

Abstract
EEF2K (eukaryotic elongation factor-2 kinase), also known as Ca2+/calmodulin-dependent protein kinase III, functions in downregulating peptide chain elongation through inactivation of EEF2 (eukaryotic translation elongation factor 2). Currently, there is a limited amount of information on the promotion of autophagic survival by EEF2K in breast and glioblastoma cell lines. However, the precise role of EEF2K in carcinogenesis as well as the underlying mechanism involved is still poorly understood. In this study, contrary to the reported autophagy-promoting activity of EEF2K in certain cancer cells, EEF2K is shown to negatively regulate autophagy in human colon cancer cells as indicated by the increase of LC3-II levels, the accumulation of LC3 dots per cell, and the promotion of autophagic flux in EEF2K knockdown cells. EEF2K negatively regulates cell viability, clonogenicity, cell proliferation, and cell size in colon cancer cells. Autophagy induced by EEF2K silencing promotes cell survival and does not potentiate the anticancer efficacy of the AKT inhibitor MK-2206. In addition, autophagy induced by silencing of EEF2K is attributed to induction of protein synthesis and activation of the AMPK-ULK1 pathway, independent of the suppression of MTOR activity and ROS generation. Knockdown of AMPK or ULK1 significantly abrogates EEF2K silencing-induced increase of LC3-II levels, accumulation of LC3 dots per cell as well as cell proliferation in colon cancer cells. In conclusion, silencing of EEF2K promotes autophagic survival via activation of the AMPK-ULK1 pathway in colon cancer cells. This finding suggests that upregulation of EEF2K activity may constitute a novel approach for the treatment of human colon cancer.
Keywords: elongation factor-2 kinase, autophagy, colon cancer, AMPK, ULK1, ATG, MTOR, ROS, cancer growth, cell survival
Introduction
Cells consume a tremendous amount of metabolic energy for survival, most of which is used in peptide chain elongation. The rate of peptide chain elongation is modulated by EEF2 which in turn is regulated by EEF2K.1,2 Under stress or starvation conditions, in order to reduce energy consumption, EEF2K is normally activated by phosphorylation at Ser398 via Ca2+-calmodulin or AMPK/AMP-activated protein kinase, or at Ser499 by cAMP-PRKA (protein kinase A).3-6 The activated EEF2K in turn phosphorylates its target EEF2 at Thr56.7 This phosphorylation inactivates EEF2 resulting in termination of peptide elongation by decreasing the affinity of the elongation factor toward the ribosome.7,8 Other stimuli such as stress and growth factors that promote protein synthesis must inhibit the activity of EEF2K. Recent studies demonstrate that signaling pathways such as those modulated by RPS6KA1/p90 (ribosomal protein S6 kinase, 90 kDa, polypeptide 1), MAPK13/p38δ/SAPK4 (mitogen-activated protein kinase 13), MTOR (mechanistic target of rapamycin) and RPS6KB (ribosomal protein S6 kinase, 70 kDa, polypeptide) can directly phosphorylate EEF2K at specific sites that inactivate EEF2K leading to increased protein translation.9-11
Apart from regulating the activity of EEF2K, AMPK can activate autophagy.12 Autophagy is a self-degradative process by the removal of damaged proteins and organelles to promote a cell survival response to nutritional starvation or stress conditions. Certain examples have demonstrated that AMPK is activated by an elevated AMP/ATP ratio due to cellular and environmental stress such as nutritional deprivation.13 Autophagy can also be activated in response to many forms of cellular stress beyond nutritional deprivation including reactive oxygen species (ROS) and DNA damage.14,15 Recent studies have shown that autophagy could also act as an apoptosis-independent programmed cell death.14 The precise role of autophagy in cellular responses to stress is far from fully elucidated.
Previous studies have demonstrated that EEF2K-mediated EEF2 phosphorylation at Thr56 could induce autophagy as a survival mechanism in glioma cells and breast cancer cells, and inhibition of EEF2K could potentiate the efficacy of anticancer agents against cancers.16-20 Silencing of EEF2K expression by siRNA could reduce both basal and starvation-induced autophagy levels in glioma cells, as characterized by a decrease in autophagic marker MAP1LC3B-II/LC3-II (microtubule-associated protein 1 light chain 3 β-II) levels.21,22 Eef2k knockout mouse embryonic fibroblasts (MEFs) also show a decrease of basal and nutrient deprivation-induced autophagy levels.22 However, Chen et al.23 report that the EEF2K inhibitor A-484954 cannot significantly inhibit cancer cell growth in lung and prostate cancer cells. This finding is consistent with the effect of silencing of EEF2K in both lung and prostate cancer cells.23 Ryazanov also has found that eef2k knockout mice grow and reproduce normally.24 Although different effects of EEF2K on cell survival have been observed, the exact mechanisms by which EEF2K regulates cell growth or autophagy are still unclear. Therefore, studies to reveal the role of EEF2K in cancer growth as well as the molecular mechanisms involved in regulating autophagy are highly warranted.
To address this issue, we silenced or overexpressed EEF2K in human colon cancer cells to characterize the role of EEF2K in cancer growth and to reveal the molecular mechanism involved in the regulation of autophagy. Our results indicate that autophagy is induced by knockdown of EEF2K in human colon cancer cells. This response is mediated by activation of the AMPK-ULK1 (unc-51 like autophagy activating kinase 1) pathway independent of MTOR inhibition in a fashion different from that during nutritional deprivation.
Results
Silencing of EEF2K induces autophagy in human colon cancer cells
Previous studies have shown that EEF2K is effective in inducing autophagy in glioma and breast cancer cells. We have therefore investigated whether EEF2K could also induce autophagy in human colon cancer cells. As shown in Figure 1A, silencing of EEF2K using a single siRNA could completely block its downstream target EEF2 phosphorylation at Thr56 in human colon cancer HT-29 and HCT-116 cells, consistent with the fact that reduction of EEF2K activity can reduce the phosphorylation of EEF2 at Thr56.21,22 However, silencing of EEF2K markedly increased but did not reduce the amount of LC3-II levels in both HT-29 and HCT-116 cells, suggesting that the increased protein synthesis can induce autophagy (Fig. 1A). The same result was obtained using multiple siRNAs targeting different regions of EEF2K (Fig. 1B). These findings were further substantiated by the increase of LC3 dots accumulation in EEF2K-depleted cells (Fig. 1C). As shown in Figure 1C, EEF2K silencing significantly increased LC3 puncta accumulation in both the cytoplasm and nucleus, and most of these LC3 puncta were concentrated in the nucleus. The amount of LC3 dots per cell was significantly increased by more than 6-fold in EEF2K knockdown cells as compared with the control group (Fig. 1D). Furthermore, to distinguish between induction of autophagy and inhibition of autophagic vesicles degradation in EEF2K silenced cells, we analyzed autophagic flux in EEF2K-silenced cells in the absence or presence of lysosomal protease inhibitors E64d and pepstatin A. As shown in Figure 1E, protease inhibitors could further increase both LC3-II and mammalian autophagy-specific substrate SQSTM1/p62 levels in EEF2K-silenced cells when compared with vehicle treatment, suggesting that LC3-II accumulation in EEF2K-silenced cells was attributable to promotion of autophagy but not to impairment of autophagic degradation. Taken together, these results indicate that knockdown of EEF2K induces autophagy in human colon cancer cells.

