

Supplemental Digital Content is available in the text.
Keywords: familial hypercholesterolemia, IDOL, lipid-lowering therapy, MYLIP, pharmacogenetic
Abstract
Background
A previous study reported that the myosin regulatory light chain interacting protein (MYLIP) might serve as a novel therapeutic class for treating dyslipidemia. It contributes to variations in the levels of circulating low-density lipoprotein cholesterol (LDL-C), promoting the degradation of LDL–LDLR, thus limiting absorption. The effect of genetic variation in the MYLIP gene in a disease scenario characterized by mutations in the LDLR gene has not been previously evaluated.
Objective
The aim of this study was to assess the effect of the p.N342S variant on the response to lipid-lowering therapy in Brazilian patients with heterozygous familial hypercholesterolemia (FH).
Patients and methods
A total of 156 patients with heterozygous FH were followed up for 12 months and received lipid-lowering therapy (different doses of atorvastatin with the addition of ezetimibe in over half the patients of each genotype group). Cholesterol data were assessed, and analysis of the MYLIP rs9370867 (p.N342S) genotypes was carried out by melting curve analysis.
Results
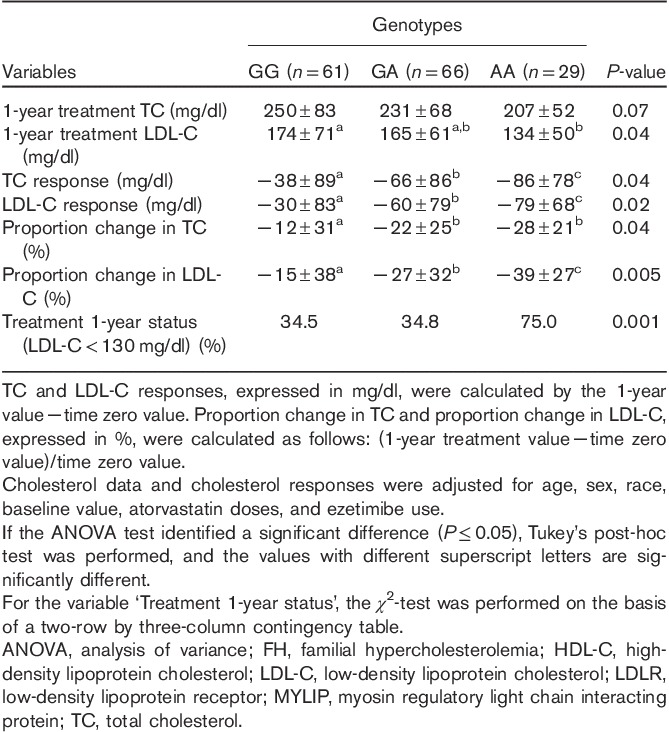
Baseline total cholesterol and baseline LDL-C levels were not different between genotypes. After 1 year of treatment, LDL-C responses (expressed as mg/dl and as %) were significantly different among genotypes (AA: −79±68 and −39±27, GA: −60±79 and −27±32, and GG: −30±83 and −15±38; P=0.02 and 0.005, respectively). In addition, FH patients carrying the AA genotype were more likely to achieve LDL-C levels of less than 130 mg/dl after 1 year of treatment (75.0%) compared with patients with the GG and GA genotypes (34.5 and 34.8%, respectively; P=0.001).
Conclusion
Our study indicates that MYLIP p.N342S might be a pharmacogenetic marker for lipid-lowering therapy in patients with FH.
Introduction
Statins, competitive inhibitors of HMG-CoA reductase, are widely prescribed to reduce cardiovascular morbidity and mortality. Although several types of statins and nonstatin lipid-lowering drugs are available, a proportion of dyslipidemic patients actually do not achieve therapeutic goals 1.
Familial hypercholesterolemia (FH) is an autosomal dominant disease caused primarily by mutations in the LDLR gene. FH patients, despite being a high-risk group for cardiovascular disease, are usually undertreated 2. The identification of new markers of treatment response would allow for more effective treatment delivery, reducing the risk for cardiovascular disease in this population.
Current studies provide evidence that genetic factors, such as polymorphisms in the APOE, SLCO1B1, CYP3A4, CYP3A5, KIF6, CETP, HMGCR, and ABCB1 genes, can contribute to interindividual variations in response to lipid-lowering statin therapy and/or toxicity from statins. However, the total variability due to genetic variants is still unknown 3–12.
The myosin regulatory light chain interacting protein (MYLIP; also known as IDOL) has been indicated as a possible target for a new and useful strategy in the pharmacological treatment of dyslipidemia. The protein is coded for by the MYLIP gene and works as a regulator of the low-density lipoprotein receptor (LDLR) pathway for cellular cholesterol absorption 13. Recent studies have identified MYLIP genomic regions to be associated with low-density lipoprotein cholesterol (LDL-C) levels 14–16. Later, a study identified a nonsynonymous polymorphism (p.N342S, rs9370867) as the accounting basis for the functional variation observed in previous studies. Furthermore, it has been shown that the N342 allele is associated with higher levels of LDLR degradation and decreased LDL-C uptake 17.
