacturonate chains |
| Pectin methyl esterases | 3.1.1.11 | Demethoxylation of pectin, forming pectate |
| Pectin acetyl esterases | 3.1.1.6 | Deacetylation of pectin, forming pectate |
| Exo-poly-α-galacturonosidase | 3.2.1.82 | Hydrolysis of pectic acid from the non-reducing end, releasing digalacturonate |
| Rhamnogalacturonan hydrolase | 3.2.1.171 | Endohydrolysis of α-d-GalA-(1→2)-α-l-rha-glycosidic bonds in the rhamnogalacturonan I backbone, releasing oligosaccharides with β-d-GalA at the reducing end. |
| α-l-rhamnosidase | 3.2.1.40 | Hydrolysis of the glycosidic bonds between rhamnose and galacturonic acid residues |
| Arabinan endo-1,5-α-l-arabinanase | 3.2.1.99 | Endohydrolysis of (1→5)-α-arabinofuranosidic linkages in (1→5)-arabinans |
| Arabinogalactanendo-β-1,4-galactanase | 3.2.1.89 | Hydrolysis of (1→4)-β-d-galactosidic linkages in type I arabinogalactans. |
| α-l-arabinofuranidases | 3.2.1.55 | Hydrolysis of terminal non-reducing α-l-arabinofuranoside residues in α-l-arabinosides. |
In order to evaluate the success of screening and strain optimization programs, it is necessary to compare the results obtained for pectinolytic activity with those reported in the literature. However, different authors carry out the hydrolysis step quite differently, with either pectin or polygalacturonic acid being used as the substrate and with diverse values being used for the pH, temperature, reaction time and substrate concentration (Table 2) [20]–[36]. These different assay conditions have consequences for comparing results, but these consequences have not yet been explored. Further, although some efforts have been made to measure initial velocities in the characterization of the kinetics of individual enzymes [37], the exact shape of the early reaction profile during assays for the liberation of reducing sugars by pectinase complexes is not clear: the profiles that have been obtained involve measurements that are rather widely spaced in time and initial rates are either not characterized or the profiles themselves not reported [38], [39]. We address these two issues in the current paper, firstly, by obtaining detailed profiles for the liberation of reducing sugars in reactions carried out under different conditions of temperature and initial pectin concentration and, secondly, by comparing reaction profiles for two commercial preparations and two crude extracts, using citric pectin and polygalacturonic acid. We identify, for the first time, the existence of a significant deceleration over the first ten minutes of the reaction and discuss the implication of this early deceleration, and other results, for the assaying of pectinolytic activities.
Table 2. Hydrolysis conditions used in assays for pectinase activities reported in the literature.
| Microorganism (cultivation mode)* | Pectic substrate & concentration (% m/v)# | T (°C) | t (min) | pH | Activity (SSF: U/g; SLF: U/mL) | Reference |
| Thermomucor indicae-seudaticae (SSF) | 1% pectin | 60 | 10 | 5.5 | 108 | [20] |
| Penicillium viridicatum (SSF) | 0.8% pectin | 50 | 10 | 5.5 | 71 | [21] |
| Thermoascus aurantiacus (SSF) | 0.8% pectin | 55 | 10 | 5.0 | 43 | [22] |
| Fusarium moniliforme (SSF) | 0.5% pectin | 40 | 30 | 4.5 | 43 | [23] |
| Aspergillus niger (SSF) | 0.45% pectin | 45 | 30 | 4.5 | 25 | [24] |
| Aspergillus awamori (SSF) | 0.33% pectin | 45 | 10 | 5.0 | 9 | [25] |
| Streptomyces sp. (SLF) | 1% pectin | 60 | - | 3 | 162 | [26] |
| Penicillium occitanis (SLF) | 0.45% pectin | 50 | 60 | 4.8 | 64 | [27] |
| Aspergillus niger (SLF) | 0.5% pectin | - | 15 | 4.5 | 34 | [28] |
| Aspergillus flavipes (SLF) | 1% pectin | 45 | 20 | 5 | 20 | [29] |
| Aspergillus niger (SLF) | 0.2% pectin | 30 | - | 5.3 | 1.3 | [30] |
| Trichoderma viridae (SlSF) | 0.2% PGA | 30 | - | 5 | 10 | [31] |
| Aspergillus sojae (SSF) | 0.2% PGA | 26 | - | 6.6 | 30 | [32] |
| Bacillus subtilis (SSF) | 0.24% PGA | 65 | 10 | 9.5 | 6592 | [33] |
| Aspergillus niger (SSF) | - | 40 | - | 3.5 | 135 | [34] |
| Aspergillus sp. (SSF) | 0.17% PGA | - | 10 | 5.3 | 390 | [35] |
| Penicillium grisereoserum (SLF) | 0.75% PGA | 40 | 20 | 4.8 | 3490 | [36] |
*SSF: solid-state fermentation; SLF: submerged fermentation; SlSF: slurry-state fermentation.
PGA: polygalacturonic acid.
-: not specified.
Materials and Methods
Strains
Aspergillus niger CH4 is maintained in the culture collection of the Biomedical Research Institute of the Universidad Nacional Autónoma de México (Mexico City, Mexico) and was kindly provided by Prof. Dr. Jesus Cordova and Prof. Dr. Gustavo Viniegra-Gonzalez. Aspergillus oryzae CPQBA 394-12 DRM 01 was isolated from a passion fruit peel and is maintained in the culture collection of the University of Campinas (Campinas, Brazil).
Substrates for solid-state fermentations
Wheat bran was purchased from the municipal market of Curitiba, located in the state of Paraná, Brazil, and was used as obtained. Sugarcane bagasse was obtained from Destilarias Melhoramentos S/A (Jussara, Brazil) and was sieved to recover particles between 1.7 and 1 mm. Citrus pulp was obtained from the agro-industrial cooperative Corol (Rolândia, Brazil) and was dried to 13% (w/w, wet basis) moisture.