Figure 1. Silencing of EEF2K induces autophagy in human colon cancer cells. (A and B) HT-29 or HCT-116 cells were transfected with nontargeting control siRNA (siCTL), a single siRNA duplex targeting EEF2K (siEEF2K; A) or multiple siRNAs targeting different regions of EEF2K (siEEF2K; B) for 48 h. EEF2K, phospho-EEF2 (Thr56; p-EEF2), EEF2, LC3, and ACTB/β-actin were analyzed by western blot. Representative western blot and densitometric analysis normalized to ACTB demonstrating the effect of EEF2K silencing on LC3-II levels. (C and D) HT-29 or HCT-116 cells were transfected with control siRNA or a single siRNA duplex targeting EEF2K for 48 h. (C) Representative immunofluorescent images showing redistribution of autophagic marker LC3 in EEF2K knockdown cells were taken on a confocal microscope. Cells were fixed with 3.5% formaldehyde for 10 min, permeabilized with 0.1% Triton X-100 for 10 min, and stained with LC3 antibody and DAPI. Scale bar: 10 μm. (D) The average number of LC3 dots per cell was counted in more than 5 fields with at least 100 cells for each group. (E) Representative western blot and densitometric analysis normalized to ACTB demonstrating the effect of lysosomal protease inhibitors E64d plus pepstatin A on EEF2K silencing induced LC3-II accumulation. HT-29 cells were transfected with nontargeting control siRNA or EEF2K siRNA. At 3 h after transfection, cells were treated with 10 μg/ml E64d and pepstatin A (Pep A) for 45 h. All quantitative data shown represent the means ± SEM of at least 3 independent experiments. *P < 0.05, $P < 0.01, and #P < 0.001, vs. the siCTL group (A, B, and D) or vehicle treatment only (E).
BECN1 and ATG7 are required for autophagy in response to EEF2K silencing
A series of autophagy-related (ATG) genes are involved in the process of autophagy. We would like to know whether autophagy induced by silencing of EEF2K contributes to regulation of specific proteins of the ATG family. ATG5 and ATG7 (a ubiquitin-activating enzyme homolog), are required for initiation of autophagy. BECN1 is required for the initiation of autophagosome formation. Previous studies show that autophagy can be induced through ATG5-, BECN1-, or ATG7-dependent or independent signaling pathways.14,25 To determine whether induction of autophagy by EEF2K silencing is related to ATG5, BECN1, or ATG7, we first analyzed the expression levels of ATG5, BECN1, and ATG7 separately by western blot. As shown in Figure 2A, knockdown of EEF2K significantly increased the protein levels of BECN1 and ATG7, but not ATG5. The increase in BECN1 and ATG7 levels in EEF2K-depleted cells is attributed to protein synthesis but not to transcriptional increase (Fig. 2B). In order to further validate the increased BECN1 and ATG7 due to protein synthesis, we blocked protein degradation by MG132. The result showed that protein levels of BECN1 and ATG7 were significantly accumulated in EEF2K-depleted cells after exposure to MG132, suggesting EEF2K silencing does not block protein degradation of BECN1 and ATG7 (Fig. 2C). Taken together, the increase of both BECN1 and ATG7 in EEF2K knockdown cells is not due to blockage of degradation but to protein synthesis. We silenced BECN1 using siRNA in HT-29 cells. The result showed that knockdown of BECN1 could significantly block the accumulation of LC3-II in EEF2K-silenced cells (Fig. 2D). Similar to the effect of BECN1 knockdown, silencing of ATG7 also markedly attenuated the accumulation of LC3-II (Fig. 2E). Moreover, the number of LC3 dots per cell was significantly reduced after silencing of BECN1 or ATG7 in EEF2K knockdown cells (Fig. 2F). Taken together, these results indicate that the upregulation of BECN1 and ATG7 is responsible for autophagy induced by EEF2K silencing.

Figure 2. BECN1 and ATG7 are required for autophagy in response to EEF2K silencing. (A) Silencing of EEF2K upregulates the protein levels of BECN1 and ATG7, but not ATG5. HT-29 or HCT-116 cells were transfected with nontargeting control siRNA (siCTL) or a single EEF2K siRNA duplex (siEEF2K) for 48 h. ATG5, BECN1, ATG7, and ACTB were analyzed by western blot. Data shown are representative of more than 3 independent experiments. (B) Silencing of EEF2K does not change the mRNA levels of ATG5, BECN1, and ATG7. Cells were transfected as in (A). The mRNA levels of ATG5, BECN1, and ATG7 were analyzed by RT-PCR. (C) Effect of MG132 on the protein levels of ATG5, BECN1, and ATG7 in EEF2K knockdown cells. HCT-116 cells were transfected with nontargeting control siRNA (siCTL) or a single EEF2K siRNA duplex (siEEF2K) for 48. Before harvested for western blot, cells were treated with MG132 (10 μM) for 12 h. (D and E) Representative western blot and densitometric analysis normalized to ACTB demonstrating the effects of BECN1 siRNA (D) and ATG7 siRNA (E) on LC3-II levels induced by EEF2K silencing. HT-29 cells were transfected with nontargeting siRNA, siEEF2K, BECN1 siRNA (siBECN1), ATG7 siRNA (siATG7), siEEF2K plus siBECN1, or siEEF2K plus siATG7 for 48 h. All quantitative data shown represent the means ± SEM of at least 3 independent experiments. *P < 0.05 and $P < 0.01, vs. the siEEF2K group. (F) The effects of BECN1 siRNA and ATG7 siRNA on LC3 dots accumulation induced by EEF2K silencing. HT-29 cells were treated with siRNAs against EEF2K, BECN1, or ATG7 as in (D and E). Cells were fixed, stained for LC3, and imaged. The average number of LC3 dots per cell was counted in more than 5 fields with at least 100 cells for each group and expressed as the means ± SEM of 3 independent experiments. #P < 0.001, vs. the EEF2K siRNA group (siEEF2K).
EEF2K silencing-induced autophagy functions to promote colon cancer cell survival
It has been reported that targeting EEF2K by siRNA reduces cancer growth in glioma and breast cancer cells.17,21 Contrary to this, silencing of EEF2K significantly promoted colon cancer cell viability and colony formation, suggesting that EEF2K negatively regulates cell proliferation in human colon cancer cells (Fig. 3A, B, and D). These findings were confirmed by the decrease of cell viability and colony formation in EEF2K-overexpressing cells as compared with control (Fig. 3A, C, and E). In order to further validate the observation on cell survival in EEF2K-silenced cells, we analyzed cell size and cell number in EEF2K knockdown cells as well as in EEF2K overexpressed cells. Both cell size and cell number in EEF2K-depleted cells were significantly increased compared with the control group (Fig. 3F and H), while the cell size and cell number were decreased in EEF2K-overexpressing cells (Fig. 3G and I). Taken together, EEF2K silencing promotes cell growth and cell proliferation in human colon cancer cells. This finding is further substantiated by the result that EEF2K silencing significantly attenuated the antitumor efficacy of oxaliplatin against colon cancer cells (Fig. 3J). Our findings in colon cancer cells are in accordance with other reports that knockdown of EEF2K by siRNA as well as reduction of EEF2 phosphorylation at effective concentrations by the EEF2K inhibitor A-484954 has little inhibitory effect on cancer cell growth in certain cancer cells including lung cancer and prostate cancer under both serum and serum-free conditions.23 In addition, overexpression of EEF2K could significantly enhance the antitumor efficacy of oxaliplatin against colon cancer cells, indicating that increase of EEF2K activity can be used to treat colon cancer (Fig. 3K).

Figure 3.EEF2K silencing promotes cell survival in human colon cancer cells. (A) Representative western blot demonstrating the knockdown efficiency of EEF2K siRNA and the overexpression efficiency of EEF2K in both HT-29 and HCT-116 cells. Cells were transfected with control siRNA (siCTL), EEF2K siRNA (siEEF2K), empty vector (Vector), or EEF2K plasmids (EEF2K) for 48 h. (B and C) The effects of EEF2K knockdown and EEF2K overexpression on cell viability. HT-29 or HCT-116 cells were transfected as in (A), and then assessed by the MTT assay. (D and E) The effects of EEF2K knockdown and EEF2K overexpression on colony formation. HT-29 or HCT-116 cells were transfected as in (A). After 48 h transfection, cells were seeded into 6-well plates at the density of 150 cells per well for control siRNA and EEF2K siRNA groups (D) and 200 cells per well for the empty vector and EEF2K overexpression groups (E), incubated at 37 °C for 12 to 14 d, stained with crystal violet (0.5% w/v) and imaged. Colonies with 50 or more cells were counted. (F and G) The effects of EEF2K knockdown and EEF2K overexpression on cell size. HT-29 or HCT-116 cells were transfected as in (A). After 48 h transfection, cells were imaged using a Nikon fluorescence microscope. Scale bar: 20 μm. Cell size was analyzed using the MetaMorph software. The amounts of cell size in more than 50 cells for each group were quantified. (H and I) The effects of EEF2K knockdown and EEF2K overexpression on cell number. HT-29 or HCT-116 cells were transfected as in (A). Cell number was quantified after 48 h transfection. (J) The effect of EEF2K siRNA on oxaliplatin induced apoptosis. HCT-116 cells were transfected with control siRNA or EEF2K siRNA for 24 h, and then treated with vehicle (0.1% DMSO) or oxaliplatin (25 μM) for 48 h. Cells were stained with ANXA5-PI. The percentage of apoptotic cells (ANXA5+) was analyzed by flow cytometry. (K) The effect of EEF2K overexpression on oxaliplatin-induced apoptosis. HT-29 cells were transfected with empty vector (Vector), or EEF2K plasmids (EEF2K) for 24 h, and then treated with vehicle (0.1% DMSO) or oxaliplatin (25 μM) for 48 h. Cells were stained with ANXA5-PI and analyzed by flow cytometry as in (J). All quantitative data shown represent the means ± SEM of at least 3 independent experiments. *P < 0.05, $P < 0.01 and #P < 0.001, vs. the siCTL group (B, D, F, and H), the vector group (C, E, G, and I), or the oxaliplatin treatment only (J and K).