In this scenario, the main aim of this study was to evaluate the effect of p.N342S on the response to lipid-lowering therapy in Brazilian patients with FH.
Patients and methods
Clinical and laboratory evaluation of patients
This study included 156 index Brazilian patients with a heterozygous FH phenotype ascertained by the Lipid Clinic, Heart Institute (InCor), University of São Paulo Medical School, São Paulo, Brazil. These patients were diagnosed on the basis of biochemical and clinical data 18,19 and were followed up for at least 12 months, from January 2006 to December 2012, while receiving lipid-lowering therapy. Patients were categorized into self-declared ‘racial/color’ subgroups, according to the criteria used by the Brazilian Census, as White, Brown (Pardo in Portuguese), or Black 20. The Institutional Ethics Committee approved the study protocol (CAPPesq numbers 022/11 and 191/04), and written informed consent was obtained from all participants before entering the study.
Treatment was administered by the attending physician without any knowledge of the kind of genetic defect causing the FH phenotype. Atorvastatin dosage and coadministration of ezetimibe, an adjuvant cholesterol-lowering medication, were at physician discretion, to attain the greatest possible LDL-C reduction. All patients were assessed at least three times during follow-up. Plasma total cholesterol (TC) and LDL-C levels were measured using enzymatic methods during the following periods: at baseline (first value), at the initiation of atorvastatin use (immediately before), and, on average, after 1-year of treatment onset (the first two measured values were not necessarily the same).
Genetic analyses
Genomic DNA was extracted from the peripheral blood of patients following a standard salting-out procedure. Coding sequences of the LDLR gene (18 exons) were amplified by PCR. PCR products were bidirectionally sequenced using the ABI Terminator Sequencing Kit and the ABI 3500XL Sequencer (Applied Biosystems, Foster City, California, USA) according to the manufacturer’s instructions. In addition, the APOB and PCSK9 genes were sequenced and the multiplex ligation-dependent probe amplification technique was used to identify LDLR gene deletions/insertions. LDLR gene mutations were classified according to their probable functional class as previously reported in the literature. For multivariable analysis, we grouped individuals with null or a defective mutation into the same group and compared them with individuals with no identified mutation (we defined this variable as the presence of an LDLR mutation) 21–24. Genotypes for the MYLIP rs9370867 (p.N342S, c.G1025A) polymorphism were detected by PCR followed by high-resolution melting (HRM) analysis using the Rotor Gene 6000 instrument (Qiagen, Courtaboeuf, France) 25. Amplification of the fragment was performed using the sense 5′-TTGTGGACCTCGTTTCAAGA-3′ and antisense 5′-GCTGCAGTTCATGCTGCT-3′ (80 bp) primers for rs9370867. A 40-cycle PCR was carried out under the following conditions: denaturation of the template DNA in the first cycle at 94°C for 120 s, denaturation at 94°C for 20 s, annealing at 53.4°C for 20 s, and extension at 72°C for 22 s. PCR was performed using a 10-μl reactive solution (10 mmol/l tris-HCl, 50 mmol/l KCl, pH 9.0, 2.0 mmol/l MgCl2, 200 μmol/l of each dNTP, 0.5 U Taq DNA polymerase, 200 nmol/l of each primer, 10 ng genomic DNA template) with the addition of fluorescent DNA-intercalating SYTO9 (1.5 μmol/l; Invitrogen, Carlsbad, California, USA).
In the HRM phase, the Rotor Gene 6000 measured the fluorescence at each 0.1°C temperature increase in the range of 73–85°C. Melting curves were generated from the decrease in fluorescence with the increase in the temperature and, on analysis, nucleotide changes resulted in three different curve patterns. Samples from the three observed curves were analyzed using bidirectional sequencing as a validation procedure (ABI Terminator Sequencing Kit and ABI 3500XL Sequencer; Applied Biosystems). The GG, GA, and AA genotypes for rs9370867 were easily discernible by HRM analysis. In addition, 15% of the samples were reanalyzed as a quality control measure and gave identical results 26.