Preparation of crude extracts from solid-state fermentations
The medium contained 1.5 g dry sugarcane bagasse and 3.5 g of either dry wheat bran (for A. niger CH4) or dry citrus pulp (for A. oryzae CPQBA 394-12 DRM 01). The mixture was placed in a 250-mL Erlenmeyer flask and autoclaved (121°C, 15 min). Saline solution (containing, in g/L, K2HPO4 3, (NH4)2SO4 13, MgSO4.7H2O 5, KCl 10, FeSO4.7H2O 0.09) was autoclaved (121°C, 15 min) and added to each flask to obtain a final moisture content of 70% (w/w, wet basis). The solid media were inoculated with 107 spores per gram of dry substrate and incubated at 30°C for 24 h. The fermented solids were then lyophilized and stored at 4°C. For the extraction, 100 mL of acetate buffer (0.2 M, pH 4.5) was added per gram of lyophilized fermented solids and the mixture was incubated on an orbital shaker at 30°C for 30 min at 180 rpm. The crude extracts were vacuum filtered using Whatman n°1 filter paper. The filtrate was stored at 4°C.
Commercial enzymes and pectic substrates
The commercial enzyme preparations used were Pectinex Ultra SPL (Novozymes, Denmark), from Aspergillus aculeatus, and Pectinase P4716 (Sigma-Aldrich, USA), from Aspergillus niger. These preparations were diluted 2500-fold in acetate buffer (0.2 M, pH 4.5) before adding to the substrate mixture. Citric pectin (D-galacturonic acid content>74% and methoxy content>6.7%) and polygalacturonic acid (∼95%, molecular weight 25000–50000), both from Sigma-Aldrich (product numbers P9135 and 81325, respectively), were solubilized in acetate buffer (0.2 M, pH 4.5).
Determination of the profiles for the release of reducing sugars
The studies of the effect of temperature and pectin concentration on the reaction profile were undertaken using the crude extract of A. niger CH4. For this, multiple identical tubes were prepared, each containing 0.25 mL of crude extract and 0.25 mL of citric pectin in acetate buffer (0.2 M, pH 4.5). The stated pectin concentration represents the value for the mixture at zero time. The samples were incubated at the stated temperature. At 30-s intervals, 0.5 mL of DNS was added to the mixture to stop the reaction. All the profiles were obtained with aliquots taken from the same crude extract.
In the study of initial hydrolysis profiles undertaken with different pectinase complexes and pectic substrates, 15 mL of the appropriate substrate solution was incubated in a 100-mL jacketed reactor at 30°C. To start the reaction, 15 mL of the appropriate pectinase preparation was added, giving an initial substrate concentration in the reaction mixture of 0.5% (m/v). Samples of 0.5 mL were taken at intervals of 10 s (0–2 min), 30 s (2–15 min) or 1 min (15–37 min). Each sample was added to 0.5 mL of DNS in an ice bath to stop the reaction.
In the study of long hydrolysis times that was undertaken with different pectinase complexes and pectic substrates, 75 mL of the appropriate substrate solution was incubated in a 250-mL Erlenmeyer flask at 30°C. To start the reaction, 75 mL of the appropriate enzyme preparation was added, giving an initial substrate concentration of 0.5% (m/v). The mixture was incubated on an orbital shaker at 200 rpm and 30°C for 72 h. Samples of 0.5 mL were taken at various times and added to 0.5 mL of DNS in an ice bath to stop the reaction.
The liberation of reducing sugars was followed using the DNS method [16]. Zero time absorbances were subtracted from the readings. Reducing sugar concentrations were estimated as D-galacturonic acid equivalents, using a calibration curve constructed with D-galacturonic acid (Sigma-Aldrich, ≥98.0% purity) concentrations from 0.94 to 9.4 mmol/L.
Determination of corresponding single-time-point activities from reaction profiles
A fifth-order polynomial was fitted to the reaction profile obtained under each set of conditions. For all fits, the value of R2 was ≥0.90. The fitted curve is only shown in Figure 1; for the sake of clarity, other fitted curves are not shown, since the graphs already contain several different plots. The activity that would have been obtained in a single-time-point assay undertaken using a particular time under a particular set of reaction conditions, denominated A(t) and with units of U/g, was then calculated as:
| (1) |
where C(t) is the reducing sugar concentration (µmol/mL, which is numerically equivalent to the value plotted on the graph as mmol/L) at time t (min), obtained by substituting t into the corresponding fitted polynomial, D is the dilution factor for the extract in the assay (equal to 2) and B is the ratio of buffer to dry solids used in the extraction step (mL/g, equal to 100). Note that 1 U corresponds to the production of 1 µmol of D-galacturonic acid equivalents per min.
Figure 1. Detailed profile for the hydrolysis of pectin during the first 40 min of the reaction.
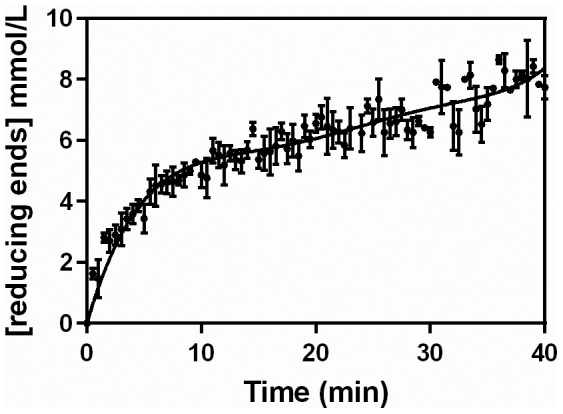
The hydrolysis was performed at 40°C, with an initial pectin concentration of 0.5% (w/v), using the pectinase complex produced by Aspergillus niger CH4 in solid-state fermentation. The reaction was undertaken in triplicate. The mean values for each time were plotted, with the error bars representing the standard error of the mean. The curve represents the best fitting fifth-order polynomial.