Furthermore, given the fact that intracellular autophagy promotes cell survival or induces programmed cell death, we investigated the role of autophagy in EEF2K knockdown colon cancer cells. Previous studies report that inhibition of EEF2K-mediated autophagy by silencing of BECN1 can significantly enhance the efficacy of anticancer agents against glioma and breast cancer cells, suggesting autophagy functions as a cell-survival mechanism.26,27 In line with this finding, we found that disruption of autophagy by knockdown of BECN1 or ATG7 reduced cell viability and clonogenicity in EEF2K-silenced colon cancer cells (Fig. 4A and B). Similar to the effect of BECN1 and ATG7 knockdown, the blockage of autophagic flux by protease inhibitors E64d and pepstatin A attenuated the increase of cell viability induced by EEF2K silencing (Fig. 4C). These results indicate that the increase of cell viability by EEF2K silencing in colon cancer cells is attributed to induction of the cell-survival mechanism of autophagy. Considering the fact that autophagy induced by EEF2K silencing acts as a cell-survival mechanism in colon cancer cells, upregulation of EEF2K as an anticancer approach might be feasible in human colon cancer.
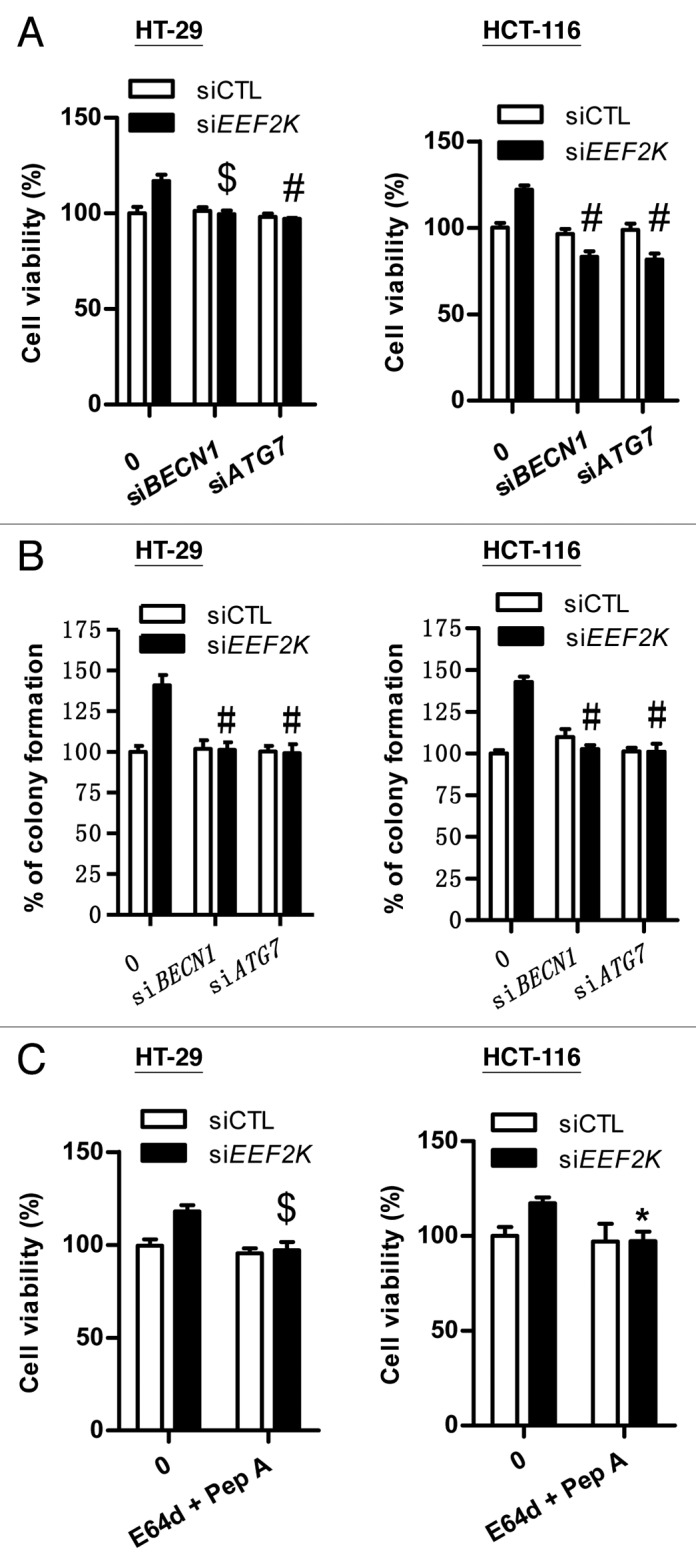
Figure 4. Cell survival induced by EEF2K silencing is attributed to induction of autophagy. (A) The effects of BECN1 siRNA and ATG7 siRNA on the increase of cell viability induced by EEF2K silencing. HT-29 or HCT-116 cells were transfected with BECN1 siRNA (siBECN1), ATG7 siRNA (siATG7), EEF2K siRNA (siEEF2K), siBECN1 plus siEEF2K, or siATG7 plus siEEF2K for 48 h. Cell viability was assessed by the MTT assay. (B) The effects of BECN1 siRNA and ATG7 siRNA on the increase of colony formation induced by EEF2K silencing. HT-29 or HCT-116 cells were transfected with siBECN1, siATG7, siEEF2K, siBECN1 plus siEEF2K, or siATG7 plus siEEF2K. After 48 h transfection, cells were seeded into 6-well plates at the density of 150 cells per well, incubated at 37 °C for 12 to 14 d and then stained with crystal violet (0.5% w/v). Colonies with 50 or more cells were counted. (C) The effects of E64d and pepstatin A on cell viability in EEF2K silenced cells. HT-29 or HCT-116 cells were transfected with nontargeting control siRNA or EEF2K siRNA. After 3 h transfection, cells were treated with vehicle (0.1% DMSO), or 10 μg/ml E64d and pepstatin A for 45 h and then assessed by the MTT assay. All data shown are expressed as the means ± SEM of 3 independent experiments. *P < 0.05, $P < 0.01, and #P < 0.001, vs. the EEF2K siRNA group (siEEF2K; A–C).