Statistical analysis
The χ2-test or Fisher’s exact test (if an expected cell frequency was <5) was used for comparative analysis of the general characteristics and the LDL-C status according to MYLIP rs9370867 genotypes. The χ2-test was also used for estimating Hardy–Weinberg equilibrium 27. The Shapiro–Wilk test showed that cholesterol data had a normal distribution. Analysis of variance (ANOVA) was carried out to compare the age, BMI, baseline cholesterol data, and cholesterol responses according to genotypes. If ANOVA identified a significant difference (P≤0.05), Tukey’s post-hoc test was performed, and the different significance levels for these hypothesis tests are shown in the tables as superscript codes. TC and LDL-C responses, expressed in mg/dl, were calculated by subtracting the time zero value from the 1-year follow-up value. The proportion change of TC and the proportion change of LDL-C, expressed as percentages, were calculated as follows: (1-year follow-up treatment value−time zero)/time zero value. For multivariate analysis, baseline cholesterol data were adjusted for age, sex, and race (defined as White or non-White). LDL-C and TC responses and 1-year treatment data were also adjusted for baseline value, atorvastatin doses, and ezetimibe use. Regression models were developed using MYLIP genotypes in an additive model (0, 1, or 2 for AA, GA, and GG, respectively). Logistic regression multivariate analysis was carried out to evaluate factors associated with not achieving target LDL-C levels (LDL-C<130 mg/dl vs. LDL-C≥130.0 mg/dl; this value was defined arbitrarily). We also tested the effect of MYLIP genotypes in a multiple linear regression model including the following variables: number of variant alleles for MYLIP rs9370867 (0, 1, or 2 for GG, GA, or AA, respectively), age, sex, BMI, race, smoking status, atorvastatin dose, ezetimibe use, baseline LDL-C level, and the presence of LDLR mutations (null plus defective mutations vs. no mutation). All statistical analyses were carried out using SPSS software (version 16.0; IBM, New York, New York, USA), with the level of significance set at a P-value of 0.05 or lower.
Results
Characteristics of studied FH patients
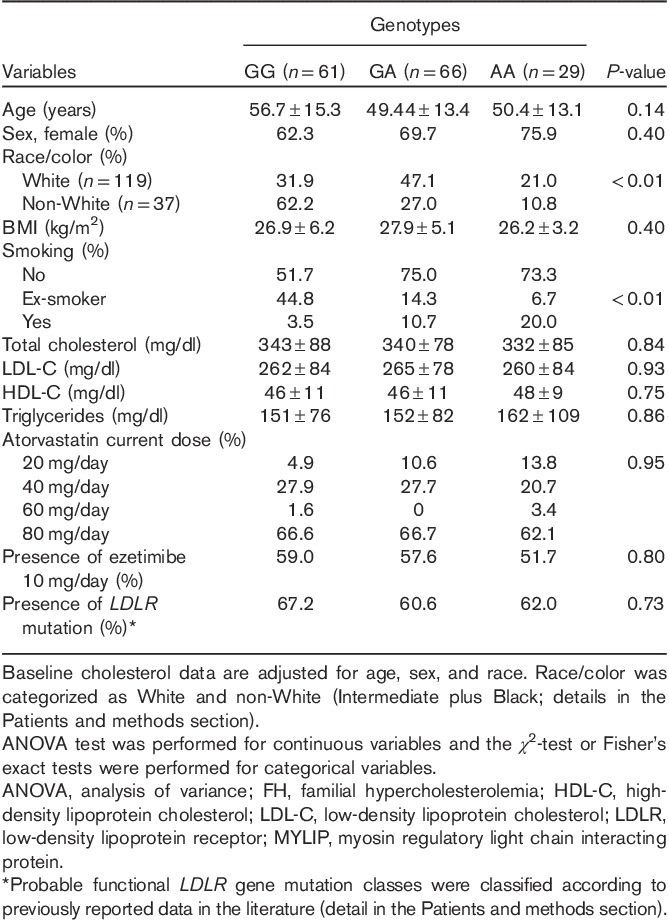
Of the 156 patients (mean age 52.5+14.5), 106 (67.9%) were female and 50 (32.1%) were male. All patients received atorvastatin for at least 12 months (mean±SD 14±3 months of follow-up on therapy). The current dose of atorvastatin and the use of 10 mg/day ezetimibe according to the MYLIP polymorphism are shown in Table 1. Only 10 (6.4%) patients had their doses modified during follow-up, and none of the study participants discontinued the drug. All dosage changes among the study participants were to uptitrate atorvastatin to a higher dosage in individuals who did not receive the maximum dosage at the first medical visit. All prescriptions were made at the physician discretion, who had no knowledge of the MYLIP genotype or the type of LDLR mutation.
Table 1.
Baseline characteristics of FH patients according to MYLIP rs9370867 genotypes
Frequencies of the MYLIP rs937086 genotypes
The frequency of the MYLIP rs937086 A variant allele was 39.7%, and the distribution of the genotypes was 18.6% (n=29) for the AA genotype, 42.3% (n=66) for the GA genotype, and 39.1% (n=61) for the GG genotype. The genotypic distribution for the MYLIP rs937086 polymorphism was in accordance with Hardy–Weinberg equilibrium (χ2=2.12, P=0.14). Table 1 shows the genotypic distribution for the MYLIP rs937086 polymorphism according to race.