Stability assays
The crude extract obtained from solid-state fermentation of A. niger CH4 was incubated for 10, 30 and 60 min at 30, 40, 50 and 55°C. The assay for residual activity was undertaken at 50°C, for 10 min, using 0.5% (w/v) pectin (initial concentration in the reaction mixture). The residual activity was calculated relative to the activity obtained in an assay undertaken under the same conditions but without previous incubation.
Results
Reaction profiles under different conditions
Since pectin degradation profiles that have been published in the literature typically only provide data points at intervals of several minutes, the first experiment involved the characterization of the degradation of pectin by a crude extract from A. niger CH4, using intervals of only 30 s. It was done at 40°C and with a pectin concentration of 0.5% (w/v). A key result, which has not previously been reported in the literature, was that the rate of liberation of reducing sugars decelerated significantly over the first 10 min (Figure 1). Then, from 10 to 40 min, there was an almost linear increase in reducing sugar concentration.
Reaction profiles were also followed at various different temperatures, using a pectin concentration of 0.5% (w/v). As in the previous experiment, in all cases there was a significant deceleration over the first 10 min (Figure 2). The effect of temperature on the liberation of reducing sugars was greatest over this 10 min period, with the amount of reducing sugars at 40 and 50°C being slightly higher than that at 30°C. From 10 min onwards, the profiles had similar slopes.
Figure 2. Reaction profiles obtained over the first 40 min at different temperatures.
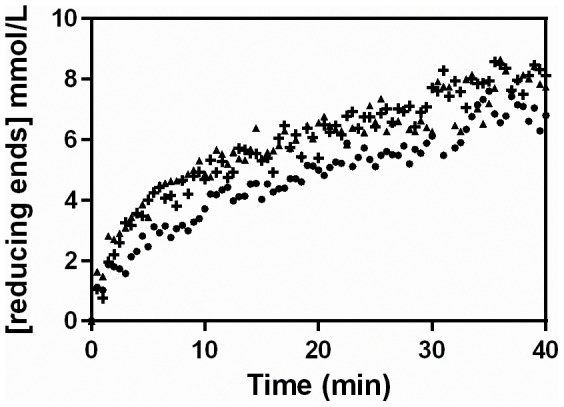
The hydrolysis was performed using 0.5% (m/v) pectin, with the pectinase complex produced by Aspergillus niger CH4 in solid-state fermentation. Symbols: (•) 30°C, (▴) 40°C and (+) 50°C.
Reaction profiles were then followed, at 45°C, with different initial pectin concentrations (Figure 3). The initial reaction rate was highest with 1% (w/v) pectin. However, this concentration led to a viscous solution, which made pipetting difficult, and undissolved pectin was visible suspended in the reaction medium. These problems are probably responsible for the greater dispersion of data points in the 1% (w/v) profile in Figure 3, compared to the profiles obtained at lower initial pectin concentrations. Again, in all the conditions, a significant deceleration occurred over the first 10 min.
Figure 3. Reaction profiles obtained over the first 40 min with different initial pectin concentrations.
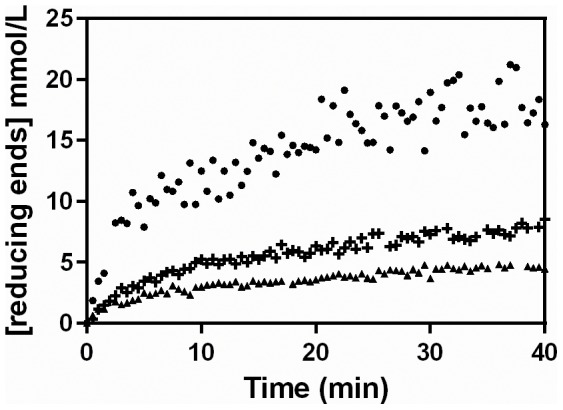
The hydrolysis was performed at 45°C, with the pectinase complex produced by Aspergillus niger CH4 in solid-state fermentation. Symbols: Initial pectin concentrations of (▴) 0.25% m/v, (+) 0.5% m/v and (•) 1% m/v.
Effect of temperature on loss of pectinolytic activity
Increasing temperatures not only increase the intrinsic rate of enzyme-catalyzed reactions, but also increase the rate of enzyme denaturation. In order to investigate the degree to which denaturation might be occurring in the profiles in Figure 2, the extract was incubated at various temperatures (Figure 4). The pectinase activity was stable over 60 min at 30°C and 40°C. At 50°C, the residual activity fell significantly, reaching only 25% at 60 min. At 55°C, denaturation was significantly faster, with only 6% residual activity at 10 min. These results suggest that thermal denaturation is not the cause of the early deceleration in Figure 1, since that reaction was carried out at 40°C, but that some denaturation probably occurred in the reactions carried out at higher temperatures in Figures 2 and 3.
Figure 4. Residual activity curves for the extract incubated over 60 min at different temperatures.
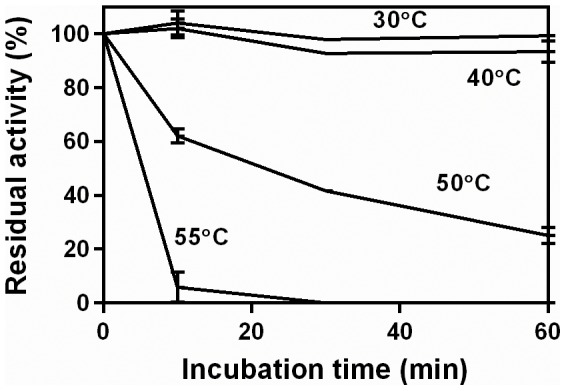
The pectinase complex produced by Aspergillus niger CH4 in solid-state fermentation was used. Residual activities were obtained at 50°C, in a 10-min assay, using 0.5% pectin (w/v).