Silencing of EEF2K cannot potentiate the anticancer efficiency of MK-2206 against colon cancer cells
AKT is an important anticancer target. The AKT inhibitor MK-2206 has been well studied for its role in promoting autophagy through activation of EEF2K and inactivation of EEF2 in glioma cells, as indicated by the increase of LC3-II.16 Consistent with this finding, MK-2206 at effective concentrations such as 0.1–5 μM also significantly promoted autophagy as indicated by the accumulation of LC3-II levels in human colon cancer cells, while EEF2 phosphorylation at Thr56 was not markedly increased in cells treated with MK-2206 at the same concentration range (Fig. 5A). These results indicate that autophagy induced by MK-2206 in colon cancer cells could not be completely attributed to activation of the EEF2K. In order to further validate the effect of EEF2K on AKT inhibition-induced autophagy in colon cancer cells, cells were transfected with siRNA against EEF2K before exposure to MK-2206. As shown in Figure 5B, knockdown of EEF2K could not block the autophagic response triggered by the AKT inhibitor MK-2206 in human colon cancer cells, suggesting that EEF2K does not correlate with MK-2206-induced autophagy. This result contradicts the conventional notion that inhibition of AKT by MK-2206 activates EEF2K-dependent autophagy. Although MK-2206 at 10 μM increased EEF2 phosphorylation at Thr56, knockdown of EEF2K could not potentiate the anticancer efficacy of MK-2206, implying that EEF2K cannot serve as an anticancer target for colon cancer therapy (Fig. 5A and C). EEF2K has been demonstrated to play a critical role in induction of autophagy in glioma cells in response to cellular stress such as AKT inhibition by MK-2206 and nutrient deprivation. However, the underlying molecular mechanisms by which EEF2K controls autophagy remain unknown. Our findings on the role of EEF2K in autophagic response induced by AKT inhibition add new insight to the AKT-mediated autophagy pathway. In addition, our results also suggest that upregulation of EEF2K activity might constitute a therapeutic option for the treatment of certain cancers including human colon cancer.

Figure 5. Silencing of EEF2K does not potentiate the anticancer efficacy of MK-2206 against colon cancer cells. (A) Representative western blot analysis of phospho-EEF2 (Thr56; p-EEF2) and LC3-II expression in HT-29 cells treated with MK-2206 (0, 0.1, 0.5, 1, 5, 10 μM) for 24 h. The amounts of LC3-II and ACTB were quantified. (B) Representative western blot and densitometric analysis normalized to ACTB demonstrating the effect of EEF2K silencing on MK-2206 induced accumulation of LC3-II. HT-29 cells were transfected with nontargeting control siRNA (siCTL) or EEF2K siRNA (siEEF2K) for 24 h and then treated with the vehicle (0.1% DMSO) or MK-2206 (5 μM) for 24 h. (C) HT-29 cells were transfected with control siRNA or EEF2K siRNA for 24 h and then treated with the vehicle (0.1% DMSO), 5 μM MK-2206, or 10 μM MK-2206 for 24 h. Cell viability was analyzed by the MTT assay. Results shown are expressed as the means ± SEM of 3 independent experiments. All quantitative data shown represent the means ± SEM of at least 3 independent experiments. #P < 0.001, vs. the control group (0 μM; A) or the siCTL plus vehicle group (B).
The AMPK-ULK1 pathway is required for autophagy in response to EEF2K silencing
Knockdown of EEF2K stimulates protein synthesis, which results in ATP consumption and then leads to an increase of AMP/ATP ratio.28 In line with this finding, ATP was markedly reduced in colon cancer cells after EEF2K silencing (Fig. 6A). Previous studies show that an increased AMP/ATP ratio can activate AMPK by phosphorylation at Thr172.29 Consistent with this finding, AMPK was significantly activated in both EEF2K-depleted HT-29 and HCT-116 cells (Fig. 6B). It has been demonstrated that AMPK can activate autophagy by activation of ULK1 or by inactivation of MTOR.30-32 In this study, we found that ULK1 was activated by phosphorylation at Ser555 and dephosphorylation at Ser757, but MTOR was not inactivated in EEF2K-depleted cells (Fig. 6B). These findings suggest that AMPK may enhance autophagy through activation of ULK1 but not via inhibition of MTOR pathway. In order to validate that EEF2K silencing leads to protein synthesis, which depletes ATP levels and then activates AMPK-ULK1-mediated autophagy, we blocked protein synthesis by cycloheximide in EEF2K-depleted cells and then detected the active form of ULK1 and LC3 levels. As shown in Figure 6C, cycloheximide could completely block both ULK1 phosphorylation at Ser555 and LC3-II accumulation induced by EEF2K silencing. This result indicated that protein synthesis induced by EEF2K silencing is responsible for ULK1 activation and autophagy accumulation. In order to further validate whether AMPK and its downstream target ULK1 are involved in autophagy induced by EEF2K knockdown, the effects of PRKAA1 and PRKAA2 (AMPKα) siRNA and ULK1 siRNA on autophagy in EEF2K-depleted cells were analyzed. As shown in Figure 6D, AMPKα siRNA could block ULK1 phosphorylation at Ser555 and significantly reduce LC3-II accumulation in EEF2K knockdown cells, suggesting that AMPK is responsible for EEF2K knockdown-induced autophagy. Furthermore, silencing of ULK1 significantly reduced LC3-II levels, indicating that ULK1 is also involved in autophagy induced by EEF2K silencing (Fig. 6E). These findings were further substantiated by quantification of the amount of LC3 dots per cell, showing that knockdown of AMPKα or ULK1 could completely block LC3 dots accumulation induced by EEF2K silencing (Fig. 6F). In addition, we found that knockdown of AMPKα and ULK1 could significantly inhibit cell growth in EEF2K-depleted human colon cancer cells (Fig. 6G). ROS production is one form of cellular stress that plays a critical role in the induction of autophagy. ROS levels were therefore analyzed using DCFDA staining in HT-29 cells after EEF2K silencing, followed by fluorescence microscopy and flow cytometry. Our results demonstrated that ROS levels were not significantly increased in EEF2K-depleted cells as compared with control or H2O2 treatment, suggesting that ROS production is not involved in autophagy induced by silencing of EEF2K (Fig. 6H and I). Taken together, these results indicate that autophagy induced by silencing of EEF2K is attributed to activation of AMPK-ULK1 pathway, independent of the suppression of MTOR activity and stimulation of ROS production.

Figure 6. The AMPK-ULK1 pathway is required for autophagy induced by EEF2K silencing. (A and B) HT-29 or HCT-116 cells were transfected with nontargeting control siRNA (siCTL) or EEF2K siRNA (siEEF2K) for 48 h. (A) Silencing of EEF2K reduces ATP level. After transfection, the ATP level was analyzed using the ATPlite Luminescence Assay Kit. (B) Silencing of EEF2K activates AMPK by phosphorylation at Thr172 and activates ULK1 by phosphorylation at Ser555 and dephosphorylation at Ser757, but does not inactivate MTOR. The protein levels of EEF2K, phospho-AMPKα (Thr172; p-AMPKα), AMPKα, phospho-ULK1 (Ser555; p-ULK1 (Ser555)), phospho-ULK1 (Ser757; p-ULK1 (Ser757)), ULK1, phospho-MTOR (Ser2448; p-MTOR), and ACTB were analyzed by western blot. (C) Representative western blot and densitometric analysis normalized to ACTB demonstrating the effect of cycloheximide on EEF2K silencing-induced LC3-II accumulation. HT-29 or HCT-116 cells were transfected with nontargeting control siRNA or EEF2K siRNA. At 3 h after transfection, cells were treated with 10 μg/ml cycloheximide (CHX) for 45 h. (D–G) HT-29 cells were transfected with control siRNA, EEF2K siRNA, PRKAA1 and PRKAA2/AMPKα siRNA (siAMPKα), ULK1 siRNA (siULK1), siEEF2K plus siAMPKα, or siEEF2K plus siULK1 for 48 h. (D) Representative western blot demonstrating the effects of AMPKα siRNA on phospho-ULK1 (Ser555; p-ULK1) and LC3-II levels induced by EEF2K silencing. Densitometric analysis normalized to ACTB demonstrating the effect of AMPKα siRNA on LC3-II levels induced by EEF2K silencing. (E) Representative western blot and densitometric analysis normalized to ACTB demonstrating the effect of ULK1 siRNA on LC3-II levels induced by EEF2K silencing. (F) The effects of AMPKα siRNA and ULK1 siRNA on LC3 dots accumulation induced by EEF2K silencing. The average number of LC3 dots per cell was counted in more than 5 fields with at least 100 cells for each group and expressed as the means ± SEM of 3 independent experiments. #P < 0.001, vs. the EEF2K siRNA group (siEEF2K). (G) The effects of AMPKα siRNA and ULK1 siRNA on the increase of cell viability induced by EEF2K silencing. Cell viability was analyzed by MTT assay. (H and I) The effect of EEF2K silencing on ROS generation. HT-29 cells were transfected with control siRNA or EEF2K siRNA for 48 h, and treated with 20 μM DCFDA for 30 min. ROS levels were detected by fluorescence microscopy (H) or by flow cytometry (I). H2O2 (1 mM) treatment for 2 h was used as a positive control group. Scale bar: 20 μm. All quantitative data shown represent the means ± SEM of at least 3 independent experiments. *P < 0.05, $P < 0.01 and #P < 0.001, vs. the siCTL group (for A), or the EEF2K siRNA group (siEEF2K; for C, D, E, and G).