Baseline characteristics according to MYLIP rs937086 genotypes
Table 1 shows the baseline characteristics of the study participants according to genotype. The variables age, sex, BMI, baseline cholesterol level, and the presence of LDLR mutations were not significantly different according to genotypes. White patients more frequently had GA and AA genotypes, whereas patients who smoked more frequently had the AA genotype.
Cholesterol data and response to lipid-lowering therapy according to MYLIP rs937086 genotypes
Table 2 shows data on cholesterol and response to lipid-lowering therapy. After 1-year of treatment, LDL-C values were different among MYLIP rs937086 genotypes adjusted for age, sex, race, baseline value, atorvastatin doses, and ezetimibe use (P=0.04, ANOVA test). In the Tukey’s post-hoc test, patients carrying the AA genotype were found to have lower LDL-C levels (134±50 mg/dl) compared with patients carrying the GG genotype (174±71 mg/dl). In addition, FH patients carrying the AA genotype had a greater LDL-C response (−79±68 mg/dl) and a higher frequency of LDL-C less than 130.0 mg/dl (75.0%) compared with GG genotype carriers (−30±83 mg/dl and 34.5%; P=0.02 and 0.001, respectively). The proportion change of LDL-C, expressed in percentage (%), was also different among genotypes (Table 2). In addition, LDL-C values according to the MYLIP polymorphism and time periods are shown in Supplementary Figure 1 (Supplemental digital content 1, http://links.lww.com/FPC/A773).
Table 2.
Cholesterol data and responses to lipid-lowering therapy of FH patients according to MYLIP rs9370867 genotypes
Figures 1 and 2 show stratified analyses for the presence of the LDLR mutation. Both LDL-C response (P=0.02) and the frequency of patients with LDL-C levels less than 130.0 mg/dl (P=0.002) statistically differed according to the rs937086 genotype in the patient group with the LDLR mutation (n=99). In the group of patients without the LDLR mutation (n=57), the P-value was either borderline (P=0.11) or significant (P=0.04; Figs 1 and 2).
Fig. 1.
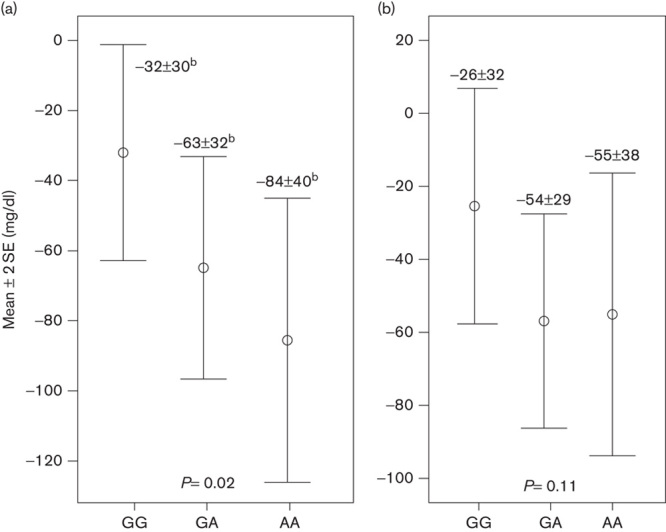
LDL-C response (mg/dl) according to genotype for the MYLIP polymorphism and the presence of the LDLR mutation. (a) Patients with the LDLR mutation (n=99) and (b) patients without the LDLR mutation (n=57). If the analysis of variance test identified a significant difference, Tukey’s post-hoc test was performed, and values with different superscript letters are significantly different. LDL-C, low-density lipoprotein cholesterol; LDLR, low-density lipoprotein receptor; MYLIP, myosin regulatory light chain interacting protein.
Fig. 2.
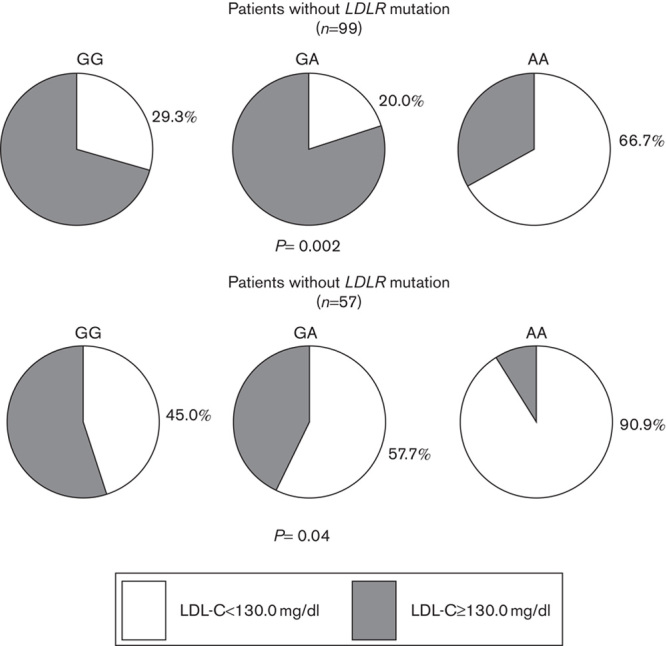
Frequencies of patients with LDL-C less than 130.0 mg/dl according to genotype for the MYLIP polymorphism and the presence of the LDLR mutation. The χ2-test was performed using a two-row by three-column contingency table. LDL-C, low-density lipoprotein cholesterol; LDLR, low-density lipoprotein receptor; MYLIP, myosin regulatory light chain interacting protein.