Consequences of determining pectinolytic activity using single-time-point assays
The reaction profiles obtained above were used to calculate the pectinolytic activities that would be obtained in single-time-point (STP) assays, carried out under various combinations of temperature and pectin concentration that are typically used in the literature (see Table 2). These will be referred to as “STP-activities”. STP activities were calculated for each of three commonly used reaction times, 10, 15 and 30 min, using the reaction profiles in Figures 2 and 3 (Table 3). It is important to note that all the assays in these two figures involved the same amount of enzyme, with the differences in STP-activity being due to the conditions and the time chosen for the reaction. The STP-activity was influenced most by the reaction time and the concentration of pectin. Importantly, the highest value of STP-activity, obtained for 1.0% (w/v) pectin, 45°C and 10 min, was 8-fold greater than the lowest value of STP-activity, obtained for 0.25% (w/v) pectin, 45°C and 30 min.
Table 3. Single-time-point pectinase activities that would be obtained for the same enzymatic extract if incubated under different reaction conditions for different times.
| Reaction conditions | “Single-time-point activity” (STP-activity) (U/g) at the stated reaction times* | |||
| [pectin] (%, w/v) | T (°C) | 10 min | 15 min | 30 min |
| 0.25 | 45 | 60 | 44 | 29 |
| 0.5 | 30 | 77 | 59 | 40 |
| 0.5 | 40 | 106 | 76 | 47 |
| 0.5 | 45 | 95 | 72 | 47 |
| 0.5 | 50 | 100 | 73 | 47 |
| 1.0 | 45 | 242 | 176 | 119 |
Early profiles for different pectinase preparations acting on different pectic substrates
To evaluate how the type of pectic substrate affects the activity, profiles for the liberation of reducing sugars were obtained for both polygalacturonic acid and citric pectin. Based on the results in Figures 2 to 4, the conditions chosen for the reactions were 0.5% (m/v) pectic substrate and 30°C. Profiles were obtained with four different pectinase complexes: a crude extract from A. niger CH4 (maintained as a reference), a crude extract from A. oryzae, Pectinase P4716 and Pectinex Ultra SPL. These pectinase complexes were diluted so as to give similar profiles on polygalacturonic acid (Figure 5A). The same dilutions were then used for the degradation of citric pectin (Figure 5B).
Figure 5. Short hydrolysis profiles (40 min), obtained using different pectinase complexes and either polygalacturonic acid (A) or citric pectin (B).
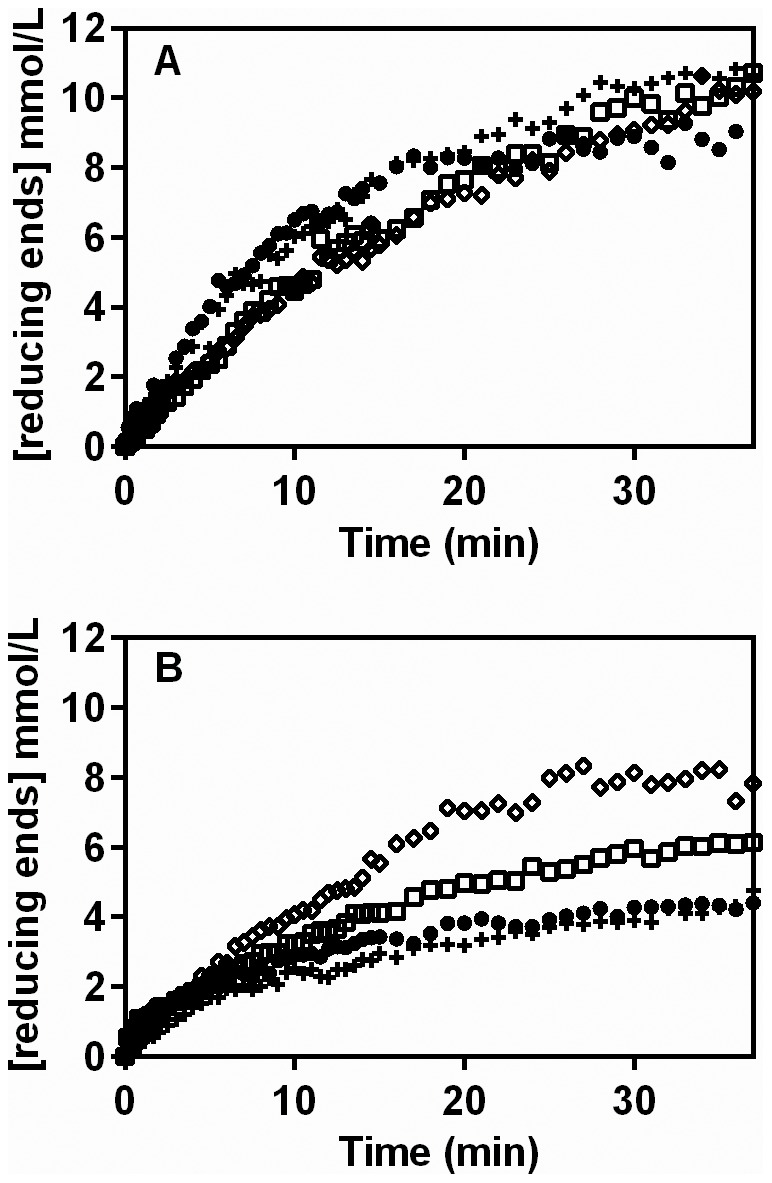
The hydrolysis was performed at 40°C, with an initial substrate concentration of 0.5% (w/v). The pectinase complexes used were the extracts of Aspergillus niger (•) and Aspergillus oryzae (+), produced in solid-state fermentation, and the commercially available Pectinex Ultra SPL (Novozymes) (□) and Pectinase P4716 (Sigma-Aldrich) (⋄).