Discussion
EEF2K is well known for its role in the negative regulation of protein translation through inactivation of EEF2 by phosphorylation at Thr56. Previous studies report that the activated EEF2K can induce autophagy in glioma and breast cancer cells. However, the effect of EEF2K on growth of colon cancer cells as well as the underlying mechanism involved is not understood. In this study, we demonstrate that silencing of EEF2K induces autophagy in colon cancer cells and the AMPK-ULK1 pathway is required for this autophagy.
Previous studies report that knockdown of EEF2K by siRNA abrogates autophagy and then results in inhibition of tumor growth, augmentation of apoptosis, and sensitization of glioma or breast cancer to the anticancer agents doxorubicin or MK-2206.16,17 In contrast, silencing of EEF2K by siRNA enhances autophagy instead of blocking autophagy in human colon cancer cells. The anticancer efficiency of MK-2206 is not further enhanced in colon cancer cells after silencing of EEF2K. This finding in colon cancer cells is in line with accumulating reports that knockdown of EEF2K by siRNA does not inhibit cell growth of lung cancer and prostate cancer cells under both serum and serum-free conditions.23 In addition, mice lacking EEF2K do not exhibit delays in development and reproduction, indicating that disruption of EEF2K is not sufficient for the inhibition of cell growth.24 Knockdown of EEF2K can activate EEF2 by reduction of EEF2 phosphorylation at Thr56 resulting in promotion of protein synthesis. It has also been reported that inhibition of EEF2 rapidly arrests protein synthesis and leads to cancer cell growth inhibiton.33 It is therefore conceivable that activation of protein synthesis by silencing of EEF2K does not arrest cancer growth in colon cancer cells. Taken together, EEF2K performs 2 apparently opposite functions in either promoting or inhibiting both autophagy and cancer growth in cell type-dependent manners.
Besides the cytoplasmic LC3-positive autophagosomes during autophagy, recent studies have demonstrated that LC3 punctate signals can also concentrate in the nucleus. For example, the picornavirus foot-and-mouth disease virus can induce LC3 puncta signal to concentrate close to the nucleus in > 95% of the cells within 2 h after infection of CHO cells.34 C2-ceramide, temozolomide, and arsenic trioxide can induce cytoplasmic and nucleus localization of LC3B in glioblastoma cells U373-MG.35 In addition, positive staining of LC3B is observed in the cytoplasm and nucleus of glioblastoma tissue in patients. These findings are consistent with the LC3 localization in EEF2K knockdown colon cancer cells. A recent study shows that CR 3294 significantly induces the accumulation of LC3-II in both the nucleus and cytosol of breast cancer cells. Such LC3-II nucleus accumulation is more abundant when the cells are treated with CR 3294 and hypoxia,36 indicating HIF1A may promote LC3 puncta to concentrate to the nuclear membrane or into the nucleus. However, the underlying mechanism of LC3 nucleus localization is not completely understood. We subsequently investigated whether certain proteins lead autophagosomes induced by EEF2K silencing into the nucleus.
Autophagy is associated with either cell survival or cell death. Under conditions of severe stress, autophagy would increase cell death.14 However, in some instances, autophagy occurs as part of normal metabolism to remove damaged proteins or organelles and some degree of autophagy formation is necessary for maintaining normal physiology.37 Therefore, cell survival requires an appropriate degree of autophagy. To date, the role of autophagy in carcinogenesis has not been completely understood. The role of autophagy in cancer in general is quite complex and is likely dependent on the tumor tissue of origin, stage, and the constellation of genetic mutations and epigenetic changes.38 Resistance of cancer cells to treatment can be associated with both autophagy and inhibition of the more common apoptotic cell death pathway. Under nutrient-deprivation condition, high levels of EEF2K could be activated to block translation elongation to adapt to the stress condition, suggesting that activated EEF2K functions to promote cell survival.39 Inhibition of EEF2K potentiates the anticancer efficacy of the AKT inhibitor MK-2206 in glioma cells.16 According to these findings, blockage of EEF2K could represent a treatment option for breast cancer and glioblastoma. Contrary to this finding, we found that inhibition of EEF2K by knockdown could activate autophagy and then promote cell survival under nutrient condition or in the presence of the antitumor drug oxaliplatin, indicating that EEF2K plays an important role in negatively regulating cell growth in human colon cancer cells. Our finding is consistent with the effect of EEF2K inhibitor A-484954 on cell growth in lung and prostate cancer cells.23 Cancer cells grow and divide much more rapidly than normal cells, thus they have a much higher demand for nutrients and oxygen than nutrient deprivation. Therefore, upregulation of EEF2K could represent an approach to treat certain cancers such as human colon cancer. Knowledge of the mechanisms and molecules involved will help us to understand the role of EEF2K on the growth and survival of different cancers. Taken together, modulation of EEF2K activity in different cancers might constitute a feasible therapeutic method for cancer treatment.
The signaling pathways that lead to autophagy under nutrient-deprivation conditions have been clearly characterized. MTOR is a central cell growth regulator that links nutrient signals and autophagy. Under starvation conditions, MTOR, which functions as a critical negative regulator of autophagy, is inhibited.40,41 However, mammalian cells rarely experience nutrient deprivation under normal physiological conditions. Inactivation of MTOR is not necessary for autophagy under nutrient conditions.42 Guo et al.32 report that lipopolysaccharide could induce autophagy via activation of AMPK but not via inhibition of MTOR. Consistent with this finding, autophagy induced by silencing of EEF2K is attributed to activation of AMPK, independent of MTOR inhibition in colon cancer cells under normal nutrient condition.
It appears that EEF2K plays opposite roles in either inducing or inhibiting autophagy in different cancer types. The signaling pathway downstream of EEF2K-EEF2 in controlling autophagy remains unknown. In this study, we report for the first time that silencing of EEF2K enhances autophagy-related genes and promotes cell survival via the AMPK-ULK1-dependent pathway (Fig. 7). This finding indicates that the increase of EEF2K activity might reduce the expression of autophagy-related genes such as BECN1 and ATG7, and inactivate the autophagic AMPK-ULK1 pathway. These, in turn, attenuate autophagy and block the growth of human colon cancer cells. We also report that increase of EEF2K activity can suppress autophagy and enhance the efficacy of drugs against colon cancer cells. In addition, it has been demonstrated that EEF2K is downregulated in human colorectal carcinoma patients (Fig. S1).43 Therefore, upregulation of EEF2K can be a novel strategy for the treatment of human colon cancer. Considering multiple autophagy pathways regulated by EEF2K, attenuation of autophagy by direct increase of EEF2K activity would be a better method than directly targeting a single autophagy pathway in human colon cancer cells. The approach of targeting against EEF2K has gained some attention for treating glioma and breast cancer. However, the general applicability of this approach is questionable in view of our findings. Cancer tissue typing in terms of its autophagic response toward EEF2K inhibition should be performed to assess whether a specific cancer would benefit from this approach. According to our findings, upregulation of EEF2K activity may be developed as a novel approach for the treatment of human colon cancer.