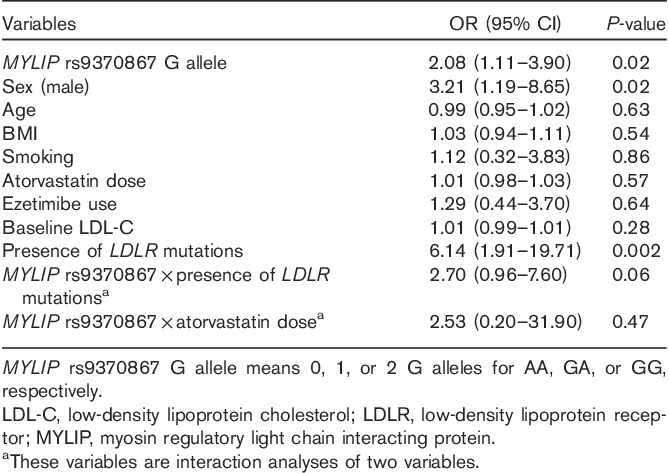
Furthermore, the presence of the G allele was associated with a greater odds of not achieving the LDL-C target in a multivariate model [odds ratio (OR)=2.08 per G allele, 95% confidence interval (CI)=1.11–3.90, P=0.02]. No interaction between MYLIP rs937086 and atorvastatin dose was found (OR=2.53, 95% CI=0.20–31.90, P=0.47), whereas a trend toward a significant interaction was observed between MYLIP rs937086 and the presence of LDLR mutations (OR=2.70, 95% CI=0.96–7.60, P=0.06; Table 3). In a multiple linear regression model, we showed that the number of variant alleles for MYLIP rs9370867 (GG=0, GA=1, and AA=2) was significant in terms of the LDL-C response (Supplementary Table 1, Supplemental digital content 2, http://links.lww.com/FPC/A774).
Table 3.
Logistic regression multivariate analysis of factors associated with not achieving target LDL-C levels (LDL-C≥130.0 mg/dl)
In a stratified analysis by race, we were able to identify significant differences even with small sample sizes. In the White group (n=119), the presence of the G allele was associated with a greater odds of not achieving the LDL-C target in a multivariate model (OR 1.98 per G allele, 95% CI 1.22–3.21, P=0.006; Supplementary Table 2, Supplemental digital content 3, http://links.lww.com/FPC/A775). In addition, patients with the AA genotype had a greater LDL-C response (−87±73 mg/dl) compared with patients with the GG genotype (−24±89 mg/dl; P=0.04; ANOVA plus Tukey’s tests, data not shown).
Discussion
The main findings of the present study were that patients carrying the homozygous genotype for p.N342S (AA genotype) had lower LDL-C mean levels after 1-year of treatment, a higher LDL-C response, and a higher frequency of LDL-C levels less than 130.0 mg/dl compared with patients carrying the GG or GA genotypes. These genetic associations are functionally based on previous data that showed the G allele (N342) to be associated with more potent ubiquitination and degradation of the LDLR and decreased LDL uptake 17. Consequently, the differential effects of the G and A alleles on LDLR and LDL-C levels are very consistent with our observations on the response to lipid-lowering therapy.
LDLR has a pivotal role in LDL-C uptake into cells, and a better example is that mutations in the LDLR gene cause FH, which is characterized by reduced hepatic LDL clearance. MYLIP is a post-transcriptional regulator of LDLR abundance, which is induced by transcription factors when cellular cholesterol levels rise 13,28,29. In contrast, atorvastatin, other than being a potent HMG-CoA reductase inhibitor, is an upregulator of LDLR gene expression 30. As our cohort was composed of patients with a heterozygous FH phenotype, and assuming that most of them had only one functional allele on the LDLR gene, one could suggest that post-transcriptional action and different allelic activities of MYLIP may be more evident in this clinical scenario. Interestingly, the association between the MYLIP genotype and the presence or absence of an LDLR mutation was marginally significant (P=0.06).
In addition, we were able to carry out stratified analyses according to the LDLR mutation and race. We observed the same results irrespective of the group being compared; AA genotype carriers had higher response to lipid-lowering therapy. In these analyses, even with the sample size being small, the effect of the MYLIP variant on the response to treatment was clear.