Three key observations can be made. First, all profiles in Figures 5A and 5B show the early deceleration observed in Figures 1, 2 and 3. Second, comparing the same pectinase complex on different substrates, the rate of liberation of reducing sugars from citric pectin was lower than that obtained with polygalacturonic acid. Third, comparing different pectinase complexes on the same substrate, although the amounts of these complexes were chosen so as to give similar hydrolysis profiles with polygalacturonic acid, the profiles obtained were quite different from one another when these same amounts were added to citric pectin. The most effective hydrolysis of citric pectin was obtained with Pectinase P4716.
Extended profiles for different pectinase preparations acting on different pectic substrates
The same amounts of pectinase complex that were used for the hydrolysis of polygalacturonic acid in Figure 5A were added to 0.5% (m/v) of the two pectic substrates and the reactions were followed for 72 h at 30°C. The results are plotted for 24 h since in all cases there was no further liberation of reducing sugars after this time (Figures 6A and 6B).
Figure 6. Extended hydrolysis profiles (24 h), obtained using different pectinase complexes and either polygalacturonic acid (A) or citric pectin (B).
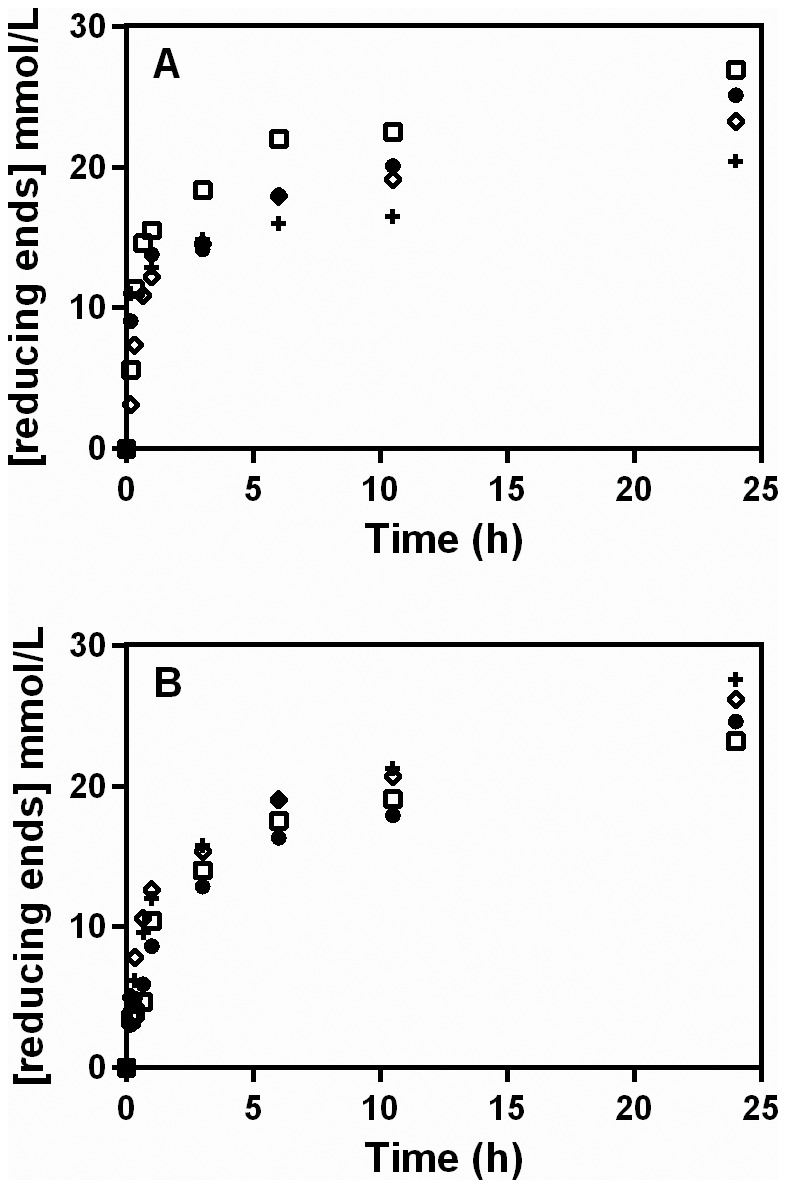
The hydrolysis was performed at 40°C, with an initial substrate concentration of 0.5% (w/v). The pectinase complexes used were the extracts of Aspergillus niger (•) and Aspergillus oryzae (+), produced in solid-state fermentation, and the commercially available Pectinex Ultra SPL (Novozymes) (□) and Pectinase P4716 (Sigma-Aldrich) (⋄).
Two key observations can be made. First, there was a continued deceleration of the reaction up to 6 h, but this deceleration was less pronounced than that which occurred over the first 10 min in Figures 1, 2, 3 and 5. This was followed by a slow, essentially constant rate of reducing sugar release from 6 to 24 h. Second, although Pectinase P4716 liberated significantly more reducing sugars from citric pectin than did the other complexes over the first 40 min of reaction (Figure 5B), this superior efficiency was not maintained. In fact, at 24 h, slightly more reducing sugars had been liberated by the extract from A. oryzae than by Pectinase P4716.
Discussion
Our work makes two main contributions. First, we have demonstrated that, during the hydrolysis of polygalacturonic acid and citric pectin by pectinase complexes from various sources, there is a significant deceleration of the rate of release of reducing sugars over the first ten minutes, followed by a longer, slower, deceleration over several hours. Second, we have identified several important issues with respect to the single-time-point assays that are commonly used for determining pectinase activities. These contributions are discussed below.