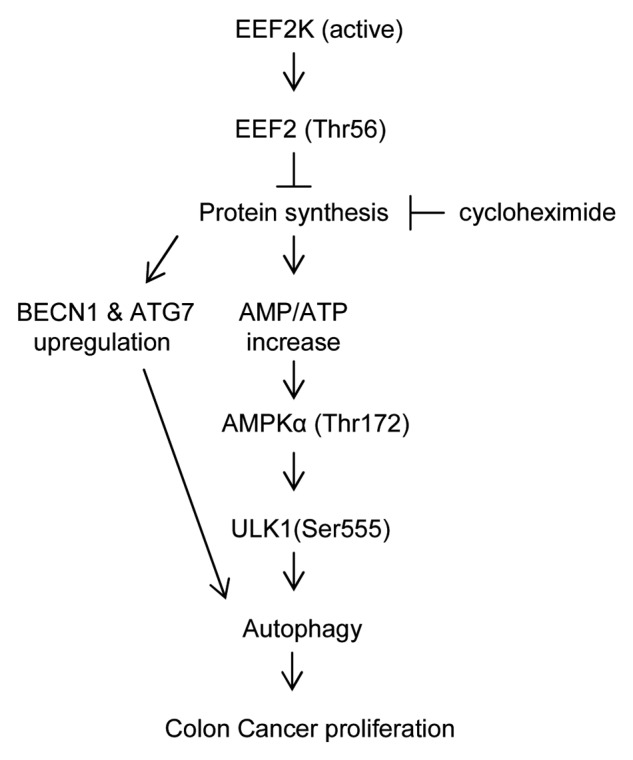
Figure 7. Signaling connections involved in autophagy pathways sensitive to EEF2K silencing in colon cancer cells. EEF2K can directly inactivate EEF2 by phosphorylation at Thr56, which negatively regulates peptide chain elongation. Silencing of EEF2K can upregulate the protein levels of BECN1 and ATG7. The upregulation of protein synthesis by EEF2K silencing can downregulate ATP level and increase AMP/ATP ratio. This in turn directly activates AMPKα by phosphorylation at Thr172 and then activates ULK1 by phosphorylation at Ser555 leading to autophagy. Autophagy induced by EEF2K silencing can promote human colon cancer cell proliferation. Inhibition of protein synthesis by cycloheximide can attenuate ULK1 activation and autophagy generation induced by EEF2K silencing. Arrows represent promotion events, blunt arrows indicate suppression events.
Materials and Methods
Reagents and antibodies
Alexa Fluor 488 donkey anti-rabbit lgG (A21206) antibody was purchased from Life Technologies Corporation. Fetal bovine serum (16000-044) was purchased from Gibco Invitrogen. McCoy’s 5A medium (M4892), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; M2128), cycloheximide (C7698), MG132 (M7449), pepstatin A (P5318) and anti-LC3B antibody (L7543) were obtained from Sigma. Anti-BECN1 (sc-10086), anti-SQSTM1/p62 (H-290) (sc-25575), anti-mouse lgG-HRP (sc-2005) and anti-rabbit lgG-HRP (sc-2004) antibodies were obtained from Santa Cruz Biotechnology. Chemiluminescence HRP substrate (WBKLS0500) was purchased from Millipore. Anti-AMPKα (#2532S), anti-phospho-AMPKα (Thr172) (#2535), anti-phospho-ULK1 (Ser757) (#6888), anti-phospho-MTOR (Ser2448) (#2971), anti-ATG5 (#2630), anti-ATG7 (#2631), anti-EEF2 (#2332), anti-phospho-EEF2 (Thr56) (#2331), anti-ULK1 (D8H5) (#8054), anti-phospho-ULK1 (Ser555) (D1H4) (#5869) and anti-EEF2K (#3692) antibodies were obtained from Cell Signaling Technology.
Overexpression of human EEF2K
A plasmid pDONR223-EEF2K containing full-length of human EEF2K coding region was obtained from Addgene (Addgene plasmid 23726, USA).44 Amplification of the coding region was performed by PCR using GeneAmp High Fidelity Enzyme Mix (Life Technologies, 4328216). The PCR conditions were denaturation at 94 °C for 3 min, followed by 20 cycles of 94 °C for 30 s, 55 °C for 30 s and 72 °C for 2 min 30 s, with a final extension step at 72 °C for 10 min. The products were purified using the QIAquick Gel Extraction Kit (Qiagen, 28706) and then inserted into a pcDNA3.1/V5-His TOPO TA expression vector (Life Technologies, K4800-01). The resultant construct encompassing EEF2K with V5 and polyhistidine epitope tags was confirmed by sequencing.
Cell culture
The human colon cancer HT-29 (HTB-38) and HCT-116 (CCL-247) cells purchased from American Type Culture Collection (ATCC) were cultured in complete McCoy’s 5A medium (supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin). All experiments were performed in HT-29 or HCT-116 cells between passage 10 and 20.
Small interfering RNA (siRNA) transfection
ON-TARGETplus SMART pool against human EEF2K siRNA (L004950-00-0005) was obtained from Dharmacon, Inc. Other siRNAs were purchased from Genepharm. EEF2K silencing was performed using an siRNA duplex targeting the following sequence: 5′- AAGCUCGAAC CAGAAUGUCA A-3′.26 BECN1 was silenced using siRNA duplexes targeting the following sequences: 5′-GAUACCGACU UGUUCCUUA-3′ and 5′-CUAAGGAGCU GCCGUUAUA-3′. ATG7 was targeted with siRNA duplexes targeting the following sequences: 5′-CCAACACACU CGAGUCUUU-3′ and 5′-GCCCACAGAU GGAGUAGCA-3′. PRKAA1 and PRKAA2/AMPKα were silenced with siRNA duplexes targeting the following sequences: GAGGAGAGCU AUUUGAUUA and GCUGUUUGGU GUAGGUAAA, respectively.45 ULK1 was targeted with siRNA duplexes targeting the following sequences: 5′-GUGGCCCUGU ACGACUUCCA GGAAA-3′ and 5′-GAGCAAGAGC ACACGGAAA-3′.46,47 A nontargeting siRNA was used as a control with sense (5′-UCUACGAGGC ACGAGACUU-3′) and antisense (5′-AAGUCUCGUG CCUCGUAGA-3′).48 In brief, cells were transfected in McCoy 5A medium with 90 nM of each siRNA duplex using DharmaFECT transfection reagent according to the manufacturer’s protocol.
Immunofluorescence staining
Cells were grown on slides and transfected with siRNAs. After 48 h transfection, cells were washed 3 times with PBS (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.46 mM KH2PO4, pH 7.4), fixed with 3.5% formaldehyde in PBS for 10 min, washed once with PBS, permeabilized with 0.1% Triton X-100 (USB, 22686) in PBS for 10 min, and blocked with 0.5% BSA (Sigma, A2153) in PBS for 15 min. Cells were incubated with LC3 antibody (1:150) for 2 h at room temperature, followed by incubation with Alexa Fluor 488 antibody (1:200) for 1 h at room temperature. All antibodies were diluted with 0.5% BSA in PBS. Slides were mounted with Vectashield mounting medium and images were taken with an Olympus FV1000 confocal microscope (Olympus, PA, USA) using a 60 × 1.35 NA oil objective.
RNA isolation and reverse transcription–polymerase chain reaction (RT-PCR)
Total RNA was isolated by Trizol (Life Technologies; 15596018). RT-PCR (reverse transcription polymerase chain reaction) was performed with PrimeScript™ RT reagent Kit (Takara; RR037A) according to the manufacturer’s instructions. To detect the mRNA levels of ATG5 and BECN1, primers used were as follows:14 ATG5 forward, 5′-AGCAACTCTG GATGGGATTG-3′; reverse, 5′-CACTGCAGAG GTGTTTCCAA-3′; BECN1 forward, 5′-GGCCAATAAG ATGGGTCTGA-3′; reverse, 5′-GCTTTTGTCC ACTGCTCCTC-3′; ATG7 forward, 5′-ACCCAGAAGA AGCTGAACGA-3′; reverse, 5′-TGCAAATGTC AAGAGGAGGA-3′; EEF2K forward, 5′-ATGTACTCGC AGTTGCCT-3′; reverse, 5′-TGCATTCCGT CGTACTCAC-3′; GAPDH forward, 5′-AAGGTCGGAG TCAACGGATT-3′; reverse, 5′-CCATGGGTGG AATCATATTG-3′. GAPDH was used as internal control.