Further, using an additive model, the number of MYLIP alleles was associated with LDL-C response as a continuous variable or with higher odds of not achieving an arbitrarily determined LDL-C cutoff (130 mg/dl). In these models, male sex was a factor associated with a worse response. Smoking and race variables were not significant predictors of the study outcome, although being differently distributed according to the genotype.
Genomewide association studies identified the MYLIP region to be associated with cholesterol levels 14–16. Other recent studies demonstrated the association of the p.N342S polymorphism with cholesterol levels in a Mexican dyslipidemic sample and a possible contribution of MYLIP variants to circulating LDL-C levels 17,31. In addition, other studies have reported an association of MYLIP polymorphisms with phenotypes such as blood pressure and obesity 32–34. Interestingly, patients from our sample had equal baseline TC and LDL-C levels according to MYLIP genotypes. One possibility is that the severity of FH may not allow us to identify a possible difference in the baseline measures. In the same sense, our group did not identify this association in Brazilian participants from the general population or in patients who underwent coronary angiography because of suspected coronary artery disease 26.
Here, we were able to observe a significant pharmacogenetic effect of an MYLIP polymorphism on lipid-lowering therapy. A previous study has reported an additional association with subgenomewide significance (P<1×10–6) for rosuvastatin-related LDL-C reduction at the MYLIP locus 35. All patients in our study took atorvastatin as the lipid-lowering therapy. Nonetheless, it is tempting to suggest that the effectiveness of statin, including inhibition of HMG-CoA reductase and upregulation of LDLR activity, might be dependent on changes in the MYLIP–LDLR scenario due to the MYLIP genetic variant.
Some aspects and study limitations must be pointed out. First, our sample size was relatively small. However, we were able to observe significant variations in the lipid profile because of the nature of our sample (FH patients), thus increasing the power of the analysis. Second, our study enrolled only clinically diagnosed FH patients; thus, further studies should assess MYLIP as a pharmacogenetic marker in patients with dyslipidemia from the general population. Third, it is not possible to completely exclude the influence of atorvastatin dose, including dose modifications for some patients, even with these frequencies being no different according to genotypes. In addition, a standard protocol to select the atorvastatin dose and ezetimibe use was not used. However, we added these as covariates in the multivariate models and the findings remained the same. Fourth, we were not able to verify information on objective measures of lifestyle changes throughout the course of follow-up, or on medication adherence. Nonetheless, we do not believe that imbalances in these variables significantly bias our conclusions. This was a single-center study, and all participants received the same (blinded to genotype status) medical assistance, nutritional and lifestyle orientations, and pharmacist assistance. Finally, our study did not include replication and other factors could still be evaluated, for example, the type of dyslipidemia, other markers associated with the pharmacogenomics of statins, the type of hypolipidemic drugs, concomitant medications, cost-effectiveness, age, sex, and ethnicity 36–40.
Conclusion
Our findings indicate that the MYLIP variant might be a pharmacogenetic marker for lipid-lowering therapy. Furthermore, genetic associations reported in this study are functionally based on previous data, and they reaffirm that pharmacologic modulation of MYLIP activity might be a potential strategy that could be translated into dyslipidemia treatment.
Acknowledgements
PCJL Santos and AC Morgan are recipients of fellowships from FAPESP, Proc. 2010-17465-8, Proc. 2013-09295-3, Proc. 2013-20614-3, and Proc. 2012-08118-8, respectively, Brazil. The authors thank the patients who participated in the study. The technical assistance of the Laboratory of Genetics and Molecular Cardiology group, Heart Institute (InCor), and Sociedade Hospital Samaritano – Ministério da Saúde (PROADI-SUS; SIPAR: 25000.180.672/2011-81) is gratefully acknowledged.
Conflicts of interest
R.D.S. has the following conflicts of interest to declare: honoraria or consulting fees from Astra Zeneca, Aegerion, Amgen, Biolab, Bristol Myers Squibb, Merck, Pfizer, Sanofi, Regeneron, Genzyme, Novo-Nordisk, Lilly, Boehringer Ingelheim, Novartis, and Nestle. For the remaining authors, there are no conflicts of interest.
Footnotes
Paulo C.J.L. Santos and Aline C. Morgan contributed equally to the writing of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website (www.pharmacogeneticsandgenomics.com).