Implications of the early deceleration
Although the release of reducing sugars from pectic substrates by pectinase complexes has already been studied [38]–[40], in these studies the reaction profiles were obtained by taking samples at intervals of up to ten minutes or more. By removing samples at 30-s intervals, we have, for the first time, demonstrated that there is a significant deceleration in the rate of release of reducing sugars over the first ten minutes of reaction.
This early deceleration has important consequences for the determination of pectinase activities by measuring the liberation of reducing sugars in STP-assays. As Table 3 shows, for the same pectinase preparation and for any given set of reaction conditions, the activity that would be determined in an STP-assay decreases significantly as the incubation time is increased. This occurs because increases in reaction time above 10 min are associated with proportionally small increases in the reducing sugar level, so the result given by Eq. (1) becomes increasingly smaller.
Interestingly, the early deceleration is not apparent in the profiles that Baciu and Jördening [41] presented for the release of D-galacturonic acid from pectin by a different Pectinex product (Pectinex 100 L, Novozymes). Their profiles cover the first 300 min and show a gradual deceleration over this time. Our experiments differ from theirs in an important aspect: we measured the liberation of reducing sugars and therefore detected not only D-galacturonic acid in itself, but also oligomers produced by endo-acting enzymes. Baciu and Jördening attributed the gradual deceleration that occurred in their studies to product inhibition by D-galacturonic acid [41]. This is likely to be the cause of the slow deceleration that occurred over longer hydrolysis times in the current work (see Figure 6). However, further research is required to determine whether the early deceleration of the rate of liberation of reducing sugars is also caused by product inhibition.
Other issues related to the use of single-time-point assays for determining pectinase activities
As Table 3 shows, in STP-assays, the same pectinase preparation could be reported as having a pectinolytic activity ranging over almost an order of magnitude, depending on the particular set of hydrolysis conditions that is chosen. Since previous researchers have used not only different reaction times in carrying out STP-assays for pectinase activity, but also different temperatures and pectin concentrations (Table 2), it is impossible to draw meaningful conclusions about which result from the literature actually represents the best pectinase production. A lower amount of pectinases, assayed under favorable conditions, could lead to an activity higher than that obtained for a higher amount of pectinases, assayed under unfavorable conditions.
This raises an important question for researchers interested in using pectin-rich agro-industrial wastes in biorefineries. One of the processing steps in such biorefineries is likely to be the saccharification of pectin, for which it is desirable to identify good pectinase-producing microorganisms: Is it possible to establish a standard assay for screening pectinase activities that will not only reflect the saccharifying potential of a pectinase preparation, but can also be used to enable direct comparisons with the results of other research groups? It is useful to compare the current situation with respect to pectinase activity determination with the situation of cellulase activity determination in the 1980's. At that time there was much interest in the production of ethanol from cellulosic biomass, but different investigators used different assays and different conditions to determine cellulase activities. Then, in 1987, Ghose [42] proposed standardized methods for the determination of cellulase activity. Most workers in the field of cellulases now use these methods, allowing meaningful comparisons to be made within the cellulase literature.
Some authors have used polygalacturonic acid as the substrate in assays for pectinolytic activity [36], [40], [43], [44]. On the basis of our results, we recommend that, in any assay that aims to evaluate the potential for pectinase preparations to saccharify pectin, pectin should be used as the substrate and not polygalacturonic acid: the activities obtained with polygalacturonic acid are higher than those obtained with pectin, so the use of polygalacturonic acid would lead to an overestimation of activities. Furthermore, the pectinase complex that hydrolyzes polygalacturonic acid most efficiently might not be the pectinase complex that hydrolyzes pectin most efficiently.
It would be a relatively simple matter to select appropriate values for pectin concentration, temperature and pH for a standard pectinase assay based on the liberation of reducing sugars. The initial pectin concentration in the reaction mixture should be as high as possible without causing problems with viscosity (e.g. 0.5% w/v). With respect to temperature, Table 2 shows that various authors are performing activity assays at temperatures of 50°C or more, but a temperature should be chosen that does not cause denaturation (e.g. a value in the range of 30 to 40°C). Temperatures above 50°C lead to high activity values over the short period of the assay, but they are unlikely to be used in saccharification processes of up to 24 h or more, unless the enzymes in the preparation are thermostable. The effect of pH on pectinase activity was not studied in the current work because it has already been well studied [38], [39], [45], [46]. Most pectinases have a pH optimum around 4.5. However, some pectinases, principally those produced by species of Bacillus, have a pH optimum between 8 and 10.5 [47], [48].
On the other hand, it is not clear what would be an appropriate reaction time for a standard pectinase assay. Several possibilities might be considered. For example, initial velocities could be determined. However, due to the non-linearity of the reaction profile, it would be necessary to obtain various data points over the first minutes of the reaction in order to be able to estimate the initial velocity with an acceptable precision. In any case, since the greater part of the saccharification process occurs over many hours, after a significant initial deceleration, initial velocities will not necessarily reflect the long-term saccharification potential.
Of course, assays could be carried out over 24 h. In this case, it would be unwise to use a single time point at 24 h, since the reaction might reach completion before this time. It would be advisable, therefore, to have various measurements during this time. However, such a long reaction time would be inconvenient in screening programs, as it would greatly reduce throughput. Researchers are likely to prefer to use such long reaction times in monitoring process development, not in initial screening programs.
One possibility might be to use a “double-time-point” assay. A first reading could be taken after the initial deceleration, at 20 min or so. A second reading could then be taken later, maybe at 40 min or even at 60 min. The activity could then be expressed on the basis of the difference between these two readings. This would not be as convenient as an STP-assay, but would have the advantage of not including the period during which the early deceleration occurs. However, it remains to be proven whether activities obtained in this manner would reflect the long-term saccharification potential.