Clonogenic assay
For clonogenic assay, cells were transfected with control siRNA (siCTL), EEF2K siRNA (siEEF2K), BECN1 siRNA (siBECN1), ATG7 siRNA (siATG7), siBECN1 plus siEEF2K, siATG7 plus siEEF2K, empty vector (Vector), or EEF2K plasmids (EEF2K) for 48 h, and then the cells were seeded out in appropriate dilutions into 6-well plates, followed by incubation at 37 °C for 12 to 14 d. Colonies were fixed with glutaraldehyde (6.0% v/v), stained with crystal violet (0.5% w/v) and imaged. Colonies with 50 or more cells were counted.
ANXA5 (annexin V) and propidium iodide (PI) staining
Cells were transfected with control siRNA, EEF2K siRNA (siEEF2K), empty vector (Vector), or EEF2K plasmids (EEF2K) for 24 h, and then the cells were treated with vehicle (0.1% DMSO) or oxaliplatin (Sigma, O9512) for 48 h, washed with PBS, incubated in the binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, 0.1% BSA, pH 7.4) containing ANXA5-FITC for 15 min.49 Cells were immediately exposed to 2 μg/ml PI (Sigma, P4170) before the analysis on a FACScan flow cytometer (Becton Dickinson, San Jose, CA).
Western blot analysis
Cell extracts were prepared for western blot. In brief, cells were lysed with lysis buffer (20 mM TRIS-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM DTT, 1 mM sodium orthovanadate [Sigma, S6508], 1 µg/ml leupeptin [Sigma, L2884], 1 mM phenylmethylsulfonyl fluoride [Sigma, P7626]) for 1 h on ice. The total proteins were analyzed on a 12% gel by SDS-PAGE, transfered onto a nitrocellulose membrane, blocked with 5% BSA in TBST buffer (Tris-buffered saline [50 mM Tris, 150 mM NaCl], pH 7.5, containing 0.1% Tween-20), incubated with primary antibodies at 4 °C overnight, washed 3 times for 15 min each in TBST at room temperature, incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature, and washed 3 times for 15 min each in TBST. The bands were detected using the Chemiluminescence HRP Substrate (Millipore, WBKLS0500). The relative band intensity was quantified using the AlphaEaseFC software version 6.0.0.
Measurement of cellular ATP
Cells were seeded into 96-well plates at 4.0 × 103 cells per well and transfected with nontargeting control siRNA or siRNA targeting EEF2K for 48 h. ATP contents were measured using the ATPlite Luminescence Assay Kit (PerkinElmer, 6016943) according to the manufacturer's protocol. The relative ATP level was normalized to control samples.26
ROS analysis
ROS generation in cells after EEF2K silencing was analyzed. In brief, cells were transfected with nontargeting control siRNA or EEF2K siRNA for 48 h, and then stained with 20 μM 2′,7′-dichlorofluorescein diacetate (DCFDA; Sigma, D6883) for 30 min in the dark. For morphological study, the cells were imaged under a Nikon TE2000 fluorescence microscope (Nikon, Melville, NY, USA). For quantifying the ROS levels, the cells were analyzed using a flow cytometer (Becton-Dickinson, CA, USA) in FL1 channel.
Statistical analysis
Statistical analysis was performed using the 2-tailed Student t test for comparison of 2 groups or one-way analysis of variance for comparison of more than 2 groups followed by the Tukey multiple comparison test. For multiple testing, the P values were determined using a 2-way analysis of variance with Bonferroni post-test. All statistical analyses were performed using the GraphPad Prism software version 5.01 (GraphPad, San Diego, CA). Data were expressed as mean ± standard error of the mean (SEM) of at least 3 independent experiments. A P value < 0.05 was considered statistically significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by a Strategic Investments Scheme from the Chinese University of Hong Kong. We thank the Chinese University of Hong Kong for the provision of Direct Grants. We also thank the depositors of the Addgene plasmid pDONR223-EEF2K (#23726), Dr William Hahn and Dr David Root of Broad Institute of Harvard and Massachusetts Institute of Technology, Massachusetts.
Glossary
Abbreviations:
- AMPK
AMP-activated protein kinase
- AMPKα
PRKAA1 and PRKAA2
- ATG
autophagy-related genes
- CHX
cycloheximide
- DCFDA
2′,7′-dichlorofluorescein diacetate
- EEF2
eukaryotic translation elongation factor 2
- EEF2K
eukaryotic elongation factor-2 kinase
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MAPK13/p38δ/SAPK4
mitogen-activated protein kinase 13
- MEFs
mouse embryonic fibroblasts
- MTOR
mechanistic target of rapamycin
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
- PRKA
protein kinase A
- ROS
reactive oxygen species
- RPS6KA1/p90/RSK1
ribosomal protein S6 kinase, 90 kDa, polypeptide 1
- RPS6KB
ribosomal protein S6 kinase, 70 kDa, polypeptide
- SEM
standard error of the mean
- ULK1
unc-51 like autophagy activating kinase 1
References
- 1.Nairn AC, Bhagat B, Palfrey HC. Identification of calmodulin-dependent protein kinase III and its major Mr 100,000 substrate in mammalian tissues. Proc Natl Acad Sci U S A. 1985;82:7939–43. doi: 10.1073/pnas.82.23.7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Browne GJ, Proud CG. Regulation of peptide-chain elongation in mammalian cells. Eur J Biochem. 2002;269:5360–8. doi: 10.1046/j.1432-1033.2002.03290.x. [DOI] [PubMed] [Google Scholar]
- 3.Diggle TA, Seehra CK, Hase S, Redpath NT. Analysis of the domain structure of elongation factor-2 kinase by mutagenesis. FEBS Lett. 1999;457:189–92. doi: 10.1016/S0014-5793(99)01034-0. [DOI] [PubMed] [Google Scholar]
- 4.Browne GJ, Finn SG, Proud CG. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J Biol Chem. 2004;279:12220–31. doi: 10.1074/jbc.M309773200. [DOI] [PubMed] [Google Scholar]
- 5.Hovland R, Eikhom TS, Proud CG, Cressey LI, Lanotte M, Døskeland SO, Houge G. cAMP inhibits translation by inducing Ca2+/calmodulin-independent elongation factor 2 kinase activity in IPC-81 cells. FEBS Lett. 1999;444:97–101. doi: 10.1016/S0014-5793(99)00039-3. [DOI] [PubMed] [Google Scholar]
- 6.Diggle TA, Subkhankulova T, Lilley KS, Shikotra N, Willis AE, Redpath NT. Phosphorylation of elongation factor-2 kinase on serine 499 by cAMP-dependent protein kinase induces Ca2+/calmodulin-independent activity. Biochem J. 2001;353:621–6. doi: 10.1042/0264-6021:3530621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carlberg U, Nilsson A, Nygård O. Functional properties of phosphorylated elongation factor 2. Eur J Biochem. 1990;191:639–45. doi: 10.1111/j.1432-1033.1990.tb19169.x. [DOI] [PubMed] [Google Scholar]
- 8.Ryazanov AG, Shestakova EA, Natapov PG. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature. 1988;334:170–3. doi: 10.1038/334170a0. [DOI] [PubMed] [Google Scholar]
- 9.Knebel A, Morrice N, Cohen P. A novel method to identify protein kinase substrates: eEF2 kinase is phosphorylated and inhibited by SAPK4/p38delta. EMBO J. 2001;20:4360–9. doi: 10.1093/emboj/20.16.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001;20:4370–9. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Browne GJ, Proud CG. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol Cell Biol. 2004;24:2986–97. doi: 10.1128/MCB.24.7.2986-2997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L, Chen Y, Gibson SB. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal. 2013;25:50–65. doi: 10.1016/j.cellsig.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 13.Bhutia SK, Kegelman TP, Das SK, Azab B, Su ZZ, Lee SG, Sarkar D, Fisher PB. Astrocyte elevated gene-1 induces protective autophagy. Proc Natl Acad Sci U S A. 2010;107:22243–8. doi: 10.1073/pnas.1009479107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie CM, Chan WY, Yu S, Zhao J, Cheng CH. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic Biol Med. 2011;51:1365–75. doi: 10.1016/j.freeradbiomed.