References
- 1.Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Cholesterol Treatment Trialists' (CTT) Collaborators . Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278 [DOI] [PubMed] [Google Scholar]
- 2.Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. European Atherosclerosis Society Consensus Panel . Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 2013; 34:3478–3490a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hubacek JA, Vrablik M. Effect of apolipoprotein E polymorphism on statin-induced decreases in plasma lipids and cardiovascular events. Drug Metabol Drug Interact 2011; 26:13–20 [DOI] [PubMed] [Google Scholar]
- 4.SEARCH Collaborative Group. Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, et al. SLCO1B1 variants and statin-induced myopathy – a genomewide study. N Engl J Med 2008; 359:789–799 [DOI] [PubMed] [Google Scholar]
- 5.Willrich MA, Hirata MH, Genvigir FD, Arazi SS, Rebecchi IM, Rodrigues AC, et al. CYP3A53A allele is associated with reduced lowering-lipid response to atorvastatin in individuals with hypercholesterolemia. Clin Chim Acta 2008; 398:15–20 [DOI] [PubMed] [Google Scholar]
- 6.Rosales A, Alvear M, Cuevas A, Saavedra N, Zambrano T, Salazar LA. Identification of pharmacogenetic predictors of lipid-lowering response to atorvastatin in Chilean subjects with hypercholesterolemia. Clin Chim Acta 2012; 413:495–501 [DOI] [PubMed] [Google Scholar]
- 7.Koroleva OS, Pushkov AA, Blagodatskikh KA, Baranova OA, Azizova OA, Nosikov VV, et al. Association of a polymorphic marker Trp719Arg of KIF6 gene with effects of atorvastatin and simvastatin in patients with early ischemic heart disease. Kardiologiia 2011; 51:4–12 [PubMed] [Google Scholar]
- 8.Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH. Influence of genetic variation in CYP3A4 and ABCB1 on dose decrease or switching during simvastatin and atorvastatin therapy. Pharmacoepidemiol Drug Saf 2010; 19:75–81 [DOI] [PubMed] [Google Scholar]
- 9.Bercovich D, Friedlander Y, Korem S, Houminer A, Hoffman A, Kleinberg L, et al. The association of common SNPs and haplotypes in the CETP and MDR1 genes with lipids response to fluvastatin in familial hypercholesterolemia. Atherosclerosis 2006; 185:97–107 [DOI] [PubMed] [Google Scholar]
- 10.Medina MW. The relationship between HMGCR genetic variation, alternative splicing, and statin efficacy. Discov Med 2010; 9:495–499 [PubMed] [Google Scholar]
- 11.Charland SL, Agatep BC, Herrera V, Schrader B, Frueh FW, Ryvkin M, et al. Providing patients with pharmacogenetic test results affects adherence to statin therapy: results of the Additional KIF6 Risk Offers Better Adherence to Statins (AKROBATS) trial. Pharmacogenomics J 2014; 14:272–280 [DOI] [PubMed] [Google Scholar]
- 12.Chasman DI, Posada D, Subrahmanyan L, Cook NR, Jr, Stanton VP, Ridker PM. Pharmacogenetic study of statin therapy and cholesterol reduction. JAMA 2004; 291:2821–2827 [DOI] [PubMed] [Google Scholar]
- 13.Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 2009; 325:100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chasman DI, Paré G, Mora S, Hopewell JC, Peloso G, Clarke R, et al. Forty-three loci associated with plasma lipoprotein size, concentration, and cholesterol content in genome-wide analysis. PLoS Genet 2009; 5:e1000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010; 466:707–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waterworth DM, Ricketts SL, Song K, Chen L, Zhao JH, Ripatti S, et al. Wellcome Trust Case Control Consortium . Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler Thromb Vasc Biol 2010; 30:2264–2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weissglas-Volkov D, Calkin AC, Tusie-Luna T, Sinsheimer JS, Zelcer N, Riba L, et al. The N342S MYLIP polymorphism is associated with high total cholesterol and increased LDL receptor degradation in humans. J Clin Invest 2011; 121:3062–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santos PC, Gagliardi AC, Miname MH, Chacra AP, Santos RD, Krieger JE, Pereira AC. SLCO1B1 haplotypes are not associated with atorvastatin-induced myalgia in Brazilian patients with familial hypercholesterolemia. Eur J Clin Pharmacol 2012; 68:273–279 [DOI] [PubMed] [Google Scholar]
- 19.Williams RR, Hunt SC, Schumacher MC, Hegele RA, Leppert MF, Ludwig EH, Hopkins PN. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol 1993; 72:171–176 [DOI] [PubMed] [Google Scholar]
- 20.Soares RA, Santos PC, Machado-Coelho GL, do Nascimento RM, Mill JG, Krieger JE, Pereira AC. CYP2C9 and VKORC1 polymorphisms are differently distributed in the Brazilian population according to self-declared ethnicity or genetic ancestry. Genet Test Mol Biomarkers 2012; 16:957–963 [DOI] [PubMed] [Google Scholar]