Conclusions
We have shown, for the first time, that there is a significant early deceleration in the liberation of reducing sugars from pectic substrates by pectinase complexes. On the basis of this phenomenon, as well as results regarding the effects of temperature and substrate concentration on the reaction profile, we have shown that it is not possible to make meaningful comparisons between pectinase activities reported by different authors who have used different assay conditions, substrates and reaction times in single-time-point assays. There is an urgent need for the establishment of a standardized assay procedure for evaluating the saccharification potential of pectinase complexes.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.
Funding Statement
Funding provided by a research grant from CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico (Brasil), National Council of Scientific and Technological Development (Brazil).) http://www.cnpq.br, Grant number: 482131-2012-8 to: DAM NK, an International Cooperation Project Grant, CNPq (Brazil) & AKA (Academy of Finland) (Finland), www.aka.fi/eng, CNPq grant number: 490236-2012-0 granted to: DAM NK, AKA grant number: Sustainable Energy (SusEn) program, grant 271025 granted to: PR and Research Scholarships. Research scholarship from CNPq, Research scholarships received by FCF FAM DHdP NK DAM and CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Brasil), Coordination for the Development of Personnel in Higher Education (Brazil)), www.capes.gov.br, research scholarship received by AB. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Schubert E (1952) Differentiation of the polygalacturonases of Aspergillus niger . Nature 169: 931–932. [DOI] [PubMed] [Google Scholar]
- 2. Alkorta I, Garbisu C, Llama MJ, Serra JL (1997) Industrial applications of pectic enzymes: a review. Process Biochem 33: 21–28. [Google Scholar]
- 3. Kashyap DR, Vohra PK, Chopra S, Tewari R (2001) Applications of pectinases in the commercial sector: a review. Bioresour Technol 77: 215–227. [DOI] [PubMed] [Google Scholar]
- 4.Perry FG (1995) Biotechnology in animal feeds and animal feeding: an overview. In: Wallace RJ, Chesson A, editors. Biotechnology in animal feeds and animal feeding. Weinheim: VCH. pp. 1–15. [Google Scholar]
- 5. Edwards MC, Doran-Peterson J (2012) Pectin-rich biomass as feedstock for fuel ethanol production. Appl Microbiol Biotechnol 95: 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Maris AJ, Abbott DA, Bellissimi E, van den Brink J, Kuyper M, et al. (2006) Alcoholic fermentation of carbon sources in biomass hydrolysates by Saccharomyces cerevisiae: current status. Antonie Van Leeuwenhoek 90: 391–418. [DOI] [PubMed] [Google Scholar]
- 7. Richard P, Hilditch S (2009) D-Galacturonic acid catabolism in microorganisms and its biotechnological relevance. Appl Microb Biotechnol 82: 597–604. [DOI] [PubMed] [Google Scholar]
- 8. Lohrasbi M, Pourbafrani C, Niklasson C, Taherzadeh MJ (2010) Process design and economic analysis of a citrus waste biorefinery with biofuels and limonene as products. Bioresour Technol 101: 7382–7388. [DOI] [PubMed] [Google Scholar]
- 9. Lopez AJS, Li Q, Thompson IP (2010) Biorefinery of waste orange peel. Crit Rev Biotechnol 30: 63–69. [DOI] [PubMed] [Google Scholar]
- 10. Vries R, Visser J (2001) Aspergillus enzymes involved in degradation of plant cell wall polysaccharides. Microbiol Mol Biol Rev 65: 497–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Andersen MR, Giese M, de Vries RP, Nielsen J (2012) Mapping the polysaccharide degradation potential of Aspergillus niger . BMC Genomics 13: 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Favela-Torres E, Volke-Sepúlveda T, Viniegra-Gonzalez G (2006) Production of hydrolytic depolymerising pectinases. Food Technol Biotechnol 44: 221–227. [Google Scholar]
- 13. Collmer A, Ried JL, Mount MS (1988) Assay methods for pectic enzymes. Method Enzymol 161: 329–335. [Google Scholar]
- 14. Torres S, Sayagob JE, Ordoñez RM, Isla MI (2011) A colorimetric method to quantify endo-polygalacturonase activity. Enzyme Microb Technol 48: 123–128. [DOI] [PubMed] [Google Scholar]
- 15. Ortiz GE, Guitart ME, Albertó E, Fernández Lahore HM, Blasco M (2014) Microplate assay for endo-polygalacturonase activity determination based on ruthenium red method. Anal Biochem 454: 33–35. [DOI] [PubMed] [Google Scholar]
- 16. Bernfeld P (1955) Amylases, α and β. Methods Enzymol 1: 149–157. [Google Scholar]
- 17. Somogyi M (1945) A new reagent for the determination of sugars. J Biol Chem 160: 61–68. [Google Scholar]
- 18. Nelson N (1944) A photometric adaptation of the Somogyi method for the determination of glucose. J Biol Chem 153: 375–380. [Google Scholar]
- 20. Martin N, Guez MU, Sette LD, Silva R, Gomes E (2010) Pectinase production by a Brazilian thermophilic fungus Thermomucor indicae-seudaticae N31 in solid-state and submerged fermentation. Microbiol 79: 306–313. [PubMed] [Google Scholar]
- 21. Silva D, Tokuioshi K, Martins ES, Silva R, Gomes E (2005) Production of pectinase by solid-state fermentation with Penicillium viridicatum RFC3. Process Biochem 40: 2885–2889. [Google Scholar]
- 22. Martins ES, Silva D, Da Silva R, Gomes E (2002) Solid-state production of thermostable pectinases from thermophilic Thermoascus aurantiacus . Process Biochem 37: 949–954. [Google Scholar]