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez-Rocha H, Garcia-Garcia A, Panayiotidis MI, Franco R. DNA damage and autophagy. Mutat Res. 2011;711:158–66. doi: 10.1016/j.mrfmmm.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng Y, Ren X, Zhang Y, Patel R, Sharma A, Wu H, Robertson GP, Yan L, Rubin E, Yang JM. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by Akt Inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK-2206. Cancer Res. 2011;71:2654–63. doi: 10.1158/0008-5472.CAN-10-2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tekedereli I, Alpay SN, Tavares CD, Cobanoglu ZE, Kaoud TS, Sahin I, Sood AK, Lopez-Berestein G, Dalby KN, Ozpolat B. Targeted silencing of elongation factor 2 kinase suppresses growth and sensitizes tumors to doxorubicin in an orthotopic model of breast cancer. PLoS One. 2012;7:e41171. doi: 10.1371/journal.pone.0041171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Y, Yan L, Ren X, Yang JM. eEF-2 kinase, another meddler in the “yin and yang” of Akt-mediated cell fate? Autophagy. 2011;7:660–1. doi: 10.4161/auto.7.6.15385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hait WN, Wu H, Jin S, Yang JM. Elongation factor-2 kinase: its role in protein synthesis and autophagy. Autophagy. 2006;2:294–6. doi: 10.4161/auto.2857. [DOI] [PubMed] [Google Scholar]
- 20.Wu H, Zhu H, Liu DX, Niu TK, Ren X, Patel R, Hait WN, Yang JM. Silencing of elongation factor-2 kinase potentiates the effect of 2-deoxy-D-glucose against human glioma cells through blunting of autophagy. Cancer Res. 2009;69:2453–60. doi: 10.1158/0008-5472.CAN-08-2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu H, Yang JM, Jin S, Zhang H, Hait WN. Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res. 2006;66:3015–23. doi: 10.1158/0008-5472.CAN-05-1554. [DOI] [PubMed] [Google Scholar]
- 22.Py BF, Boyce M, Yuan J. A critical role of eEF-2K in mediating autophagy in response to multiple cellular stresses. Autophagy. 2009;5:393–6. doi: 10.4161/auto.5.3.7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Z, Gopalakrishnan SM, Bui MH, Soni NB, Warrior U, Johnson EF, Donnelly JB, Glaser KB. 1-Benzyl-3-cetyl-2-methylimidazolium iodide (NH125) induces phosphorylation of eukaryotic elongation factor-2 (eEF2): a cautionary note on the anticancer mechanism of an eEF2 kinase inhibitor. J Biol Chem. 2011;286:43951–8. doi: 10.1074/jbc.M111.301291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryazanov AG. Elongation factor-2 kinase and its newly discovered relatives. FEBS Lett. 2002;514:26–9. doi: 10.1016/S0014-5793(02)02299-8. [DOI] [PubMed] [Google Scholar]
- 25.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–8. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Y, Li H, Ren X, Niu T, Hait WN, Yang J. Cytoprotective effect of the elongation factor-2 kinase-mediated autophagy in breast cancer cells subjected to growth factor inhibition. PLoS One. 2010;5:e9715. doi: 10.1371/journal.pone.0009715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng Y, Ren X, Zhang Y, Shan Y, Huber-Keener KJ, Zhang L, Kimball SR, Harvey H, Jefferson LS, Yang JM. Integrated regulation of autophagy and apoptosis by EEF2K controls cellular fate and modulates the efficacy of curcumin and velcade against tumor cells. Autophagy. 2013;9:208–19. doi: 10.4161/auto.22801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Podrabsky JE, Hand SC. Depression of protein synthesis during diapause in embryos of the annual killifish Austrofundulus limnaeus. Physiol Biochem Zool. 2000;73:799–808. doi: 10.1086/318106. [DOI] [PubMed] [Google Scholar]
- 29.Liu Q, Gauthier MS, Sun L, Ruderman N, Lodish H. Activation of AMP-activated protein kinase signaling pathway by adiponectin and insulin in mouse adipocytes: requirement of acyl-CoA synthetases FATP1 and Acsl1 and association with an elevation in AMP/ATP ratio. FASEB J. 2010;24:4229–39. doi: 10.1096/fj.10-159723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo L, Stripay JL, Zhang X, Collage RD, Hulver M, Carchman EH, Howell GM, Zuckerbraun BS, Lee JS, Rosengart MR. CaMKIα regulates AMP kinase-dependent, TORC-1-independent autophagy during lipopolysaccharide-induced acute lung neutrophilic inflammation. J Immunol. 2013;190:3620–8. doi: 10.4049/jimmunol.1102975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wullner U, Neef I, Eller A, Kleines M, Tur MK, Barth S. Cell-specific induction of apoptosis by rationally designed bivalent aptamer-siRNA transcripts silencing eukaryotic elongation factor 2. Curr Cancer Drug Targets. 2008;8:554–65. doi: 10.2174/156800908786241078. [DOI] [PubMed] [Google Scholar]
- 34.Berryman S, Brooks E, Burman A, Hawes P, Roberts R, Netherton C, Monaghan P, Whelband M, Cottam E, Elazar Z, et al. Foot-and-mouth disease virus induces autophagosomes during cell entry via a class III phosphatidylinositol 3-kinase-independent pathway. J Virol. 2012;86:12940–53. doi: 10.1128/JVI.00846-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aoki H, Kondo Y, Aldape K, Yamamoto A, Iwado E, Yokoyama T, Hollingsworth EF, Kobayashi R, Hess K, Shinojima N, et al. Monitoring autophagy in glioblastoma with antibody against isoform B of human microtubule-associated protein 1 light chain 3. Autophagy. 2008;4:467–75. doi: 10.4161/auto.5668. [DOI] [PubMed] [Google Scholar]
- 36.Tafani M, Schito L, Anwar T, Indelicato M, Sale P, Di Vito M, Morgante E, Beraldi R, Makovec F, Letari O, et al. Induction of autophagic cell death by a novel molecule is increased by hypoxia. Autophagy. 2008;4:1042–53. doi: 10.4161/auto.7070. [DOI] [PubMed] [Google Scholar]
- 37.Uchiyama Y, Shibata M, Koike M, Yoshimura K, Sasaki M. Autophagy-physiology and pathophysiology. Histochem Cell Biol. 2008;129:407–20. doi: 10.1007/s00418-008-0406-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol. 2012;30:671–8. doi: 10.1038/nbt.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leprivier G, Remke M, Rotblat B, Dubuc A, Mateo AR, Kool M, Agnihotri S, El-Naggar A, Yu B, Somasekharan SP, et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell. 2013;153:1064–79. doi: 10.1016/j.cell.2013.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 41.Chang YY, Juhász G, Goraksha-Hicks P, Arsham AM, Mallin DR, Muller LK, Neufeld TP. Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem Soc Trans. 2009;37:232–6. doi: 10.1042/BST0370232. [DOI] [PubMed] [Google Scholar]
- 42.Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, Hsu E, Liu X, Eisenberg J, Liu J, Blenis J, et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell. 2010;18:1041–52. doi: 10.1016/j.devcel.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skrzypczak M, Goryca K, Rubel T, Paziewska A, Mikula M, Jarosz D, Pachlewski J, Oledzki J, Ostrowski J. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS One. 2010;5:e13091. doi: 10.1371/journal.pone.0013091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakano A, Kato H, Watanabe T, Min KD, Yamazaki S, Asano Y, Seguchi O, Higo S, Shintani Y, Asanuma H, et al. AMPK controls the speed of microtubule polymerization and directional cell migration through CLIP-170 phosphorylation. Nat Cell Biol. 2010;12:583–90. doi: 10.1038/ncb2060. [DOI] [PubMed] [Google Scholar]
- 46.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ganley IG, Lam H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie CM, Liu XY, Yu S, Cheng CH. Cardiac glycosides block cancer growth through HIF-1α- and NF-κB-mediated Plk1. Carcinogenesis. 2013;34:1870–80. doi: 10.1093/carcin/bgt136. [DOI] [PubMed] [Google Scholar]
- 49.Mok CF, Xie CM, Sham KW, Lin ZX, Cheng CH. 1,4-dihydroxy-2-naphthoic Acid Induces Apoptosis in Human Keratinocyte: Potential Application for Psoriasis Treatment. Evid Based Complement Alternat Med. 2013;2013:792840. doi: 10.1155/2013/792840. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.