- 21.Miltiadous G, Xenophontos S, Bairaktari E, Ganotakis M, Cariolou M, Elisaf M. Genetic and environmental factors affecting the response to statin therapy in patients with molecularly defined familial hypercholesterolaemia. Pharmacogenet Genomics 2005; 15:219–225 [DOI] [PubMed] [Google Scholar]
- 22.Alonso R, Mata N, Castillo S, Fuentes F, Saenz P, Muñiz O, et al. Spanish Familial Hypercholesterolaemia Group . Cardiovascular disease in familial hypercholesterolaemia: influence of low-density lipoprotein receptor mutation type and classic risk factors. Atherosclerosis 2008; 200:315–321 [DOI] [PubMed] [Google Scholar]
- 23.Leigh SE, Foster AH, Whittall RA, Hubbart CS, Humphries SE. Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database. Ann Hum Genet 2008; 72:485–498 [DOI] [PubMed] [Google Scholar]
- 24.Santos PC, Morgan AC, Jannes CE, Turolla L, Krieger JE, Santos RD, Pereira AC. Presence and type of low density lipoprotein receptor (LDLR) mutation influences the lipid profile and response to lipid-lowering therapy in Brazilian patients with heterozygous familial hypercholesterolemia. Atherosclerosis 2014; 233:206–210 [DOI] [PubMed] [Google Scholar]
- 25.Santos PC, Soares RA, Santos DB, Nascimento RM, Coelho GL, Nicolau JC, et al. CYP2C19 and ABCB1 gene polymorphisms are differently distributed according to ethnicity in the Brazilian general population. BMC Med Genet 2011; 12:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santos PC, Oliveira TG, Lemos PA, Mill JG, Krieger JE, Pereira AC. MYLIP p.N342S polymorphism is not associated with lipid profile in the Brazilian population. Lipids Health Dis 2012; 11:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rohlfs RV, Weir BS. Distributions of Hardy–Weinberg equilibrium test statistics. Genetics 2008; 180:1609–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sorrentino V, Scheer L, Santos A, Reits E, Bleijlevens B, Zelcer N. Distinct functional domains contribute to degradation of the low density lipoprotein receptor (LDLR) by the E3 ubiquitin ligase inducible Degrader of the LDLR (IDOL). J Biol Chem 2011; 286:30190–30199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest 2006; 116:607–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pocathikorn A, Taylor RR, Mamotte CD. Atorvastatin increases expression of low-density lipoprotein receptor mRNA in human circulating mononuclear cells. Clin Exp Pharmacol Physiol 2010; 37:471–476 [DOI] [PubMed] [Google Scholar]
- 31.Sorrentino V, Fouchier SW, Motazacker MM, Nelson JK, Defesche JC, Dallinga-Thie GM, et al. Identification of a loss-of-function inducible degrader of the low-density lipoprotein receptor variant in individuals with low circulating low-density lipoprotein. Eur Heart J 2013; 34:1292–1297 [DOI] [PubMed] [Google Scholar]
- 32.Yin RX, Wu DF, Aung LH, Yan TT, Cao XL, Long XJ, et al. Several lipid-related gene polymorphisms interact with overweight/obesity to modulate blood pressure levels. Int J Mol Sci 2012; 13:12062–12081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan TT, Yin RX, Li Q, Huang P, Zeng XN, Huang KK, et al. Association of MYLIP rs3757354 SNP and several environmental factors with serum lipid levels in the Guangxi Bai Ku Yao and Han populations. Lipids Health Dis 2012; 11:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yin RX, Wu DF, Miao L, Aung LH, Cao XL, Yan TT, et al. Several genetic polymorphisms interact with overweight/obesity to influence serum lipid levels. Cardiovasc Diabetol 2012; 11:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chasman DI, Giulianini F, MacFadyen J, Barratt BJ, Nyberg F, Ridker PM. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ Cardiovasc Genet 2012; 5:257–264 [DOI] [PubMed] [Google Scholar]
- 36.Alvim RO, Freitas SR, Ferreira NE, Santos PC, Cunha RS, Mill JG, et al. APOE polymorphism is associated with lipid profile, but not with arterial stiffness in the general population. Lipids Health Dis 2010; 9:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev 2011; 63:157–181 [DOI] [PubMed] [Google Scholar]
- 38.Santos PC, Alvim Rde O, Ferreira NE, de Sá Cunha R, Krieger JE, Mill JG, Pereira AC. Ethnicity and arterial stiffness in Brazil. Am J Hypertens 2011; 24:278–284 [DOI] [PubMed] [Google Scholar]
- 39.Santos PC, Bueno CT, Lemos PA, Krieger JE, Pereira AC. LPA rs10455872 polymorphism is associated with coronary lesions in Brazilian patients submitted to coronary angiography. Lipids Health Dis 2014; 13:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santos PC, Soares RA, Nascimento RM, Machado-Coelho GL, Mill JG, Krieger JE, Pereira AC. SLCO1B1 rs4149056 polymorphism associated with statin-induced myopathy is differently distributed according to ethnicity in the Brazilian general population: Amerindians as a high risk ethnic group. BMC Med Genet 2011; 12:136. [DOI] [PMC free article] [PubMed] [Google Scholar]