- 23. Kumar SY, Varakumar S, Reddy OVS (2010) Production and optimization of polygalacturonase from mango (Mangifera indica) peel using Fusarium moniliforme in solid-state fermentation. World J Microbiol Biotechnol 26: 1973–1980. [Google Scholar]
- 24. Patil SR, Dayanand A (2006) Production of pectinase from deseeded sunflower head by Aspergillus niger in submerged and solid-state conditions. Bioresour Technol 97: 2054–2058. [DOI] [PubMed] [Google Scholar]
- 25. Blandino A, Iqbalsyah T, Pandiella SS, Cantero D, Webb C (2002) Polygalacturonase production by Aspergillus awamori on wheat in solid-state fermentation. Appl Microbiol Biotechnol 58: 164–169. [DOI] [PubMed] [Google Scholar]
- 26. Beg QK, Bhushan B, Kapoor M, Hoondal GS (2000) Effect of amino acids on production of xylanase and pectinase from Streptomyces sp. QG-11-3. World J Microbiol Biotechnol 16: 211–213. [Google Scholar]
- 27. Hadj-Taieb N, Ayadi M, Trigui S, Bouabdallah F, Gargouri A (2002) Hyperproduction of pectinase activities by a fully constitutive mutant (CT1) of Penicillium occitanis . Enzyme Microb Technol 30: 662–666. [Google Scholar]
- 28. Taragano V, Sanchez VE, Pilosof AMR (1997) Combined effect of water activity depression and glucose addition on pectinase and protease production by Aspergillus niger . Biotechnol Lett 19: 233–236. [Google Scholar]
- 29. Martínez-Trujillo A, Arreguín-Rangel L, García-Rivero M, Aguilar-Osorio G (2011) Use of fruit residues for pectinase production by Aspergillus flavipes FP-500 and Aspergillus terreus FP-370. Lett Appl Microbiol 53: 202–209. [DOI] [PubMed] [Google Scholar]
- 30. Naidu G, Panda T (1998) Application of response surface methodology to evaluate some aspects on stability of pectolytic enzymes from Aspergillus niger . Biochem Eng J 2: 71–77. [Google Scholar]
- 31. Gregorio AD, Mandalarig G, Arena N, Nucita F, Tripodo MM, et al. (2002) SCP and crude pectinase production by slurry-state fermentation of lemon pulps. Bioresour Technol 83: 89–94. [DOI] [PubMed] [Google Scholar]
- 32. Ustok FI, Tari C, Gogus N (2007) Solid-state production of polygalacturonase by Aspergillus sojae ATCC 20235. J Biotechnol 127: 322–334. [DOI] [PubMed] [Google Scholar]
- 33. Ahlawat S, Battan B, Dhiman SS, Sharma J, Mandhan RP (2007) Production of thermostable pectinase and xylanase for their potential application in bleaching of kraft pulp. J Ind Microbiol Biotechnol 34: 763–770. [DOI] [PubMed] [Google Scholar]
- 34. Mamma D, Kourtoglou E, Christakopoulos P (2008) Fungal multienzyme production on industrial by-products of the citrus-processing industry. Bioresour Technol 99: 2373–2383. [DOI] [PubMed] [Google Scholar]
- 35. Taskin E, Eltem R, Silva ES, Souza JVB (2008) Screening of Aspergillus strains isolated from vineyards for pectinase production. J Food Agric Environ 6: 5–7. [Google Scholar]
- 36. Teixeira JA, Gonçalves DB, de Queiroz MV, de Araújo EF (2011) Improved pectinase production in Penicillium griseoroseum recombinant strains. J Appl Microbiol 111: 818–25. [DOI] [PubMed] [Google Scholar]
- 37. Niture SK (2008) Comparative biochemical and structural characterizations of fungal polygalacturonases. Biologia 63: 1–19. [Google Scholar]
- 38. Bélafi-Bakó K, Gubikza L, Kiss K, Nemestóthy N (2007) Hydrolysis of pectin by Aspergillus niger polygalacturonase in a membrane bioreactor. J Food Eng 78: 438–442. [Google Scholar]
- 39. Rodríguez-Nogalez M, Ortega N, Perez-Mateos M (2008) Pectin hydrolysis in a free enzyme membrane reactor: An approach to the wine and juice clarification. Food Biochem 107: 112–119. [Google Scholar]
- 40. Nikolić S, Mojović L (2007) Hydrolysis of apple pectin by the coordinated activity of pectic enzymes. Food Chem 101: 1–9. [Google Scholar]
- 41. Baciu IE, Jördening HJ (2004) Kinetics of galacturonic acid release from sugar-beet pulp. Enzyme Microb Technol 34: 505–512. [Google Scholar]
- 42. Ghose TK (1987) Measurement of cellulase activities. Pure Appl Chem 59: 257–268. [Google Scholar]
- 43. Heerd D, Yegin S, Tari C, Fernandez-Lahore M (2012) Pectinase enzyme-complex production by Aspergillus spp. in solid-state fermentation: A comparative study. Food Bioprod Process 90: 102–110. [Google Scholar]
- 44. Panda T, Naidu GSN, Sinha J (1999) Multiresponse analysis of pectolytic enzymes by Aspergillus niger: a statistical view. Process Biochem 35: 187–195. [Google Scholar]
- 45. Ortega N, Diego S, Perez-Mateos M, Busto MD (2004) Kinetic properties and thermal behaviour of polygalacturonase used in fruit juice clarification. Food Chem 88: 209–217. [Google Scholar]
- 46. Wilkins MR, Wilbur W, Grohmann K (2007) Simultaneous saccharification and fermentation of citrus peel waste by Saccharomyces cerevisiae to produce ethanol. Process Biochem 42: 1614–1619. [Google Scholar]
- 47. Kapoor M, Kuhad RC (2002) Improved polygalacturonase production from Bacillus sp. MG-cp-2 under submerged (SmF) and solid-state (SSF) fermentation. Lett Appl Microbiol 34: 317–322. [DOI] [PubMed] [Google Scholar]
- 48. Kashyap DR, Soni SK, Tewari (2003) Enhanced production of pectinase by Bacillus sp. DT7 using solid-state fermentation. Bioresour Technol 88: 251–254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.